

Analysis of Non-Amyloidogenic Mutations in APP Supports Loss of Function Hypothesis of Alzheimer’s Disease

Abstract
:1. Introduction
2. Results
2.1. Membrane Association of Aβ–Effect of Peptide Length
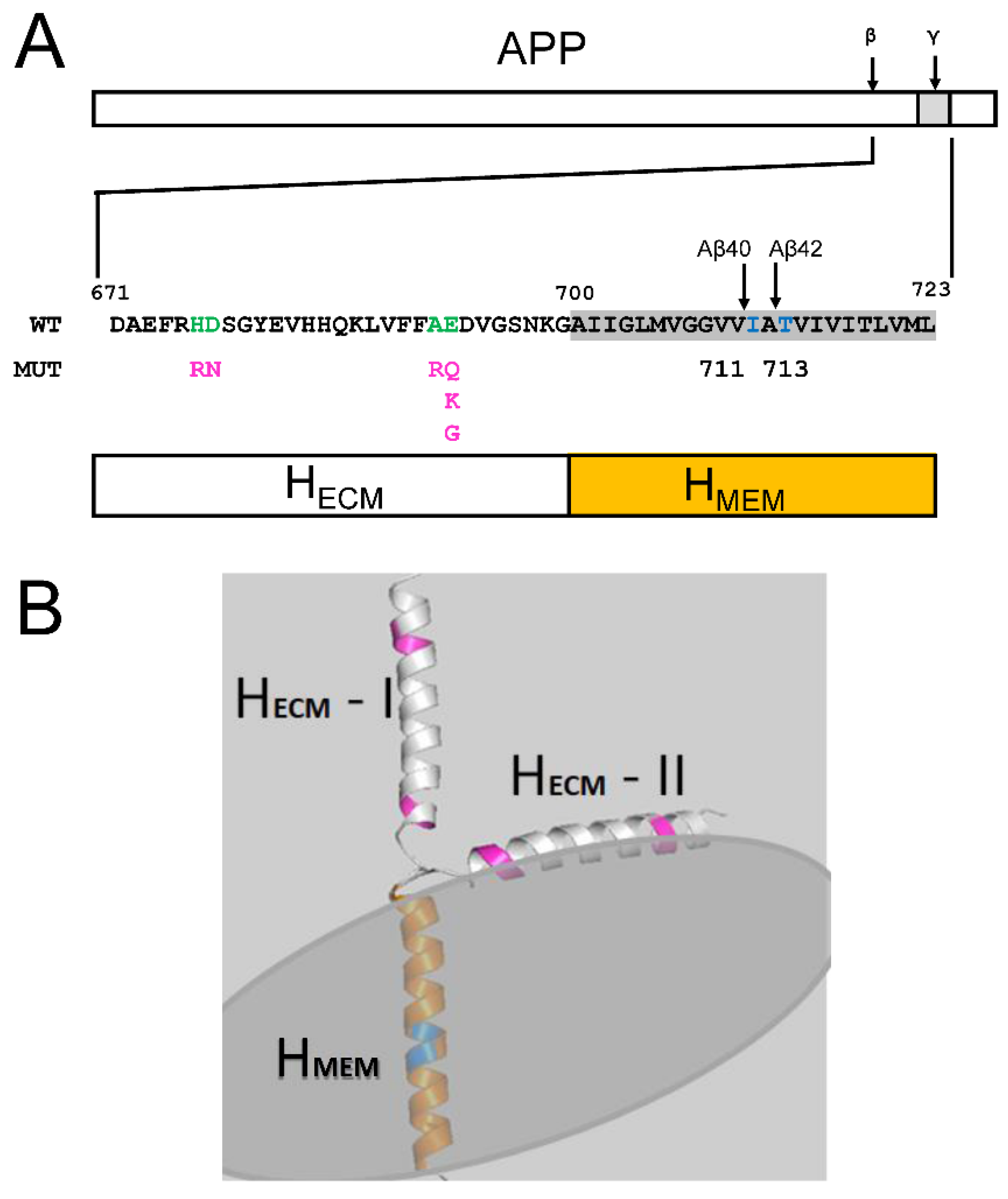
2.2. Membrane Association of Aβ–Effect of Non-Amyloidogenic FAD Mutations in APP
2.3. Reduction in Soluble Aβ as a Result of FAD Mutations in Presenilin 1
2.4. Effects of Membrane Curvature on APP Processing by γ-Secretase
3. Discussion
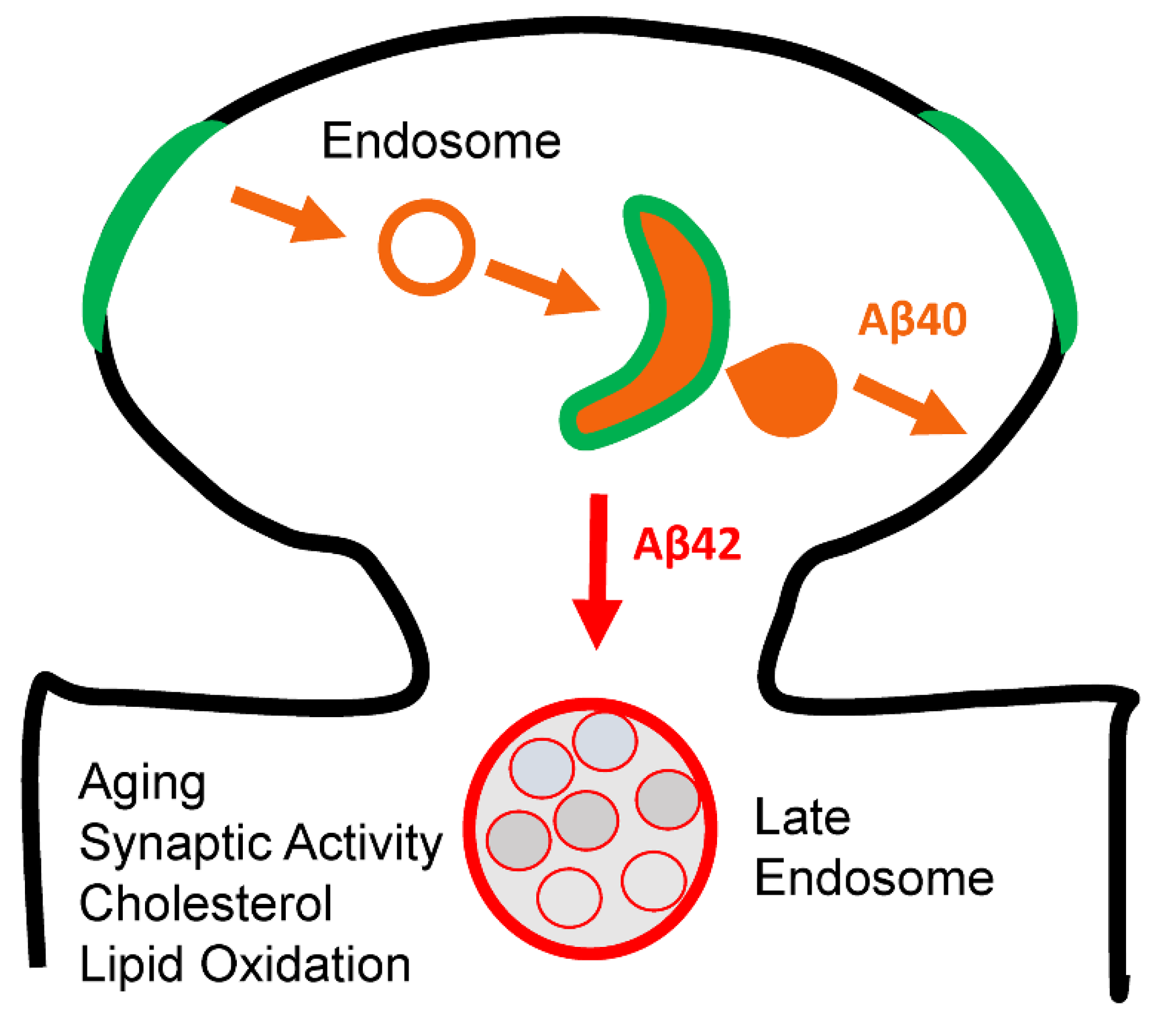
Loss of Aβ40 Function and AD
4. Materials and Methods
4.1. The Aβ Peptide Model Building and Membrane-Associating Energy (EM) Calculations
4.2. Classification of PS1-FAD Based on Generated Aβ Products
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active γ-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Tolia, A.; De Strooper, B. Structure and function of γ-secretase. Semin. Cell Dev. Biol. 2009, 20, 211–218. [Google Scholar] [CrossRef]
- Bergmans, B.A.; De Strooper, B. γ-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. γ-secretase: Proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 2004, 5, 499–504. [Google Scholar] [CrossRef]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Lessons from a failed γ-secretase Alzheimer trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Kanatsu, K.; Tomita, T. Membrane trafficking and proteolytic activity of γ-secretase in Alzheimer’s disease. Biol. Chem. 2016, 397, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Sannerud, R.; Esselens, C.; Ejsmont, P.; Mattera, R.; Rochin, L.; Tharkeshwar, A.K.; De Baets, G.; De Wever, V.; Habets, R.; Baert, V.; et al. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Abeta Pool. Cell 2016, 166, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Ubelmann, F.; Burrinha, T.; Salavessa, L.; Gomes, R.; Ferreira, C.; Moreno, N.; Guimas Almeida, C. Bin1 and CD2AP polarise the endocytic generation of β-amyloid. EMBO Rep. 2017, 18, 102–122. [Google Scholar] [CrossRef] [Green Version]
- Ahn, K.; Shelton, C.C.; Tian, Y.; Zhang, X.; Gilchrist, M.L.; Sisodia, S.S.; Li, Y.M. Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. USA 2010, 107, 21435–21440. [Google Scholar] [CrossRef] [Green Version]
- Schedin-Weiss, S.; Caesar, I.; Winblad, B.; Blom, H.; Tjernberg, L.O. Super-resolution microscopy reveals γ-secretase at both sides of the neuronal synapse. Acta Neuropathol. Commun. 2016, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Bezprozvanny, I. Conformational Models of APP Processing by Gamma Secretase Based on Analysis of Pathogenic Mutations. Int. J. Mol. Sci. 2021, 22, 13600. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.A.; Choi, S.Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chattarji, S.; Kelleher, R.J., 3rd; Kandel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Kelleher, R.J., 3rd. The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J., 3rd. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Heilig, E.A.; Gutti, U.; Tai, T.; Shen, J.; Kelleher, R.J., 3rd. Trans-dominant negative effects of pathogenic PSEN1 mutations on γ-secretase activity and Abeta production. J. Neurosci. 2013, 33, 11606–11617. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 2015, 4, e11182. [Google Scholar] [CrossRef]
- Zoltowska, K.M.; Berezovska, O. Dynamic Nature of presenilin1/γ-Secretase: Implication for Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2018, 55, 2275–2284. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Alzheimer’s disease—Where do we go from here? Biochem. Biophys. Res. Commun. 2022, 633, 72–76. [Google Scholar] [CrossRef]
- Zhou, B.; Lu, J.G.; Siddu, A.; Wernig, M.; Sudhof, T.C. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabn9380. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of intermediate steps in amyloid beta (Abeta) production under near-native conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the final cut: Pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2018, 2, 292–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kufareva, I.; Lenoir, M.; Dancea, F.; Sridhar, P.; Raush, E.; Bissig, C.; Gruenberg, J.; Abagyan, R.; Overduin, M. Discovery of novel membrane binding structures and functions. Biochem. Cell. Biol. 2014, 92, 555–563. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aβ Product | PS1 MUTANT Groups | WT | ||
|---|---|---|---|---|
| GREEN (n = 37) | ORANGE (n = 38) | YELLOW (n = 59) | ||
| Aβ40 (norm) | 0.09189 (0.189) | 0.30026 (0.037) | 0.41525 (0.490) | 1.0 |
| Aβ42 (norm) | 0.024 (0.027) | 0.11187 (0.169) | 0.0903 (0.09) | 0.1 |
| TOTAL Aβ | 0.126 | 0.412 | 0.513 | 1.1 |
| Aβ40/Aβ42 | 3.8 | 2.7 | 4.5 | 10.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Bezprozvanny, I. Analysis of Non-Amyloidogenic Mutations in APP Supports Loss of Function Hypothesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 2092. https://doi.org/10.3390/ijms24032092
Kim M, Bezprozvanny I. Analysis of Non-Amyloidogenic Mutations in APP Supports Loss of Function Hypothesis of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(3):2092. https://doi.org/10.3390/ijms24032092
Chicago/Turabian StyleKim, Meewhi, and Ilya Bezprozvanny. 2023. "Analysis of Non-Amyloidogenic Mutations in APP Supports Loss of Function Hypothesis of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 3: 2092. https://doi.org/10.3390/ijms24032092
APA StyleKim, M., & Bezprozvanny, I. (2023). Analysis of Non-Amyloidogenic Mutations in APP Supports Loss of Function Hypothesis of Alzheimer’s Disease. International Journal of Molecular Sciences, 24(3), 2092. https://doi.org/10.3390/ijms24032092