

Monomeric C-Reactive Protein in Atherosclerotic Cardiovascular Disease: Advances and Perspectives

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. NLRP3 Inflammasome and Inflammatory IL-1β/IL-6/CRP Axis in Pathogenesis of Subclinical Vascular Inflammation
3. Assessment of Residual Inflammatory Risk
3.1. C-Reactive Protein as a Biomarker of Subclinical Vascular Inflammation
3.2. Pentameric C-Reactive Protein
3.3. Monomeric C-Reactive Protein
3.4. Level of Monomeric C-Reactive Protein in Health and Disease
4. Treatment to Reduce CRP Level and Ameliorate Residual Inflammatory Risk
4.1. Lifestyle Modifications
4.2. Statins
4.3. Other Lipid-Lowering Agents
4.4. Anti-Inflammatory Agents
4.4.1. Upstream Agents
4.4.2. Anti-CRP Agents
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of Lipids on Cardiovascular Health. J. Am. Coll. Cardiol. 2018, 72, 1141–1156. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Williams, K.J.; Gidding, S.; Borén, J.; Tabas, I.; Fisher, E.A.; Packard, C.; Pencina, M.; Fayad, Z.A.; Mani, V.; et al. Eradicating the Burden of Atherosclerotic Cardiovascular Disease by Lowering Apolipoprotein B Lipoproteins Earlier in Life. JAHA 2018, 7, e009778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giugliano, R.P.; Pedersen, T.R.; Park, J.-G.; De Ferrari, G.M.; Gaciong, Z.A.; Ceska, R.; Toth, K.; Gouni-Berthold, I.; Lopez-Miranda, J.; Schiele, F.; et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Gabriel Steg, P.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Kim, Y.-U.; Li, Q.H.; Manvelian, G.; et al. Clinical Efficacy and Safety of Alirocumab After Acute Coronary Syndrome According to Achieved Level of Low-Density Lipoprotein Cholesterol: A Propensity Score–Matched Analysis of the ODYSSEY OUTCOMES Trial. Circulation 2021, 143, 1109–1122. [Google Scholar] [CrossRef]
- Dimmitt, S.B.; Stampfer, H.G.; Martin, J.H.; Warren, J.B. Clinical benefits of evolocumab appear less than hoped. Lancet 2018, 391, 933–934. [Google Scholar] [CrossRef] [Green Version]
- Lawler, P.R.; Bhatt, D.L.; Godoy, L.C.; Lüscher, T.F.; Bonow, R.O.; Verma, S.; Ridker, P.M. Targeting cardiovascular inflammation: Next steps in clinical translation. Eur. Heart J. 2020, 42, ehaa099. [Google Scholar] [CrossRef]
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur. Heart J. 2016, 37, 1720–1722. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Kiefer, J.; Braig, D.; Loseff-Silver, J.; Potempa, L.A.; Eisenhardt, S.U.; Peter, K. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front. Immunol. 2018, 9, 1351. [Google Scholar] [CrossRef]
- Rajab, I.M.; Hart, P.C.; Potempa, L.A. How C-Reactive Protein Structural Isoforms With Distinctive Bioactivities Affect Disease Progression. Front. Immunol. 2020, 11, 2126. [Google Scholar] [CrossRef] [PubMed]
- Dix, C.; Zeller, J.; Stevens, H.; Eisenhardt, S.U.; Shing, K.S.C.T.; Nero, T.L.; Morton, C.J.; Parker, M.W.; Peter, K.; McFadyen, J.D. C-reactive protein, immunothrombosis and venous thromboembolism. Front. Immunol. 2022, 13, 1002652. [Google Scholar] [CrossRef]
- Potempa, L.A.; Rajab, I.M.; Olson, M.E.; Hart, P.C. C-Reactive Protein and Cancer: Interpreting the Differential Bioactivities of Its Pentameric and Monomeric, Modified Isoforms. Front. Immunol. 2021, 12, 744129. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Zeller, J.; Potempa, L.A.; Pietersz, G.A.; Eisenhardt, S.U.; Peter, K. C-Reactive Protein and Its Structural Isoforms: An Evolutionary Conserved Marker and Central Player in Inflammatory Diseases and Beyond. In Vertebrate and Invertebrate Respiratory Proteins, Lipoproteins and other Body Fluid Proteins; Hoeger, U., Harris, J.R., Eds.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2020; Volume 94, pp. 499–520. ISBN 978-3-030-41768-0. [Google Scholar]
- Luan, Y.; Yao, Y. The Clinical Significance and Potential Role of C-Reactive Protein in Chronic Inflammatory and Neurodegenerative Diseases. Front. Immunol. 2018, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Burger, F.; Baptista, D.; Roth, A.; da Silva, R.F.; Montecucco, F.; Mach, F.; Brandt, K.J.; Miteva, K. NLRP3 Inflammasome Activation Controls Vascular Smooth Muscle Cells Phenotypic Switch in Atherosclerosis. Int. J. Mol. Sci. 2021, 23, 340. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Sig. Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Silvis, M.J.M.; Demkes, E.J.; Fiolet, A.T.L.; Dekker, M.; Bosch, L.; van Hout, G.P.J.; Timmers, L.; de Kleijn, D.P.V. Immunomodulation of the NLRP3 Inflammasome in Atherosclerosis, Coronary Artery Disease, and Acute Myocardial Infarction. J. Cardiovasc. Trans. Res. 2021, 14, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T.; et al. Inflammasome Activation by Mitochondrial Oxidative Stress in Macrophages Leads to the Development of Angiotensin II–Induced Aortic Aneurysm. ATVB 2015, 35, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome Activation of Cardiac Fibroblasts Is Essential for Myocardial Ischemia/Reperfusion Injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity: Inflammasomes in kidney disease. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jang, H.; Fang, J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 2019, 9, 850. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Yang, X.-Y.; Xu, G.; Zheng, T.; Jin, S. CRP-Induced NLRP3 Inflammasome Activation Increases LDL Transcytosis Across Endothelial Cells. Front. Pharmacol. 2019, 10, 40. [Google Scholar] [CrossRef]
- Ridker, P.M. Anticytokine Agents: Targeting Interleukin Signaling Pathways for the Treatment of Atherothrombosis. Circ Res 2019, 124, 437–450. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regener. 2019, 39, 12. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S. Interleukin-6 signalling in health and disease. F1000Research 2020, 9, 1013. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Kaplanski, G. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Roldan, V. Interleukin-6, endothelial activation and thrombogenesis in chronic atrial fibrillation. Eur. Heart J. 2003, 24, 1373–1380. [Google Scholar] [CrossRef]
- Morimoto, S.; Nabata, T.; Koh, E.; Shiraishi, T.; Fukuo, K.; Imanaka, S.; Kitano, S.; Miyashita, Y.; Ogihara, T. Interleukin-6 Stimulates Proliferation of Cultured Vascular Smooth Muscle Cells Independently of Interleukin-1β. J. Cardiovasc. Pharmacol. 1991, 17, S117–S118. [Google Scholar] [CrossRef] [PubMed]
- Gierens, H.; Nauck, M.; Roth, M.; Schinker, R.; Schürmann, C.; Scharnagl, H.; Neuhaus, G.; Wieland, H.; März, W. Interleukin-6 Stimulates LDL Receptor Gene Expression via Activation of Sterol-Responsive and Sp1 Binding Elements. ATVB 2000, 20, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousoulis, D.; Oikonomou, E.; Economou, E.K.; Crea, F.; Kaski, J.C. Inflammatory cytokines in atherosclerosis: Current therapeutic approaches. Eur. Heart J. 2016, 37, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, N.; Fahey, J.L.; Detels, R.; Butch, A.W. Analytical Performance of a Highly Sensitive C-Reactive Protein-Based Immunoassay and the Effects of Laboratory Variables on Levels of Protein in Blood. Clin. Vaccine Immunol. 2003, 10, 652–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clyne, B.; Olshaker, J.S. The C-reactive protein. J. Emerg. Med. 1999, 17, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Póvoa, P.; Almeida, E.; Moreira, P.; Fernandes, A.; Mealha, R.; Aragão, A.; Sabino, H. C-reactive protein as an indicator of sepsis. Intensive Care Med. 1998, 24, 1052–1056. [Google Scholar] [CrossRef]
- Eda, S.; Kaufmann, J.; Molwitz, M.; Vorberg, E. A new method of measuring C-reactive protein, with a low limit of detection, suitable for risk assessment of coronary heart disease. Scand. J. Clin. Lab Investig. Suppl. 1999, 230, 32–35. [Google Scholar] [CrossRef]
- Buckley, D.I.; Fu, R.; Freeman, M.; Rogers, K.; Helfand, M. C-Reactive Protein as a Risk Factor for Coronary Heart Disease: A Systematic Review and Meta-analyses for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2009, 151, 483. [Google Scholar] [CrossRef] [Green Version]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O.; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L.; et al. Markers of Inflammation and Cardiovascular Disease: Application to Clinical and Public Health Practice: A Statement for Healthcare Professionals From the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-Reactive Protein Levels and Outcomes after Statin Therapy. N. Engl. J. Med. 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Crowe, T.; Sasiela, W.J.; Tsai, J.; Orazem, J.; Magorien, R.D.; O’Shaughnessy, C.; Ganz, P. Statin Therapy, LDL Cholesterol, C-Reactive Protein, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; de Lemos, J.A.; Sabatine, M.S.; Wiviott, S.D.; Blazing, M.A.; Shui, A.; Rifai, N.; Califf, R.M.; Braunwald, E. Clinical Relevance of C-Reactive Protein During Follow-Up of Patients With Acute Coronary Syndromes in the Aggrastat-to-Zocor Trial. Circulation 2006, 114, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, Aspirin, and the Risk of Cardiovascular Disease in Apparently Healthy Men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- MRFIT Research Group; Kuller, L.H.; Tracy, R.P.; Shaten, J.; Meilahn, E.N. Relation of C-Reactive Protein and Coronary Heart Disease in the MRFIT Nested Case-Control Study. Am. J. Epidemiol. 1996, 144, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Emerging Risk Factors Collaboration C-Reactive Protein, Fibrinogen, and Cardiovascular Disease Prediction. N. Engl. J. Med. 2012, 367, 1310–1320. [CrossRef] [Green Version]
- Saito, I.; Maruyama, K.; Eguchi, E. C-Reactive Protein and Cardiovascular Disease in East Asians: A Systematic Review. Clin. Med. Insights Cardiol. 2014, 8, CMC-S17066. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.-M.; Lü, S.-Q.; Liu, Z.-P.; Zhang, J.; Gao, B.-X.; Yao, Z.-Y.; Wu, Y.-X.; Potempa, L.A.; Ji, S.-R.; Long, M.; et al. Conformational folding and disulfide bonding drive distinct stages of protein structure formation. Sci. Rep. 2018, 8, 1494. [Google Scholar] [CrossRef] [Green Version]
- Volanakis, J.E.; Wirtz, K.W.A. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature 1979, 281, 155–157. [Google Scholar] [CrossRef]
- Thompson, D.; Pepys, M.B.; Wood, S.P. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure 1999, 7, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Hack, C.E.; Wolbink, G.-J.; Schalkwijk, C.; Speijer, H.; Hermens, W.T.; van den Bosch, H. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol. Today 1997, 18, 111–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Huang, Y.H.; Elinder, L.S.; Frostegård, J. Lysophosphatidylcholine Is Involved in the Antigenicity of Oxidized LDL. ATVB 1998, 18, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Law, S.-H.; Chan, M.-L.; Marathe, G.K.; Parveen, F.; Chen, C.-H.; Ke, L.-Y. An Updated Review of Lysophosphatidylcholine Metabolism in Human Diseases. Int. J. Mol. Sci. 2019, 20, 1149. [Google Scholar] [CrossRef] [Green Version]
- Corrêa, R.; Silva, L.F.F.; Ribeiro, D.J.S.; das Neves Almeida, R.; de Oliveira Santos, I.; Corrêa, L.H.; de Sant’Ana, L.P.; Assunção, L.S.; Bozza, P.T.; Magalhães, K.G. Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front. Immunol. 2020, 10, 2927. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-Y.; Dong, Y.-Q.; Wang, P.; Zhang, X.; Yan, Y.; Sun, L.; Liu, B.; Zhang, D.; Zhang, H.; Liu, H.; et al. Adipocyte-derived Lysophosphatidylcholine Activates Adipocyte and Adipose Tissue Macrophage Nod-Like Receptor Protein 3 Inflammasomes Mediating Homocysteine-Induced Insulin Resistance. EBioMedicine 2018, 31, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Clos, T.W.; Mold, C. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcγ receptors. Curr. Opin. Organ Transplant. 2011, 16, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Du Clos, T.W. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol. Biol. Rep. 1996, 23, 253–260. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.-G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef]
- Ji, S.; Wu, Y.; Zhu, L.; Potempa, L.A.; Sheng, F.; Lu, W.; Zhao, J. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRPm. FASEB J. 2007, 21, 284–294. [Google Scholar] [CrossRef]
- Zhang, C.-M.; Tan, Y.-B.; Zhou, H.-H.; Ge, Z.-B.; Feng, J.-R.; Lv, G.-B.; Sun, Z.-Y.; Fu, Y.; Wang, M.-Y. Intra-subunit Disulfide Determines the Conversion and Structural Stability of CRP Isoforms. Inflammation 2020, 43, 466–477. [Google Scholar] [CrossRef]
- Li, H.-Y.; Wang, J.; Meng, F.; Jia, Z.-K.; Su, Y.; Bai, Q.-F.; Lv, L.-L.; Ma, F.-R.; Potempa, L.A.; Yan, Y.-B.; et al. An Intrinsically Disordered Motif Mediates Diverse Actions of Monomeric C-reactive Protein. J. Biol. Chem. 2016, 291, 8795–8804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khreiss, T.; József, L.; Hossain, S.; Chan, J.S.D.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry of C-reactive Protein Is Associated with Delayed Apoptosis of Human Neutrophils. J. Biol. Chem. 2002, 277, 40775–40781. [Google Scholar] [CrossRef] [Green Version]
- Habersberger, J.; Strang, F.; Scheichl, A.; Htun, N.; Bassler, N.; Merivirta, R.-M.; Diehl, P.; Krippner, G.; Meikle, P.; Eisenhardt, S.U.; et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc. Res. 2012, 96, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, J.R.; Trial, J.; Nambi, V.; Hoogeveen, R.C.; Taffet, G.E.; Entman, M.L. Plasma Levels of Endothelial Microparticles Bearing Monomeric C-reactive Protein are Increased in Peripheral Artery Disease. J. Cardiovasc. Trans. Res. 2016, 9, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnikov, I.; Kozlov, S.; Saburova, O.; Zubkova, E.; Guseva, O.; Domogatsky, S.; Arefieva, T.; Radyukhina, N.; Zvereva, M.; Avtaeva, Y.; et al. CRP Is Transported by Monocytes and Monocyte-Derived Exosomes in the Blood of Patients with Coronary Artery Disease. Biomedicines 2020, 8, 435. [Google Scholar] [CrossRef]
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; ten Cate-Hoek, A.J.; ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [Green Version]
- Molins, B.; Peña, E.; Vilahur, G.; Mendieta, C.; Slevin, M.; Badimon, L. C-Reactive Protein Isoforms Differ in Their Effects on Thrombus Growth. ATVB 2008, 28, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Molins, B.; Peña, E.; de la Torre, R.; Badimon, L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc. Res. 2011, 92, 328–337. [Google Scholar] [CrossRef]
- de la Torre, R.; Peña, E.; Vilahur, G.; Slevin, M.; Badimon, L. Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: Pentraxins and platelet deposition. J. Thromb. Haemost. 2013, 11, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Khreiss, T.; József, L.; Potempa, L.A.; Filep, J.G. Conformational Rearrangement in C-Reactive Protein Is Required for Proinflammatory Actions on Human Endothelial Cells. Circulation 2004, 109, 2016–2022. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ren, M.; Luo, M.; Chen, N.; Zhang, Z.; Luo, B.; Wu, J. Monomeric C-reactive protein alters fibrin clot properties on endothelial cells. Thromb. Res. 2012, 129, e251–e256. [Google Scholar] [CrossRef]
- Zouki, C.; Haas, B.; Chan, J.S.D.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry of C-Reactive Protein Is Associated with Promotion of Neutrophil-Endothelial Cell Adhesion. J. Immunol. 2001, 167, 5355–5361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhardt, S.U.; Habersberger, J.; Murphy, A.; Chen, Y.-C.; Woollard, K.J.; Bassler, N.; Qian, H.; von zur Muhlen, C.; Hagemeyer, C.E.; Ahrens, I.; et al. Dissociation of Pentameric to Monomeric C-Reactive Protein on Activated Platelets Localizes Inflammation to Atherosclerotic Plaques. Circ. Res. 2009, 105, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Thiele, J.R.; Habersberger, J.; Braig, D.; Schmidt, Y.; Goerendt, K.; Maurer, V.; Bannasch, H.; Scheichl, A.; Woollard, K.J.; von Dobschütz, E.; et al. Dissociation of Pentameric to Monomeric C-Reactive Protein Localizes and Aggravates Inflammation: In Vivo Proof of a Powerful Proinflammatory Mechanism and a New Anti-Inflammatory Strategy. Circulation 2014, 130, 35–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, N.; Ma, F.-R.; Han, J.; Liu, X.-L.; Fu, Y.; Liu, Y.-T.; Liang, Y.-L.; Ouyang, H.; Li, H.-Y. Monomeric C-reactive protein regulates fibronectin mediated monocyte adhesion. Mol. Immunol. 2020, 117, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Na, H.; Gan, Q.; Tao, Q.; Alekseyev, Y.; Hu, J.; Yan, Z.; Yang, J.B.; Tian, H.; Zhu, S.; et al. Monomeric C-reactive protein via endothelial CD31 for neurovascular inflammation in an ApoE genotype-dependent pattern: A risk factor for Alzheimer’s disease? Aging Cell 2021, 20, e13501. [Google Scholar] [CrossRef]
- Li, H.-Y.; Wang, J.; Wu, Y.-X.; Zhang, L.; Liu, Z.-P.; Filep, J.G.; Potempa, L.A.; Wu, Y.; Ji, S.-R. Topological Localization of Monomeric C-reactive Protein Determines Proinflammatory Endothelial Cell Responses. J. Biol. Chem. 2014, 289, 14283–14290. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.-R.; Ma, L.; Bai, C.-J.; Shi, J.-M.; Li, H.-Y.; Potempa, L.A.; Filep, J.G.; Zhao, J.; Wu, Y. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J. 2009, 23, 1806–1816. [Google Scholar] [CrossRef]
- Khreiss, T.; József, L.; Potempa, L.A.; Filep, J.G. Loss of Pentameric Symmetry in C-Reactive Protein Induces Interleukin-8 Secretion Through Peroxynitrite Signaling in Human Neutrophils. Circ. Res. 2005, 97, 690–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trial, J.; Potempa, L.A.; Entman, M.L. The role of C-reactive protein in innate and acquired inflammation: New perspectives. Inflamm. Cell Signal. 2016, 3, e1409. [Google Scholar] [PubMed]
- Ji, S.; Wu, Y.; Potempa, L.; Qiu, Q.; Zhao, J. Interactions of C-reactive protein with low-density lipoproteins: Implications for an active role of modified C-reactive protein in atherosclerosis. Int. J. Biochem. Cell Biol. 2006, 38, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Zha, Z.; Cheng, Y.; Cao, L.; Qian, Y.; Liu, X.; Guo, Y.; Wang, J. Monomeric CRP Aggravates Myocardial Injury After Myocardial Infarction by Polarizing the Macrophage to Pro-Inflammatory Phenotype Through JNK Signaling Pathway. JIR 2021, 14, 7053–7064. [Google Scholar] [CrossRef]
- Thiele, J.R.; Zeller, J.; Kiefer, J.; Braig, D.; Kreuzaler, S.; Lenz, Y.; Potempa, L.A.; Grahammer, F.; Huber, T.B.; Huber-Lang, M.; et al. A Conformational Change in C-Reactive Protein Enhances Leukocyte Recruitment and Reactive Oxygen Species Generation in Ischemia/Reperfusion Injury. Front. Immunol. 2018, 9, 675. [Google Scholar] [CrossRef] [Green Version]
- Boras, E.; Slevin, M.; Alexander, M.Y.; Aljohi, A.; Gilmore, W.; Ashworth, J.; Krupinski, J.; Potempa, L.A.; Al Abdulkareem, I.; Elobeid, A.; et al. Monomeric C-reactive protein and Notch-3 co-operatively increase angiogenesis through PI3K signalling pathway. Cytokine 2014, 69, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Turu, M.M.; Slevin, M.; Matou, S.; West, D.; Rodríguez, C.; Luque, A.; Grau-Olivares, M.; Badimon, L.; Martinez-Gonzalez, J.; Krupinski, J. C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signalling pathways and gene expression. BMC Cell Biol. 2008, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Krupinski, J.; Turu, M.M.; Martinez-Gonzalez, J.; Carvajal, A.; Juan-Babot, J.O.; Iborra, E.; Slevin, M.; Rubio, F.; Badimon, L. Endogenous Expression of C-Reactive Protein Is Increased in Active (Ulcerated Noncomplicated) Human Carotid Artery Plaques. Stroke 2006, 37, 1200–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Inoue, N.; Ohashi, Y.; Terashima, M.; Matsui, K.; Mori, T.; Fujita, H.; Awano, K.; Kobayashi, K.; Azumi, H.; et al. Interaction of Oxidative Stress and Inflammatory Response in Coronary Plaque Instability: Important Role of C-Reactive Protein. ATVB 2003, 23, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, I.S.; Kozlov, S.G.; Chumachenko, P.V.; Saburova, O.S.; Guseva, O.A.; Prokofyeva, L.V.; Gabbasov, Z.A. Monomeric C-reactive protein and local inflammatory reaction in the wall of the coronary arteries in patients with stable coronary artery disease. Russ. J. Cardiol. 2019, 24, 56–61. [Google Scholar] [CrossRef]
- Vainas, T.; Stassen, F.R.M.; de Graaf, R.; Twiss, E.L.L.; Herngreen, S.B.; Welten, R.J.T.J.; van den Akker, L.H.J.M.; van Dieijen-Visser, M.P.; Bruggeman, C.A.; Kitslaar, P.J.E.H.M. C-reactive protein in peripheral arterial disease: Relation to severity of the disease and to future cardiovascular events. J. Vasc. Surg. 2005, 42, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabs, W.J.; Theissing, E.; Nitschke, M.; Bechtel, J.F.M.; Duchrow, M.; Mohamed, S.; Jahrbeck, B.; Sievers, H.-H.; Steinhoff, J.; Bartels, C. Local Generation of C-Reactive Protein in Diseased Coronary Artery Venous Bypass Grafts and Normal Vascular Tissue. Circulation 2003, 108, 1428–1431. [Google Scholar] [CrossRef] [PubMed]
- Sattler, K.J.E.; Woodrum, J.E.; Galili, O.; Olson, M.; Samee, S.; Meyer, F.B.; Zhu, X.-Y.; Lerman, L.O.; Lerman, A. Concurrent Treatment With Renin-Angiotensin System Blockers and Acetylsalicylic Acid Reduces Nuclear Factor κB Activation and C-Reactive Protein Expression in Human Carotid Artery Plaques. Stroke 2005, 36, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabro, P.; Chang, D.W.; Willerson, J.T.; Yeh, E.T.H. Release of C-Reactive Protein in Response to Inflammatory Cytokines by Human Adipocytes: Linking Obesity to Vascular Inflammation. J. Am. Coll. Cardiol. 2005, 46, 1112–1113. [Google Scholar] [CrossRef] [Green Version]
- Kolb-Bachofen, V.; Puchta-Teudt, N.; Egenhofer, C. Expression of membrane-associated C-reactive protein by human monocytes: Indications for a selectin-like activity participating in adhesion. Glycoconj. J. 1995, 12, 122–127. [Google Scholar] [CrossRef]
- Ciubotaru, I.; Potempa, L.A.; Wander, R.C. Production of Modified C-Reactive Protein in U937-Derived Macrophages. Exp. Biol. Med. 2005, 230, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Haider, D.G.; Leuchten, N.; Schaller, G.; Gouya, G.; Kolodjaschna, J.; Schmetterer, L.; Kapiotis, S.; Wolzt, M. C-reactive protein is expressed and secreted by peripheral blood mononuclear cells. Clin. Exp. Immunol. 2006, 146, 533–539. [Google Scholar] [CrossRef]
- Wang, J.; Tang, B.; Liu, X.; Wu, X.; Wang, H.; Xu, D.; Guo, Y. Increased monomeric CRP levels in acute myocardial infarction: A possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis 2015, 239, 343–349. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.-Y.; Li, W.; Shen, Z.-Y.; Wang, Y.-D.; Ji, S.-R.; Wu, Y. An ELISA Assay for Quantifying Monomeric C-Reactive Protein in Plasma. Front. Immunol. 2018, 9, 511. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.D.; Moran, J.A.; Fryer, A.A.; Littlejohn, J.R.; Williams, H.M.; Greenhough, T.J.; Shrive, A.K. Monomeric C-Reactive Protein in Serum With Markedly Elevated CRP Levels Shares Common Calcium-Dependent Ligand Binding Properties With an in vitro Dissociated Form of C-Reactive Protein. Front. Immunol. 2020, 11, 115. [Google Scholar] [CrossRef]
- Wu, K.-L.; Liang, Q.-H.; Huang, B.-T.; Ding, N.; Li, B.-W.; Hao, J. The plasma level of mCRP is linked to cardiovascular disease in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res. Ther. 2020, 22, 228. [Google Scholar] [CrossRef]
- Munuswamy, R.; De Brandt, J.; Burtin, C.; Derave, W.; Aumann, J.; Spruit, M.A.; Michiels, L. Monomeric CRP is Elevated in Patients with COPD Compared to Non-COPD Control Persons. JIR 2021, 14, 4503–4507. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xu, K.; Liu, W.; Liu, X.; Yuan, P.; Xu, P.; Li, H. Monomeric C-reactive protein level is associated with osteoarthritis. Exp. Ther. Med. 2022, 23, 277. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, I.; Kozlov, S.; Pogorelova, O.; Tripoten, M.; Khamchieva, L.; Saburova, O.; Avtaeva, Y.; Zvereva, M.; Matroze, E.; Kuznetsova, T.; et al. The monomeric C-reactive protein level is associated with the increase in carotid plaque number in patients with subclinical carotid atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 968267. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Wetterö, J.; Weiner, M.; Rönnelid, J.; Fernandez-Botran, R.; Sjöwall, C. Associations of C-reactive protein isoforms with systemic lupus erythematosus phenotypes and disease activity. Arthritis Res. Ther. 2022, 24, 139. [Google Scholar] [CrossRef] [PubMed]
- Fujita, C.; Sakurai, Y.; Yasuda, Y.; Homma, R.; Huang, C.-L.; Fujita, M. mCRP as a Biomarker of Adult-Onset Still’s Disease: Quantification of mCRP by ELISA. Front. Immunol. 2022, 13, 938173. [Google Scholar] [CrossRef] [PubMed]
- Ristagno, G.; Fumagalli, F.; Bottazzi, B.; Mantovani, A.; Olivari, D.; Novelli, D.; Latini, R. Pentraxin 3 in Cardiovascular Disease. Front. Immunol. 2019, 10, 823. [Google Scholar] [CrossRef] [Green Version]
- van ’t Klooster, C.C.; van der Graaf, Y.; Ridker, P.M.; Westerink, J.; Hjortnaes, J.; Sluijs, I.; Asselbergs, F.W.; Bots, M.L.; Kappelle, L.J.; Visseren, F.L.J. The relation between healthy lifestyle changes and decrease in systemic inflammation in patients with stable cardiovascular disease. Atherosclerosis 2020, 301, 37–43. [Google Scholar] [CrossRef]
- Gaesser, G.A.; Angadi, S.S.; Ryan, D.M.; Johnston, C.S. Lifestyle Measures to Reduce Inflammation. Am. J. Lifestyle Med. 2012, 6, 4–13. [Google Scholar] [CrossRef]
- Fedewa, M.V.; Hathaway, E.D.; Ward-Ritacco, C.L. Effect of exercise training on C reactive protein: A systematic review and meta-analysis of randomised and non-randomised controlled trials. Br. J. Sports Med. 2017, 51, 670–676. [Google Scholar] [CrossRef]
- Bianchi, V.E. Weight loss is a critical factor to reduce inflammation. Clin. Nutr. ESPEN 2018, 28, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Giugliano, R.P.; Cannon, C.P.; Zhou, J.; Murphy, S.A.; White, J.A.; Tershakovec, A.M.; Blazing, M.A.; Braunwald, E. Achievement of Dual Low-Density Lipoprotein Cholesterol and High-Sensitivity C-Reactive Protein Targets More Frequent With the Addition of Ezetimibe to Simvastatin and Associated With Better Outcomes in IMPROVE-IT. Circulation 2015, 132, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissen, S.E. Effect of Intensive Lipid Lowering on Progression of Coronary Atherosclerosis: Evidence for an Early Benefit from the Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) Trial. Am. J. Cardiol. 2005, 96, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Gotto, A.M.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Bohula, E.A.; Giugliano, R.P.; Leiter, L.A.; Verma, S.; Park, J.-G.; Sever, P.S.; Lira Pineda, A.; Honarpour, N.; Wang, H.; Murphy, S.A.; et al. Inflammatory and Cholesterol Risk in the FOURIER Trial. Circulation 2018, 138, 131–140. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Aday, A.W.; Rose, L.M.; Ridker, P.M. Residual Inflammatory Risk on Treatment With PCSK9 Inhibition and Statin Therapy. Circulation 2018, 138, 141–149. [Google Scholar] [CrossRef]
- Kalkman, D.N.; Aquino, M.; Claessen, B.E.; Baber, U.; Guedeney, P.; Sorrentino, S.; Vogel, B.; de Winter, R.J.; Sweeny, J.; Kovacic, J.C.; et al. Residual inflammatory risk and the impact on clinical outcomes in patients after percutaneous coronary interventions. Eur. Heart J. 2018, 39, 4101–4108. [Google Scholar] [CrossRef]
- Guedeney, P.; Claessen, B.E.; Kalkman, D.N.; Aquino, M.; Sorrentino, S.; Giustino, G.; Farhan, S.; Vogel, B.; Sartori, S.; Montalescot, G.; et al. Residual Inflammatory Risk in Patients With Low LDL Cholesterol Levels Undergoing Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2019, 73, 2401–2409. [Google Scholar] [CrossRef]
- Ridker, P.M. Clinician’s Guide to Reducing Inflammation to Reduce Atherothrombotic Risk. J. Am. Coll. Cardiol. 2018, 72, 3320–3331. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Pagidipati, N.J.; Hellkamp, A.S.; Sharma, P.P.; Wang, T.Y.; Fonarow, G.C.; Pencina, M. High-sensitivity C-reactive protein elevation in patients with prior myocardial infarction in the United States. Am. Heart J. 2018, 204, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Ballantyne, C.M.; Veltri, E.; Shah, A.; Bird, S.; Lin, J.; Rosenberg, E.; Tershakovec, A.M. Pooled Analyses of Effects on C-Reactive Protein and Low Density Lipoprotein Cholesterol in Placebo-Controlled Trials of Ezetimibe Monotherapy or Ezetimibe Added to Baseline Statin Therapy. Am. J. Cardiol. 2009, 103, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.P.; Lins, L.C.; Fonseca, F.A.; Matos, L.N.; Aguirre, A.C.; Bianco, H.T.; Amaral, J.B.; França, C.N.; Santana, J.M.; Izar, M.C. Effects of ezetimibe on markers of synthesis and absorption of cholesterol in high-risk patients with elevated C-reactive protein. Life Sci. 2013, 92, 845–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kater, A.-L.A.; Batista, M.C.; Ferreira, S.R. Synergistic effect of simvastatin and ezetimibe on lipid and pro-inflammatory profiles in pre-diabetic subjects. Diabetol. Metab. Syndr. 2010, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.S.; Min, Y.J.; Kwon, J.E.; Cho, E.J.; Kim, J.E.; Lee, W.-S.; Lee, K.J.; Kim, S.-W.; Kim, T.H.; Kim, C.J.; et al. Effects of Ezetimibe Added to Ongoing Statin Therapy on C-Reactive Protein Levels in Hypercholesterolemic Patients. Korean Circ. J. 2011, 41, 253. [Google Scholar] [CrossRef] [Green Version]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Di Giosia, P.; Stamerra, C.A.; Grassi, D.; Pedone, C.; Ferretti, G.; Bacchetti, T.; Ferri, C.; Giorgini, P. Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: A meta-analysis of 16 randomized controlled treatment arms: PCSK9 inhibitors and hs-CRP levels. Br. J. Clin. Pharmacol. 2016, 81, 1175–1190. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.A.; Gutierrez, J.A.T.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016, 315, 1591. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Steen, D.P.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; et al. Effect of Darapladib on Major Coronary Events After an Acute Coronary Syndrome: The SOLID-TIMI 52 Randomized Clinical Trial. JAMA 2014, 312, 1006. [Google Scholar] [CrossRef]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, R.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in Patients With Acute Coronary Syndrome: The Australian COPS Randomized Clinical Trial. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Libby, P.; MacFadyen, J.G.; Thuren, T.; Ballantyne, C.; Fonseca, F.; Koenig, W.; Shimokawa, H.; Everett, B.M.; Glynn, R.J. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: Analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 2018, 39, 3499–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Ridker, P.M. From RESCUE to ZEUS: Will interleukin-6 inhibition with ziltivekimab prove effective for cardiovascular event reduction? Cardiovasc. Res. 2021, 117, e138–e140. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, Z.; Waqar, A.B.; Ning, B.; Yang, X.; Shiomi, M.; Graham, M.J.; Crooke, R.M.; Liu, E.; Dong, S.; et al. Effects of Antisense Oligonucleotides against C-Reactive Protein on the Development of Atherosclerosis in WHHL Rabbits. Mediat. Inflamm. 2014, 2014, 979132. [Google Scholar] [CrossRef]
- Jones, N.R.; Pegues, M.A.; McCrory, M.A.; Singleton, W.; Bethune, C.; Baker, B.F.; Norris, D.A.; Crooke, R.M.; Graham, M.J.; Szalai, A.J. A Selective Inhibitor of Human C-reactive Protein Translation Is Efficacious In Vitro and in C-reactive Protein Transgenic Mice and Humans. Mol. Ther. —Nucleic Acids 2012, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Noveck, R.; Stroes, E.S.G.; Flaim, J.D.; Baker, B.F.; Hughes, S.; Graham, M.J.; Crooke, R.M.; Ridker, P.M. Effects of an Antisense Oligonucleotide Inhibitor of C-Reactive Protein Synthesis on the Endotoxin Challenge Response in Healthy Human Male Volunteers. JAHA 2014, 3, e001084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugihara, C.; Freemantle, N.; Hughes, S.G.; Furniss, S.; Sulke, N. The effect of C-reactive protein reduction with a highly specific antisense oligonucleotide on atrial fibrillation assessed using beat-to-beat pacemaker Holter follow-up. J. Intervig. Card. Electrophysiol. 2015, 43, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.S.; Hughes, S.G.; Singleton, W.; Yamashita, M.; Genovese, M.C. Results of a proof of concept, double-blind, randomized trial of a second generation antisense oligonucleotide targeting high-sensitivity C-reactive protein (hs-CRP) in rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 80. [Google Scholar] [CrossRef] [Green Version]
- Pepys, M.B.; Hirschfield, G.M.; Tennent, G.A.; Ruth Gallimore, J.; Kahan, M.C.; Bellotti, V.; Hawkins, P.N.; Myers, R.M.; Smith, M.D.; Polara, A.; et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 2006, 440, 1217–1221. [Google Scholar] [CrossRef] [Green Version]
- Zeller, J.; Cheung Tung Shing, K.S.; Nero, T.L.; McFadyen, J.D.; Krippner, G.; Bogner, B.; Kreuzaler, S.; Kiefer, J.; Horner, V.K.; Braig, D.; et al. A novel phosphocholine-mimetic inhibits a pro-inflammatory conformational change in C-reactive protein. EMBO Mol. Med. 2022, 15, e16236. [Google Scholar] [CrossRef]
- Kather, M.G.; Zeller, J.; Plattner, D.; Breit, B.; Kreuzaler, S.; Krippner, G.; Peter, K.; Eisenhardt, S.U.; Kammerer, B. Pharmacokinetic study of the novel phosphocholine derivative 3-dibutylaminopropylphosphonic acid by LC-MS coupling. J. Chromatogr. B 2021, 1186, 122998. [Google Scholar] [CrossRef]
- Fujita, C.; Sakurai, Y.; Yasuda, Y.; Takada, Y.; Huang, C.-L.; Fujita, M. Anti-Monomeric C-Reactive Protein Antibody Ameliorates Arthritis and Nephritis in Mice. J. Immunol. 2021, 207, 1755–1762. [Google Scholar] [CrossRef]

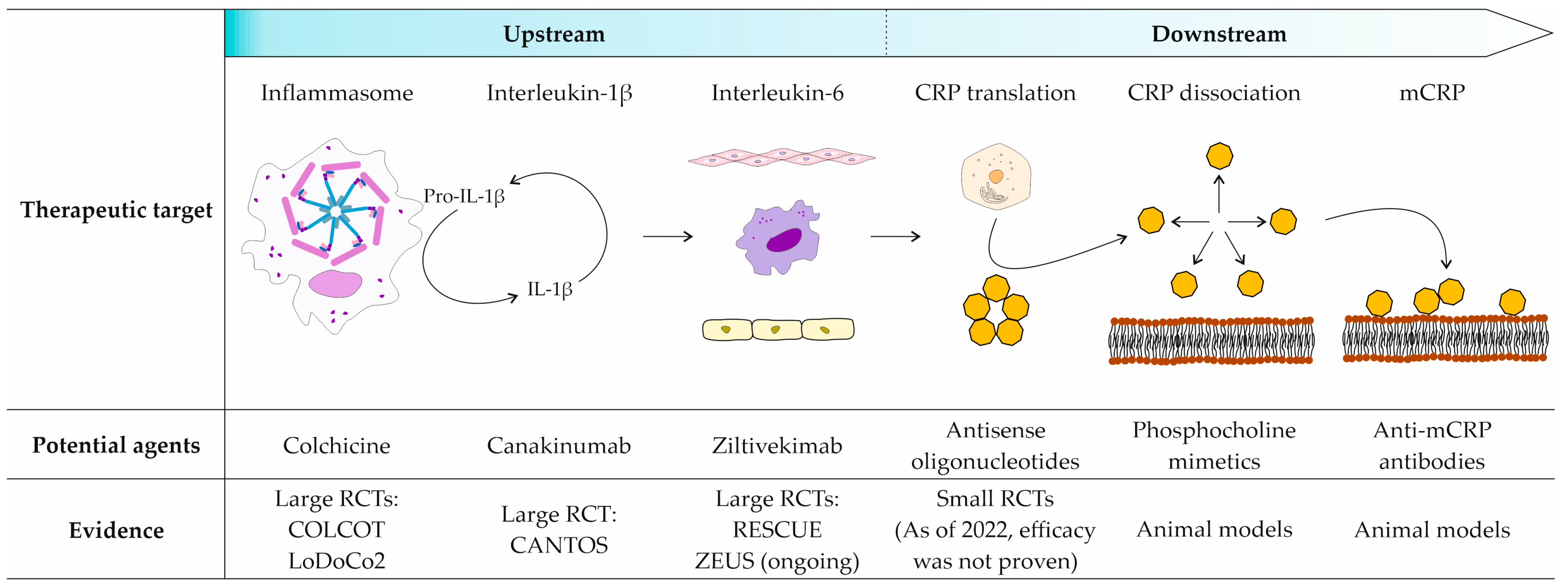

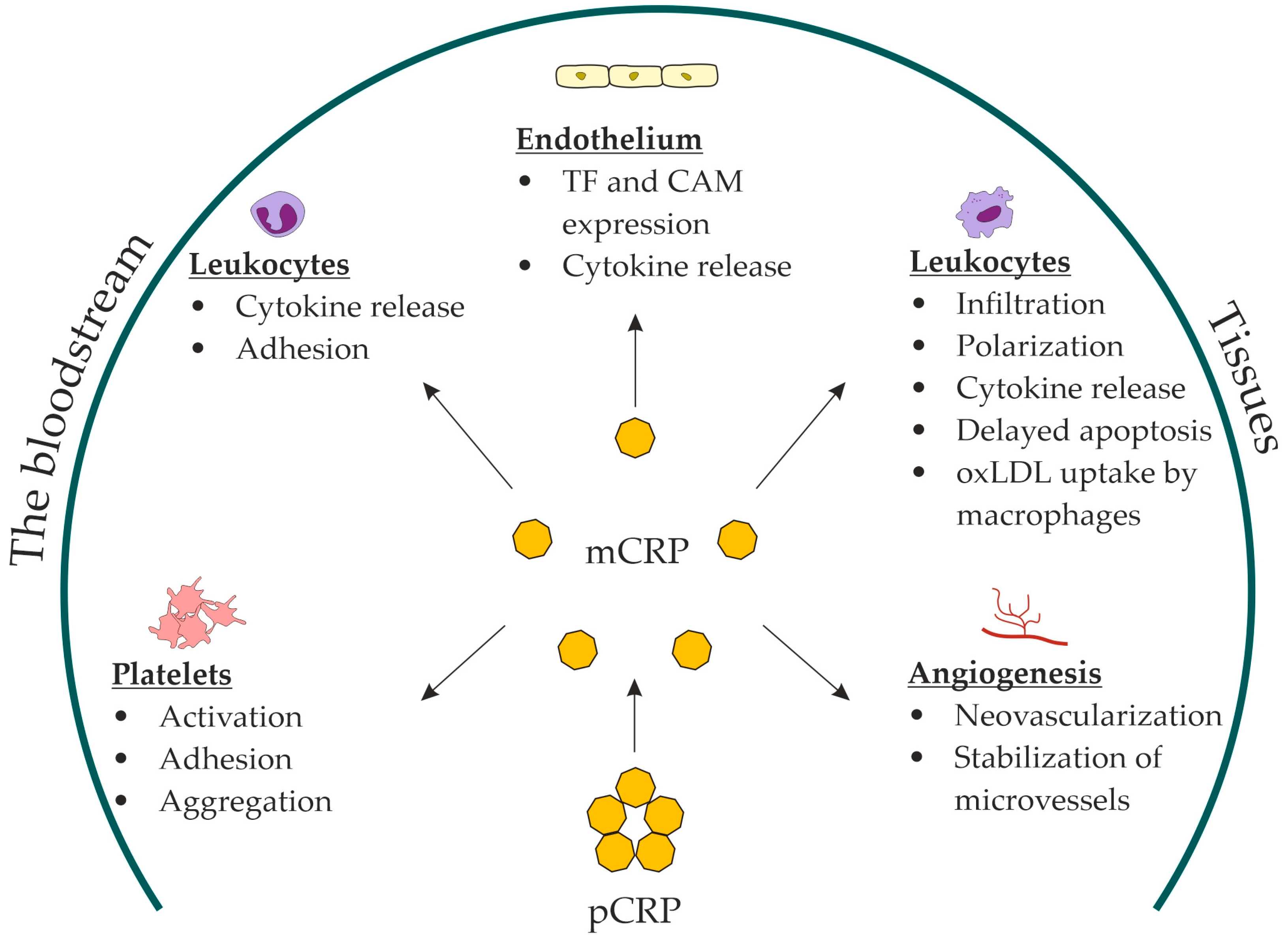{kind=link}
{kind=link}
| Assay Type | Anti-mCRP Antibody | Blood-Specimen Type | Reported mCRP Levels | Correlation (mCRP/ hsCRP) | Reference |
|---|---|---|---|---|---|
| ELISA | Unspecified mAb | Heparinized plasma | 20.96 ± 1.64 μg/L in acute myocardial infarction 0.0 μg/L in angina pectoris, unstable angina, and non-CAD patients | n/a | Wang et al., 2015 [109] |
| ELISA | CRP-8 | Plasma (unspecified anticoagulant) | 59.8 (44.5; 79.1) μg/L in urticaria 35.4 (17.0; 48.6) μg/L in psoriasis 30.0 (4.77; 36.3) in eczema 15.2 (5.05; 27.1) μg/L in healthy participants | n/a | Zhang et al., 2018 [110] |
| ELISA | CRP-8 | Serum purified by chromatography | 1.03 ± 0.11 mg/L in patients with hsCRP > 100 mg/L | None | Williams et al., 2020 [111] |
| ELISA | Unspecified mAb | EDTA plasma | 244.1 (226.1; 331.7) mg/L in AAV 170.0 (135.7; 199.3) mg/L in control participants | Negative | Wu et al., 2020 [112] |
| ELISA | Aptamer | Serum | 0.66 (0.38; 1.03) mg/L in COPD 0.0 (0.0; 0.29) mg/L in non-COPD patients | None | Munuswamy et al., 2021 [113] |
| ELISA | CRP-8 | Plasma (unspecified anticoagulant) | 12.5 (7.8; 24.8) μg/L in osteoarthritis 5.0 (3.5; 9.8) μg/L in healthy participants | Positive | Liang et al., 2022 [114] |
| CBA | CRP-8 | Citrated plasma | 5.2 (3.3; 7.1) μg/L in patients with subclinical carotid atherosclerosis | None | Melnikov et al., 2022 [115] |
| ELISA | 8C10 | Serum | 3.7 (1.3; 7.4) μg/L in systemic lupus erythematosus 11 (5.8; 22) μg/L in AAV patients | None | Karlsson et al., 2022 [116] |
| ELISA | 12C | EDTA plasma | 477 (100; 2570) μg/L in adult-onset Still’s disease 186 (0.035; 626) μg/L in polymyalgia rheumatica 77 (0.035; 501) μg/L in rheumatoid arthritis 228 (0.035; 1086) μg/L in acute infections 1.2(0.035; 10) μg/L in healthy participants | None | Fujita et al., 2022 [117] |
| Target | Agent | Stage of Development | Main Findings |
|---|---|---|---|
| CRP translation | ISIS 280290 | Animal models | No antiatherogenic effect was shown in a rabbit model of atherosclerosis. |
| ISIS 329993 | Phase II clinical trials | No benefit was shown in atrial fibrillation or rheumatoid arthritis treatment. | |
| CRP dissociation | 1,6-bisPC | Animal models | Myocardial damage and renal damage were reduced in rat models of myocardial and renal IRI, respectively. |
| C10M | Animal models | Renal damage was reduced and allograft rejection prevented in rat models of renal IRI and hindlimb transplantation, respectively. | |
| mCRP | mAb C3 | Animal models | Joint damage and renal damage were reduced in murine models of rheumatoid arthritis and lupus nephritis, respectively. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnikov, I.; Kozlov, S.; Saburova, O.; Avtaeva, Y.; Guria, K.; Gabbasov, Z. Monomeric C-Reactive Protein in Atherosclerotic Cardiovascular Disease: Advances and Perspectives. Int. J. Mol. Sci. 2023, 24, 2079. https://doi.org/10.3390/ijms24032079
Melnikov I, Kozlov S, Saburova O, Avtaeva Y, Guria K, Gabbasov Z. Monomeric C-Reactive Protein in Atherosclerotic Cardiovascular Disease: Advances and Perspectives. International Journal of Molecular Sciences. 2023; 24(3):2079. https://doi.org/10.3390/ijms24032079
Chicago/Turabian StyleMelnikov, Ivan, Sergey Kozlov, Olga Saburova, Yuliya Avtaeva, Konstantin Guria, and Zufar Gabbasov. 2023. "Monomeric C-Reactive Protein in Atherosclerotic Cardiovascular Disease: Advances and Perspectives" International Journal of Molecular Sciences 24, no. 3: 2079. https://doi.org/10.3390/ijms24032079