
Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept

 , ,
, ,  ,
, 
Abstract
:1. Introduction
- -
- Prepare, characterize, and compare different semi-IPNs using GG as the natural polysaccharide due to its mucoadhesive properties.
- -
- To achieve this, it is necessary to first explore the preparation of several hydrophilic, biodegradable homo- and copolymers by polymerization procedures compatible with the chemical nature of GG. Due to the versatility of click chemistry (CC) and in search of an orthogonal chemical procedure in IPN formation, the thiol-ene click reaction is selected to prepare and characterize various homo- and copolymers. Several batches will be synthesized to optimize polymerization conditions and, hence, choose the polymeric systems with the best performance. This is the first time that this methodology for the preparation of IPNs has been investigated.
- -
- Synthesize a monomer that can enhance the biodegradability of the final matrices under the acidic conditions of the stomach. The compound, named acetaldehyde diallylacetal, is chosen as one of the diallyl monomers to be used in semi-IPN preparation so that the presence of the acetal group in its structure will be responsible for the increase in the biodegradability of the semi-IPN-based matrices.
- -
- Conduct a proof of concept on the ability of the new materials to behave as AMOX controlled-release matrices at pHs 1.2 and 5.0.
2. Results
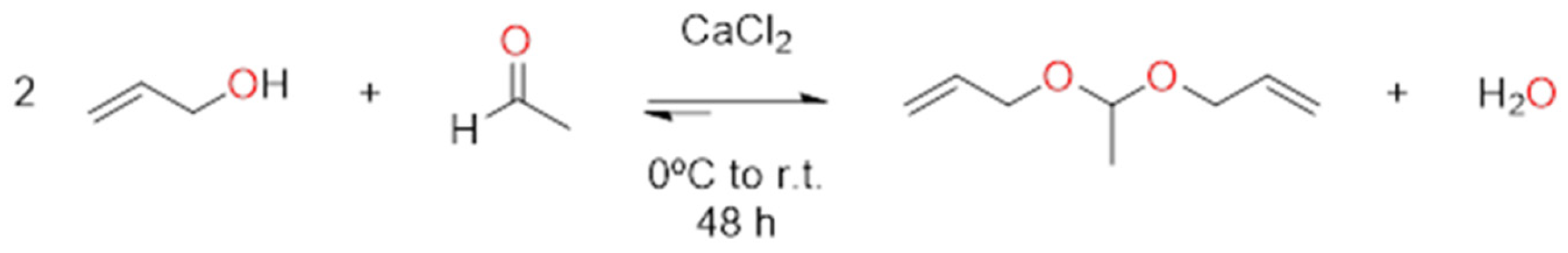
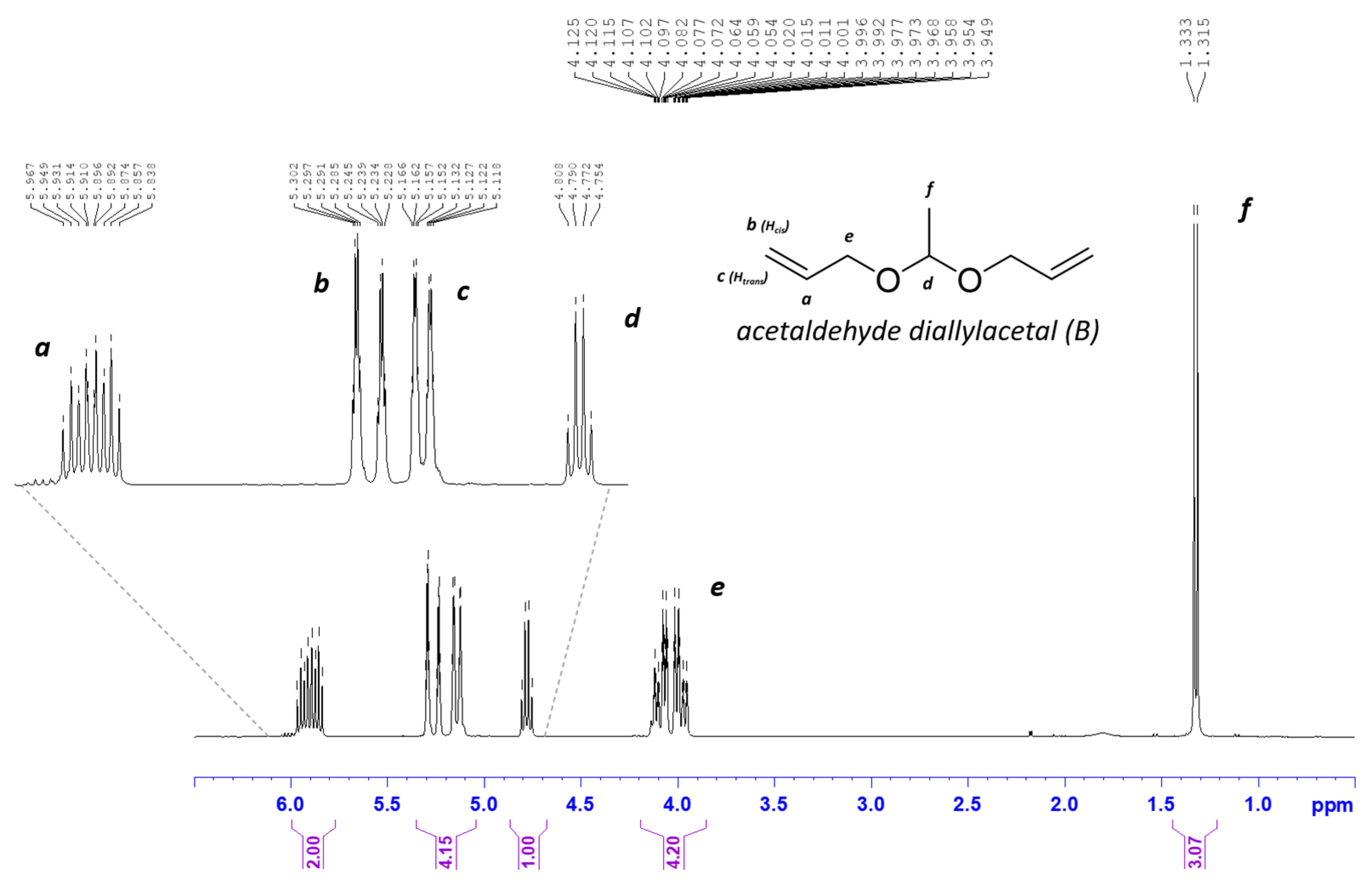
2.1. Synthesis of Acetaldehyde Diallyl Acetal (B Monomer)
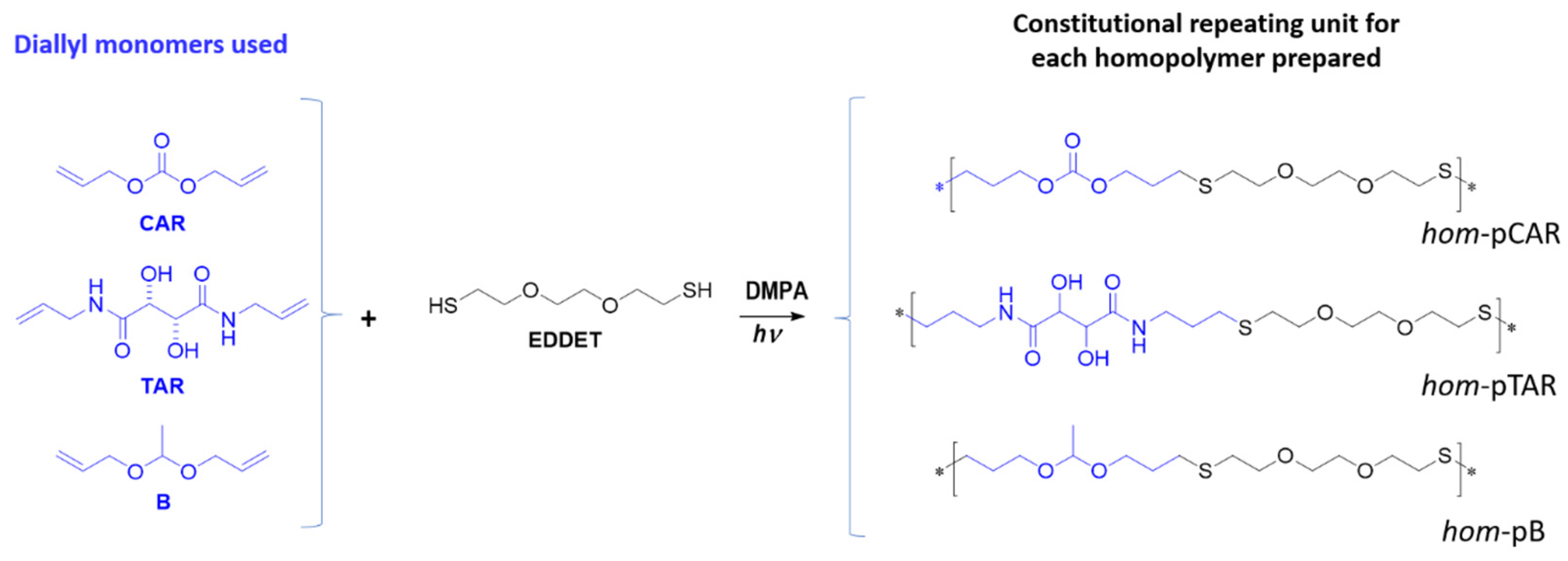
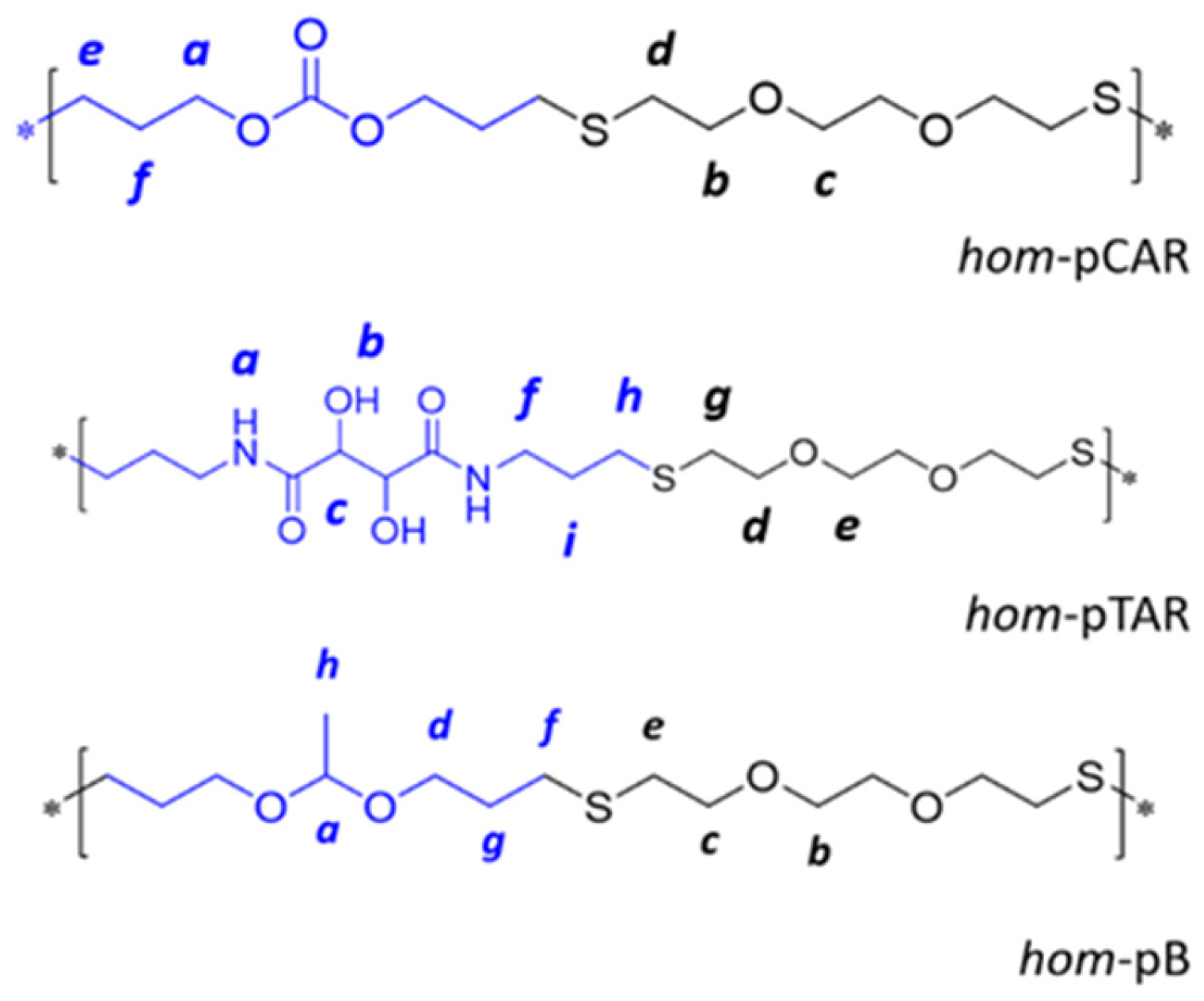
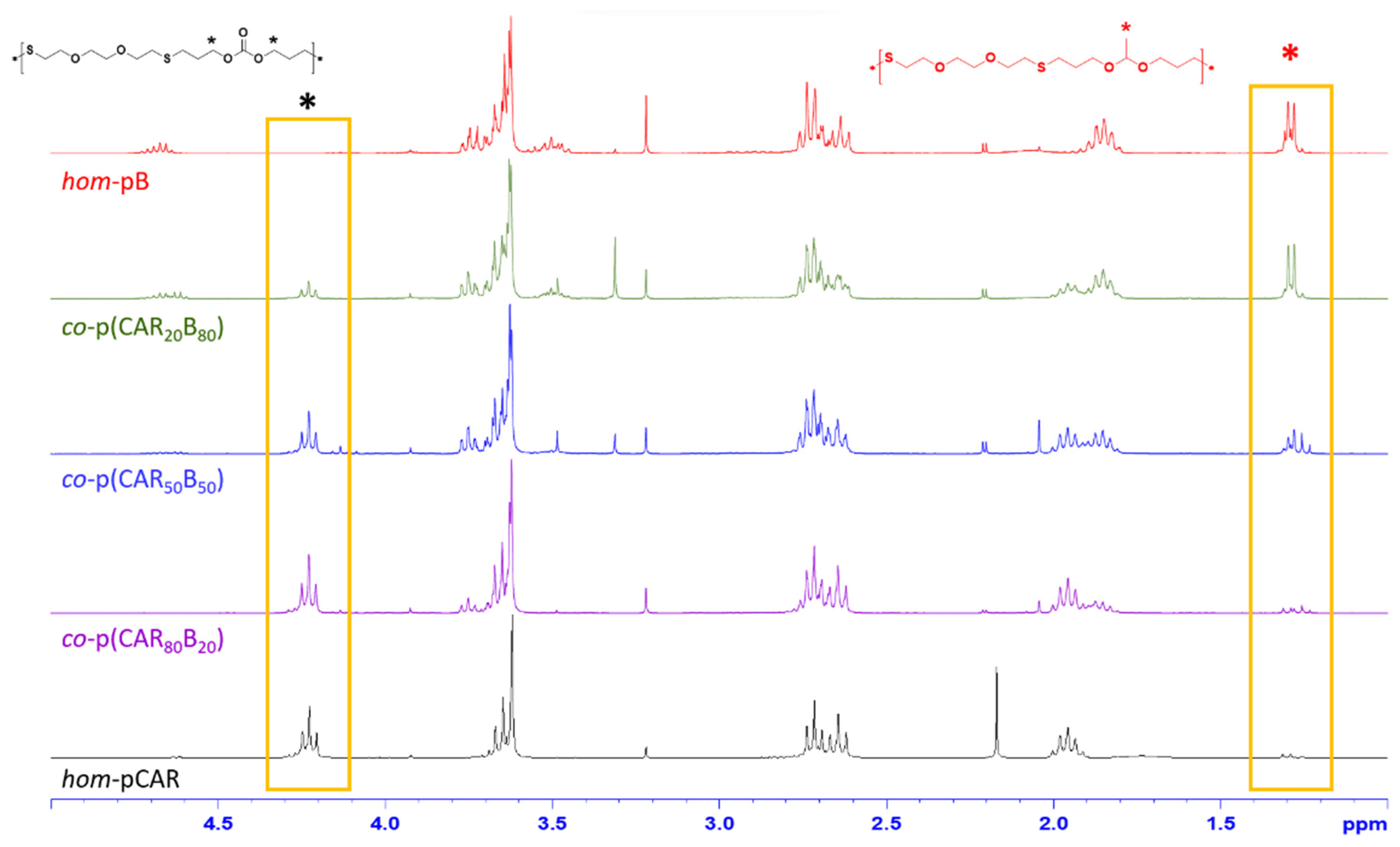
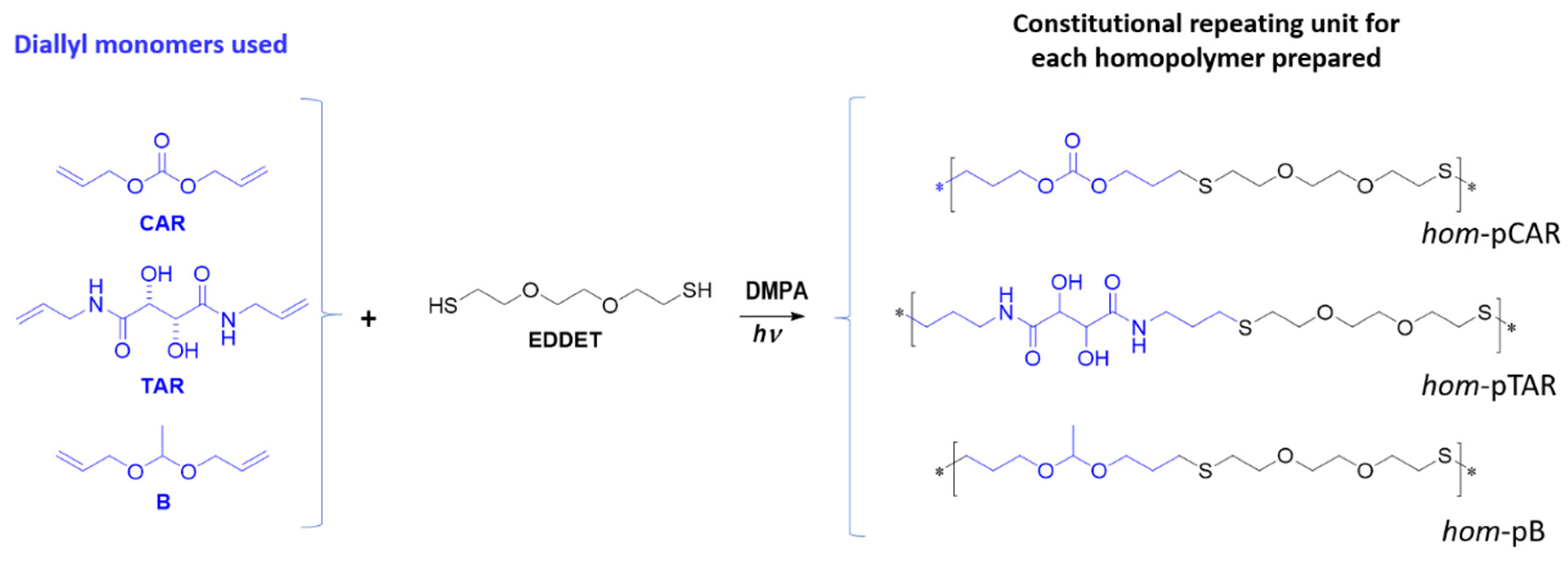
2.2. Preparation and Characterization of Homopolymers and Copolymers by Thiol-Ene Reaction
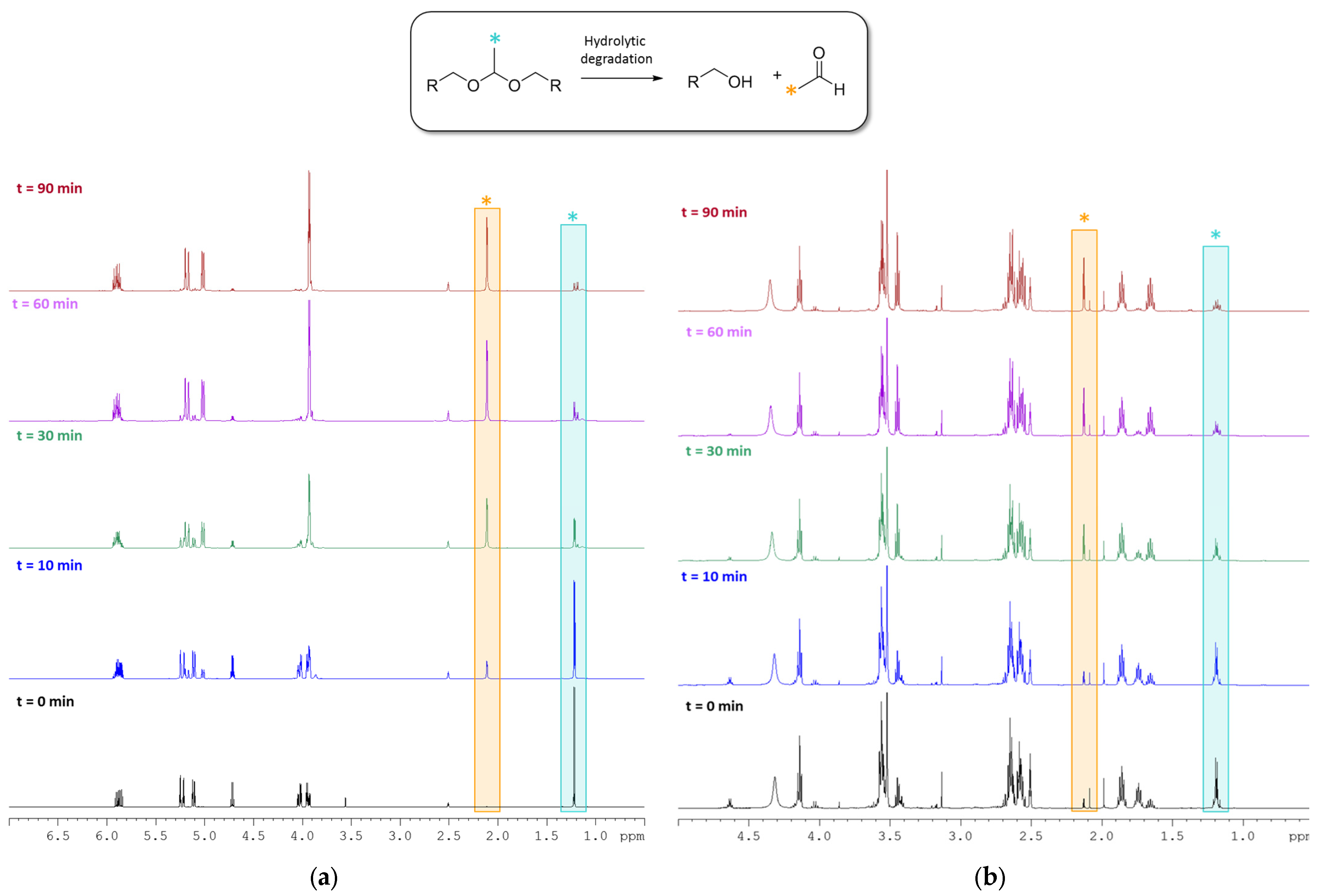
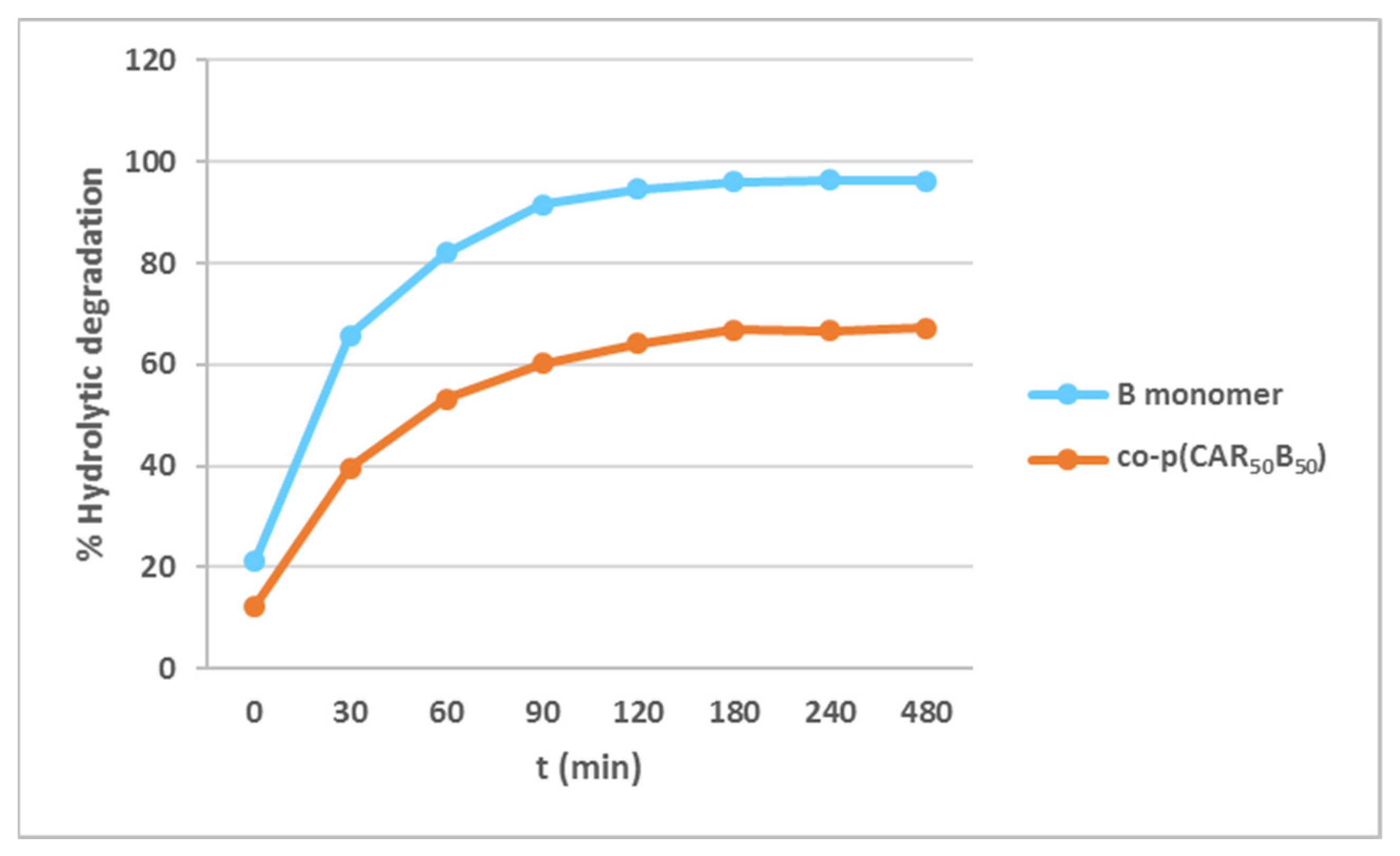
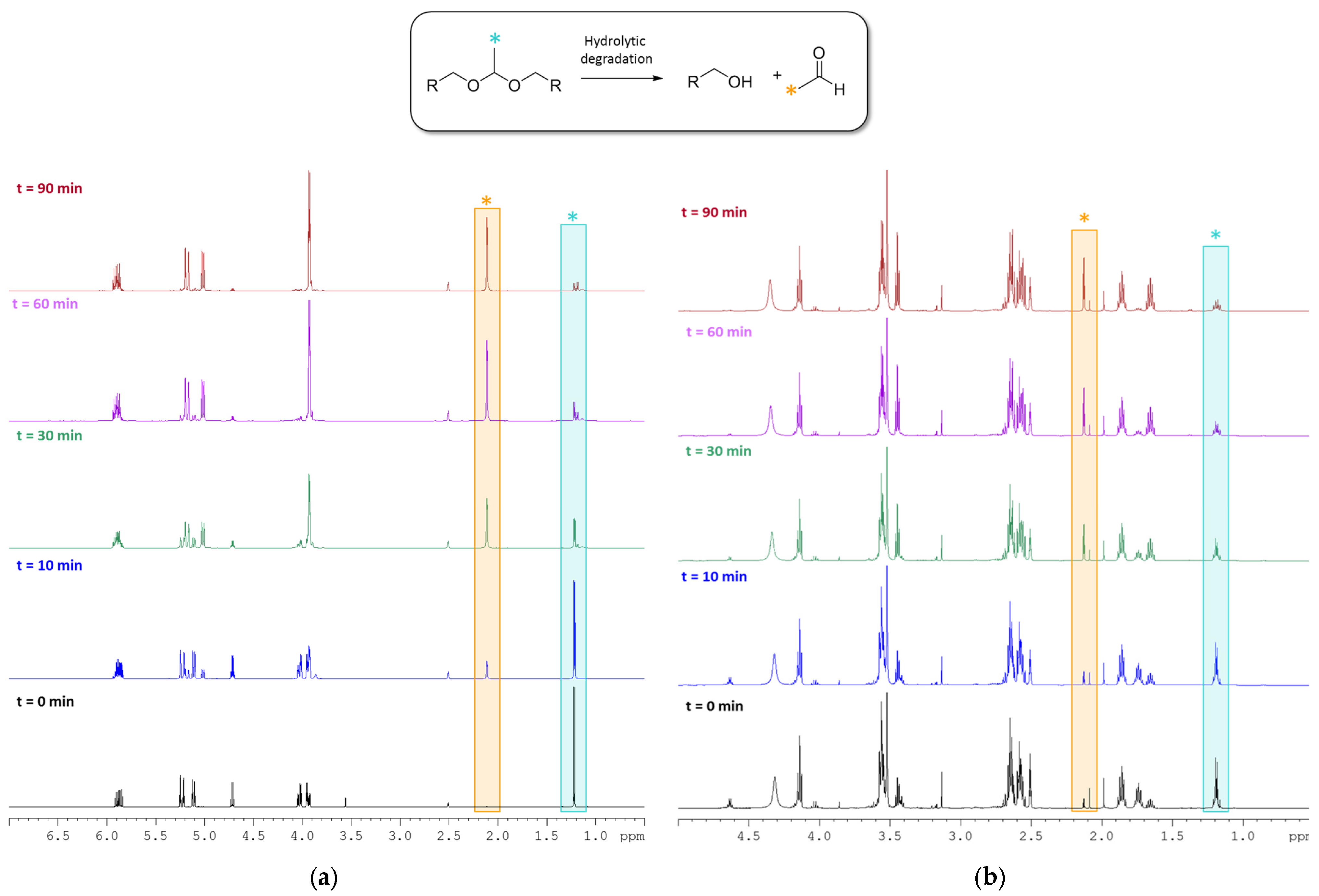
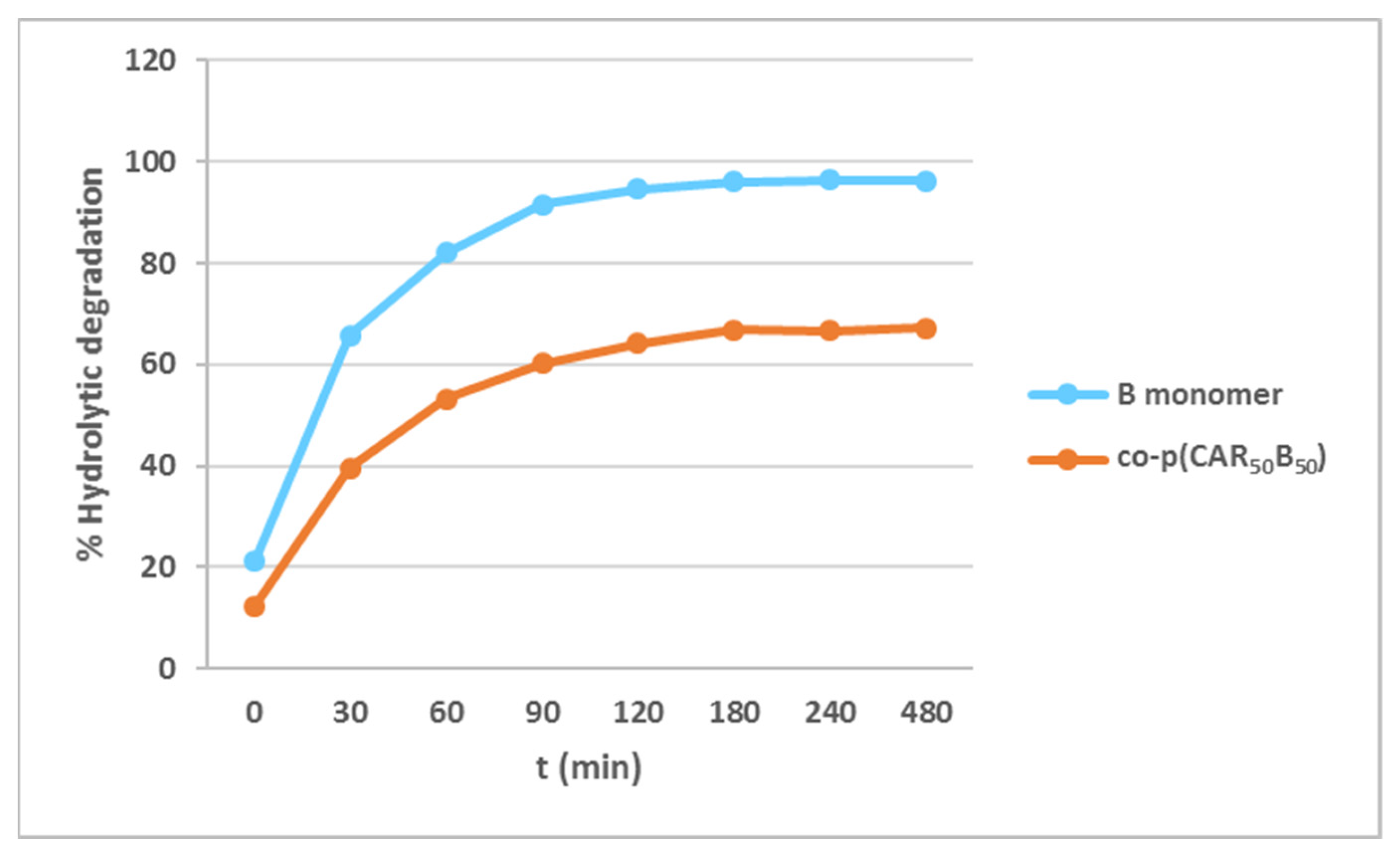
2.3. Degradability Studies of the B Monomer and co-p(CAR50B50) under Acidic Aqueous Conditions
2.4. Preparation of Guar-Gum-Based Semi-IPNs
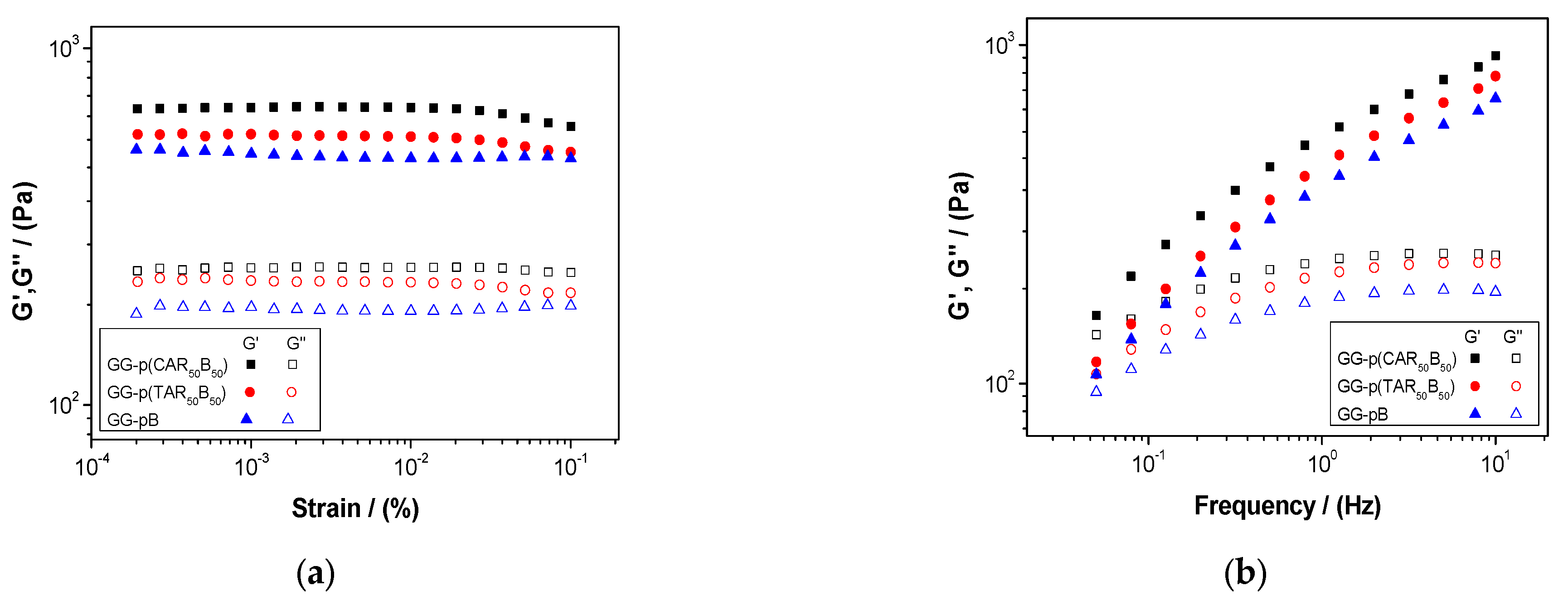
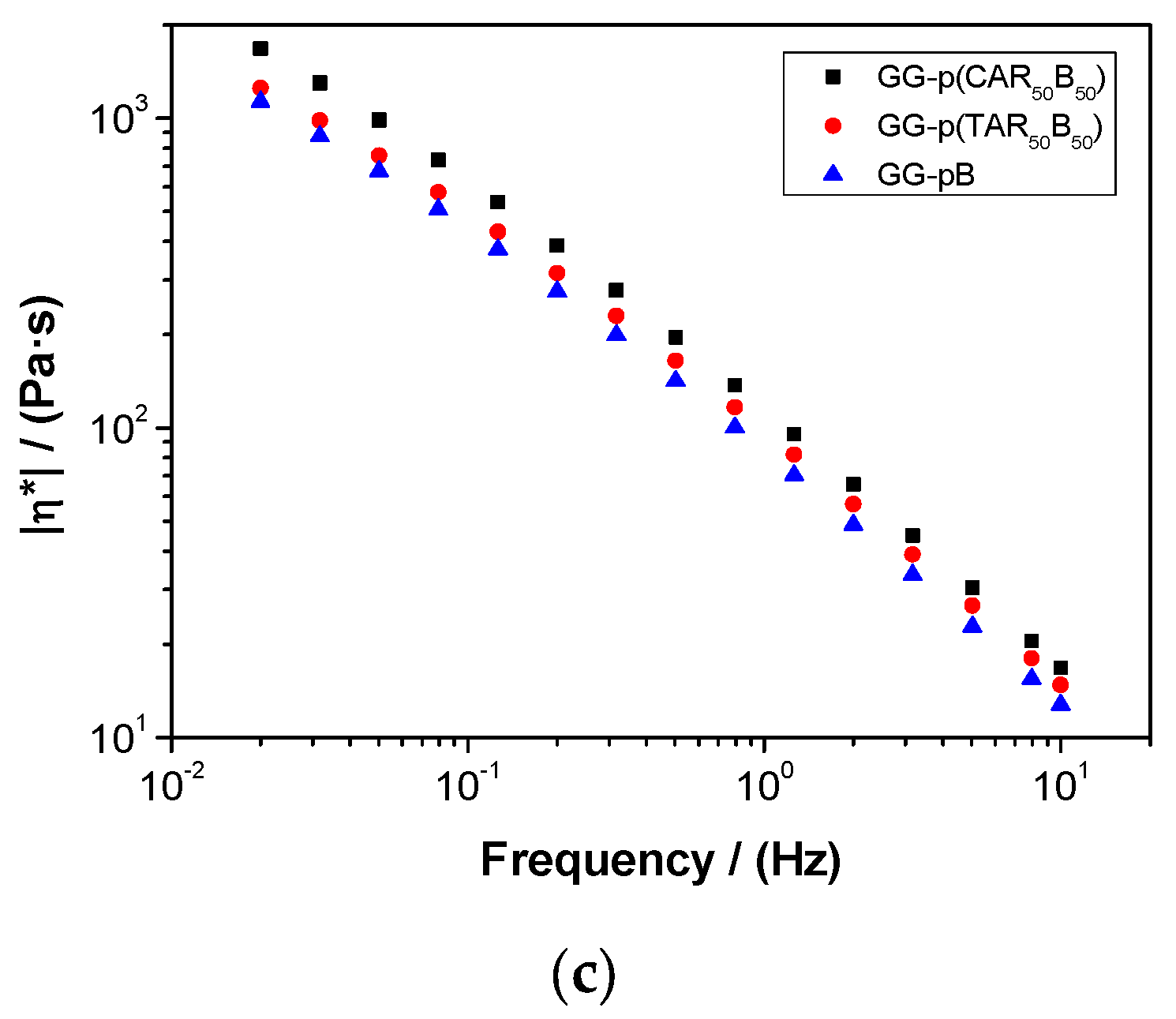
2.5. Characterization of Guar-Gum-Based Semi-IPNs
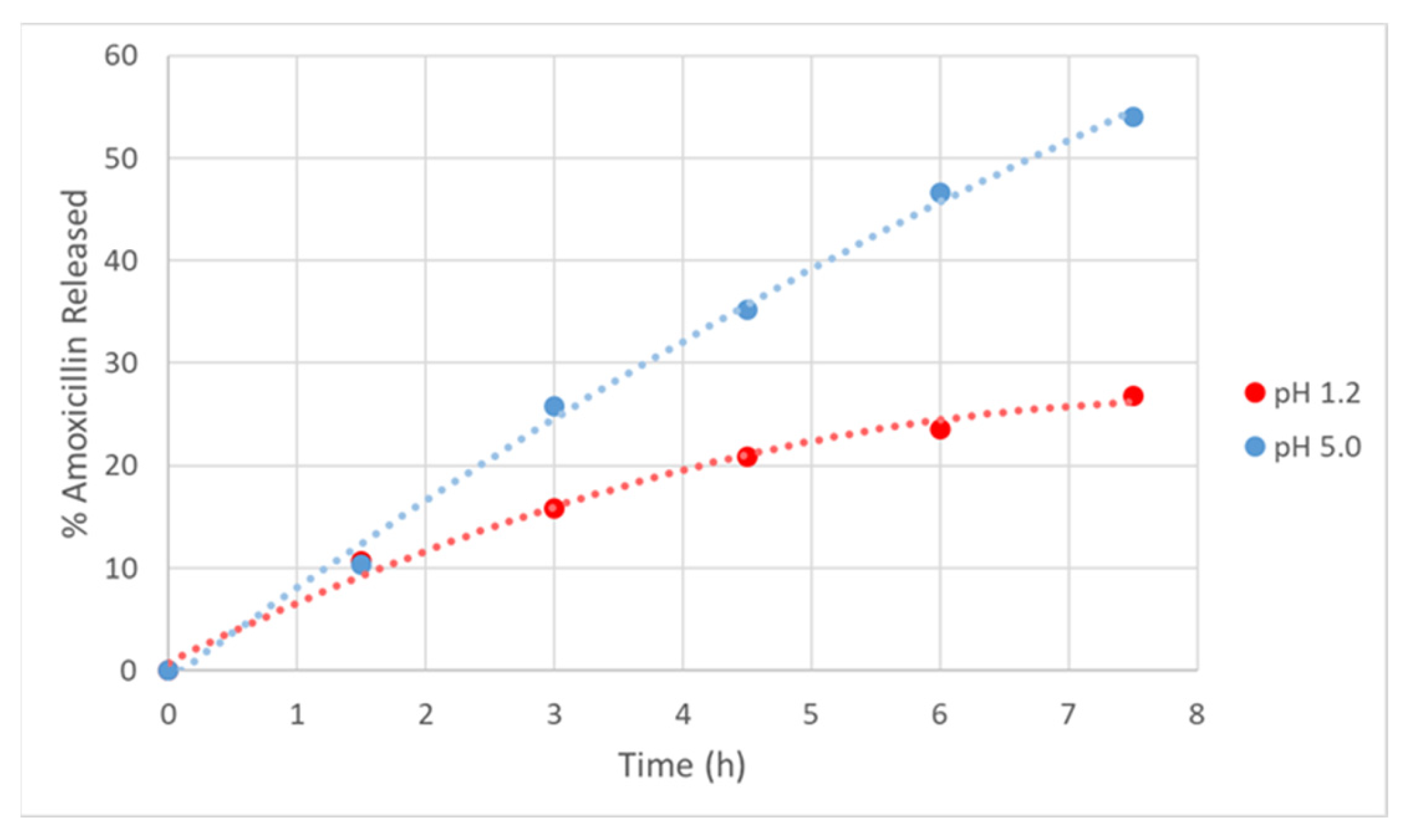
2.6. AMOX Loading and In Vitro Drug Release Studies
3. Discussion
3.1. Preparation and Characterization of Homopolymers and Copolymers
3.2. Hydrolytic Degradation of Diallyl Moieties in B Monomer and Copolymer co-p(CAR50B50)
3.3. Synthesis and Characterization of Guar-Gum-Based Semi-IPNs
3.4. AMOX Loading and In Vitro Drug Release Studies
4. Materials and Methods
4.1. Chemicals
4.2. Preparation and Characterization of Homopolymers and Copolymers
4.3. Preparation and Characterization of Guar-Gum-Based Semi-IPNs
4.4. AMOX Loading and In Vitro Drug Release Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Cancer Society: Cancer Facts & Figures. Atlanta Am. Cancer Soc. 2021, 1–72.
- Fernández-Navarro, P.; Roquette, R.; Nuñez, O.; de Sousa-Uva, M.; García-Pérez, J.; López-Abente, G.; Nunes, B.; González-Sánchez, M.; Dinis, J.; Carmona, R.; et al. Atlas of Cancer Mortality in Portugal and Spain (2003–2012). Natl. Inst. Health Doutor Ricardo Jorge (Port.) Natl. Inst. Health Carlos III (Spain) 2021. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Dore, M.P.; Graham, D.Y. Diagnosis and Treatment of Helicobacter pylori Infection. Chin. J. Gastroenterol. 2022, 73, 183–195. [Google Scholar] [CrossRef]
- Grosso, R.; De-Paz, M.V. Scope and Limitations of Current Antibiotic Therapies against Helicobacter pylori: Reviewing Amoxicillin Gastroretentive Formulations. Pharmaceutics 2022, 14, 1340. [Google Scholar] [CrossRef]
- Kiyotoki, S.; Nishikawa, J.; Sakaida, I. Efficacy of vonoprazan for helicobacter pylori eradication. Intern. Med. 2020, 59, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, N.; Galbis, E.; Romero-Azogil, L.; Benito, E.; Lucas, R.; García-Martín, M.G.; De-Paz, M.V. In-depth study into polymeric materials in low-density gastroretentive formulations. Pharmaceutics 2020, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Rajinikanth, P.S.; Balasubramaniam, J.; Mishra, B. Development and evaluation of a novel floating in situ gelling system of amoxicillin for eradication of Helicobacter pylori. Int. J. Pharm. 2007, 335, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Patel, D.K.; Patel, C.N. Formulation and Evaluation of Floating Oral In Situ Gelling System of Amoxicillin. ISRN Pharm. 2011, 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Rajinikanth, P.S.; Mishra, B. Stomach-site specific drug delivery system of clarithromycin for eradication of Helicobacter pylori. Chem. Pharm. Bull. 2009, 57, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Ranade, A.N.; Wankhede, S.S.; Ranpise, N.S.; Mundada, M.S. Development of bilayer floating tablet of amoxicillin and aloe vera gel powder for treatment of gastric ulcers. AAPS Pharm. Sci. Tech. 2012, 13, 1518–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Conti, C.; Colombo, G.; Castrati, L.; Scarpignato, C.; Barata, P.; Sandri, G.; Caramella, C.; Bettini, R.; Buttini, F.; et al. Floating modular drug delivery systems with buoyancy independent of release mechanisms to sustain amoxicillin and clarithromycin intra-gastric concentrations. Drug Dev. Ind. Pharm. 2016, 42, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Charoenying, T.; Patrojanasophon, P.; Ngawhirunpat, T.; Rojanarata, T.; Akkaramongkolporn, P.; Opanasopit, P. Fabrication of floating capsule-in-3D-printed devices as gastro-retentive delivery systems of amoxicillin. J. Drug Deliv. Sci. Technol. 2020, 55, 101393. [Google Scholar] [CrossRef]
- Kamsali, A.; Eranti, B.; Ch, M.; Manne, R.; Barghav, G.C.; Reddy, P.S. Development and Optimization of Amoxicillin Floating Raft System to effectively treat Helicobacter pylori infection. Ars Pharm. 2020, 61, 163–168. [Google Scholar] [CrossRef]
- Awasthi, R.; Kulkarni, G.T.; Pawar, V.K.; Garg, G. Optimization studies on gastroretentive floating system using response surface methodology. AAPS Pharm. Sci. Tech. 2012, 13, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gils, P.S.; Ray, D.; Sahoo, P.K. Characteristics of xanthan gum-based biodegradable superporous hydrogel. Int. J. Biol. Macromol. 2009, 45, 364–371. [Google Scholar] [CrossRef]
- Gemeinhart, R.A.; Park, H.; Park, K. Pore structure of superporous hydrogels. Polym. Adv. Technol. 2000, 11, 617–625. [Google Scholar] [CrossRef]
- Wang, B. Super Porous Hydrogel: A Promising Gastroretentive Drug Delivery System. World J. Pharm. Pharm. Sci. 2017, 6, 129–159. [Google Scholar] [CrossRef]
- Omidian, H.; Park, K.; Rocca, J.G. Recent developments in superporous hydrogels. J. Pharm. Pharmacol. 2007, 59, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Lv, Y.; Zhang, J.B.; Wang, B.; Lv, G.J.; Ma, X.J. Gastroretentive drug delivery systems for the treatment of Helicobacter pylori. World J. Gastroenterol. 2014, 20, 9321–9329. [Google Scholar] [CrossRef]
- Mandal, U.K.; Chatterjee, B.; Senjoti, F.G. Gastro-retentive drug delivery systems and their in vivo success: A recent update. Asian J. Pharm. Sci. 2016, 8, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.L.; Ahmed, T.; Mannan, M.A. Development of floating-mucoadhesive microsphere for site specific release of metronidazole. Adv. Pharm. Bull. 2016, 6, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Sandilya, A.; Patel, U.; Shukla, A.; Sadhu, S.D. A bio-inspired exploration of eco-friendly bael gum and guar gum-based bioadhesive as tackifiers for packaging applications. Int. J. Adhes. Adhes. 2021, 110, 102946. [Google Scholar] [CrossRef]
- Verdugo-Fernández, P.M. Stimuli Cleavable ABA Block Copolymers: Synthesis of Degradable Thermoplastic Elastomers from Renewable Resources [Dissertation]; Universitat Rovira i Virgili: Tarragona, Spain, 2020. [Google Scholar]
- Bray, D. Critical Point Drying of Biological Specimens for Scanning Electron Microscopy. Supercritical Fluid Methods and Protocols. Methods In Biotechnology; Williams, J.R., Clifford, A.A., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 235–243. [Google Scholar] [CrossRef]
- Tripathi, J.; Thapa, P.; Maharjan, R.; Jeong, S.H. Current State and Future Perspectives on Gastroretentive Drug Delivery Systems. Pharmaceutics 2019, 11, 193. [Google Scholar] [CrossRef] [Green Version]
- Jafar, M.; Salahuddin, M.; Khan, M.S.A.; Alshehry, Y.; Alrwaili, N.R.; Alzahrani, Y.A.; Imam, S.S.; Alshehri, S. Preparation and in vitro-in vivo evaluation of luteolin loaded gastroretentive microsponge for the eradication of helicobacter pylori infections. Pharmaceutics 2021, 13, 2094. [Google Scholar] [CrossRef]
- Carvalho, I.C.; Medeiros Borsagli, F.G.L.; Mansur, A.A.P.; Caldeira, C.L.; Haas, D.J.; Lage, A.P.; Ciminelli, V.S.T.; Mansur, H.S. 3D sponges of chemically functionalized chitosan for potential environmental pollution remediation: Biosorbents for anionic dye adsorption and ‘antibiotic-free’ antibacterial activity. Environ. Technol. 2021, 42, 2046–2066. [Google Scholar] [CrossRef]
- Biswas, A.; Mondal, S.; Das, S.K.; Bose, A.; Thomas, S.; Ghosal, K.; Roy, S.; Provaznik, I. Development and Characterization of Natural Product Derived Macromolecules Based Interpenetrating Polymer Network for Therapeutic Drug Targeting. ACS Omega 2021, 6, 28699–28709. [Google Scholar] [CrossRef]
- Hadke, J.; Khan, S. Preparation of sterculia foetida-pullulan-based semi-interpenetrating polymer network gastroretentive microspheres of amoxicillin trihydrate and optimization by response surface methodology. Turk. J. Pharm. Sci. 2021, 18, 388–397. [Google Scholar] [CrossRef]
- Raghu Kiran, C.V.S.; Gopinath, C. Development and evaluation of interpenetrating polymer network based superporous hydrogel gastroretentive drug delivery systems (SPH IPN-GRDDS). Mater. Today Proc. 2021, 46, 3056–3061. [Google Scholar] [CrossRef]
- Panahi, Y.; Gharekhani, A.; Hamishehkar, H.; Zakeri-Milani, P.; Gharekhani, H. Stomach-specific drug delivery of clarithromycin using a semi interpenetrating polymeric network hydrogel made of montmorillonite and chitosan: Synthesis, characterization and in vitro drug release study. Adv. Pharm. Bull. 2019, 9, 159–173. [Google Scholar] [CrossRef]
- Majeed, A.; Pervaiz, F.; Shoukat, H.; Shabbir, K.; Noreen, S.; Anwar, M. Fabrication and evaluation of pH sensitive chemically cross-linked interpenetrating network [Gelatin/Polyvinylpyrrolidone-co-poly(acrylic acid)] for targeted release of 5-fluorouracil. Polym. Bull. 2022, 79, 1–20. [Google Scholar] [CrossRef]
- Cardos, I.A.; Zaha, D.C.; Sindhu, R.K.; Cavalu, S. Revisiting therapeutic strategies for h. Pylori treatment in the context of antibiotic resistance: Focus on alternative and complementary therapies. Molecules 2021, 26, 6078. [Google Scholar] [CrossRef]
- Vuković, J.S.; Filipović, V.V.; Babić Radić, M.M.; Vukomanović, M.; Milivojevic, D.; Ilic-Tomic, T.; Nikodinovic-Runic, J.; Tomić, S.L. In Vitro and In Vivo Biocompatible and Controlled Resveratrol Release Performances of HEMA/Alginate and HEMA/Gelatin IPN Hydrogel Scaffolds. Polymers 2022, 14, 4459. [Google Scholar] [CrossRef]
- Tuan, H.N.A.; Nhu, V.T.T. Synthesis and properties of pH-thermo dual responsive semi-IPN hydrogels based on N,N′-diethylacrylamide and itaconamic acid. Polymers 2020, 12, 1139. [Google Scholar] [CrossRef] [PubMed]
- Dragan, E.S.; Dinu, M.V.; Ghiorghita, C.A.; Lazar, M.M.; Doroftei, F. Preparation and Characterization of Semi-IPN Cryogels Based on Polyacrylamide and Poly(N,N-dimethylaminoethyl methacrylate); Functionalization of Carrier with Monochlorotriazinyl-β-cyclodextrin and Release Kinetics of Curcumin. Molecules 2021, 26, 6975. [Google Scholar] [CrossRef] [PubMed]
- Ferris, C.; de Paz, M.V.; Galbis, J.A. L-arabinitol-based functional polyurethanes. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 1147–1154. [Google Scholar] [CrossRef]
- Ferris, C.; de Paz, M.V.; Galbis, J.A. Synthesis of Functional Sugar-Based Polyurethanes. Macromol. Chem. Phys. 2012, 213, 480–488. [Google Scholar] [CrossRef]
- Ferris, C.; de Paz, M.V.; Aguilar-de-Leyva, A.; Caraballo, I.; Galbis, J.A. Reduction-sensitive functionalized copolyurethanes for biomedical applications. Polym. Chem. 2014, 5, 2370–2381. [Google Scholar] [CrossRef]
- Iglesias, N.; Galbis, E.; Valencia, C.; Díaz-Blanco, M.J.; Lacroix, B.; De-Paz, M.-V. Biodegradable double cross-linked chitosan hydrogels for drug delivery: Impact of chemistry on rheological and pharmacological performance. Int. J. Biol. Macromol. 2020, 165, 2205–2218. [Google Scholar] [CrossRef]
- Iglesias, N.; Galbis, E.; Valencia, C.; De-Paz, M.-V.; Galbis, J.A. Reversible pH-sensitive chitosan-based hydrogels. Influence of dispersion composition on rheological properties and sustained drug delivery. Polymers 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Brüning, J.; Petereit, A.C.; Alig, E.; Bolte, M.; Dressman, J.B.; Schmidt, M.U. Characterization of a new solvate of risedronate. J. Pharm. Sci. 2011, 100, 863–873. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | CAR (μL) | TAR (mg) | B (μL) | Diallyl Monomers Molar Ratio | EDDET (μL) | Initiator (mg) | Solvent (mL) |
|---|---|---|---|---|---|---|---|
| hom-pCAR | 176.8 | - | - | - | 200.7 | 31.6 | 2 (EtOAc) |
| hom-pTAR | - | 222.4 | - | - | 158.6 | 25 | 2 (MeOH) |
| hom-pB | - | - | 202.2 | - | 200.7 | 31.6 | 2 (EtOAc) |
| co-p(CAR80B20) | 141.5 | - | 40.4 | 80:20 | 211.2 | 31.6 | 2 (EtOAc) |
| co-p(CAR50B50) | 88.4 | - | 101.1 | 50:50 | 211.2 | 31.6 | 2 (EtOAc) |
| co-p(CAR20B80) | 35.36 | - | 161.7 | 20:80 | 211.2 | 31.6 | 2 (EtOAc) |
| Polymer | Diallyl Monomer and Molar Percentage a | Mwb | Mnb | Mw/Mnb | TGA c | ||
|---|---|---|---|---|---|---|---|
| Td (°C) | ΔW(%) | ||||||
| hom-pCAR | CAR | 42,600 | 33,400 | 1.3 | 268.5 | 340.6 | 98.9 |
| hom-pTAR | TAR | 21,900 | 18,000 | 1.2 | 277.9 | 351.3 | 88.7 |
| hom-pB | B | 19,800 | 16,500 | 1.2 | 226.5 | 350.6 | 99.4 |
| co-p(CAR80B20) | CAR-B 80:20 | 21,500 | 18,000 | 1.2 | 253.4 | 339.9 | 100.0 |
| co-p(CAR50B50) | CAR-B 50:50 | 11,200 | 8800 | 1.3 | 230.9 | 334.0 | 98.3 |
| co-p(CAR20B80) | CAR-B 20:80 | 8100 | 6900 | 1.2 | 236.4 | 342.2 | 98.9 |
| Copolymer | Ratio Diallyl Monomers in the Feed (in Molar Percent) | 1H-NMR Integrals for the Peaks at δ (ppm) 4.25 (from CAR Moiety: 4 H) and 1.28 (from B Moiety: 3 H) | 1H-NMR Integrals for the Peaks at δ (ppm) 4.25 (from CAR Moiety: 4H) and 1.28 (from B Moiety: 3 H) Divided by the n.º of H for each Peak | Experimental CAR/B Molar Ratios (in %) | ||||
|---|---|---|---|---|---|---|---|---|
| CAR | B | 4.25 ppm | 1.28 ppm | 4.25 ppm | 1.28 ppm | CAR | B | |
| co-p(CAR80B20) | 80 | 20 | 4 a | 0.6494 | 1 | 0.2165 | 82.1 | 17.9 |
| co-p(CAR50B50) | 50 | 50 | 4 a | 2.7339 | 1 | 0.9113 | 52.4 | 47.6 |
| co-p(CAR20B80) | 20 | 80 | 4 a | 10.6580 | 1 | 3.5527 | 21.9 | 78.1 |
| semi-IPN | CAR (μL) | TAR (mg) | B (μL) | Diallyl Monomers Molar Ratio | EDDET (μL) | TriT (mg) | Initiator (mg) |
|---|---|---|---|---|---|---|---|
| GG-p(TAR50B50) | - | 122 | 87.7 | 50:50 | 160.1 | 22.7 | 27.4 |
| GG-pB | - | - | 198.1 | 100 | 180.9 | 25.7 | 31 |
| GG-p(CAR50B50) | 86.6 | - | 99.1 | 50:50 | 180.9 | 25.7 | 31 |
| GG-p(CAR80B20) | 138.6 | - | 39.6 | 80:20 | 190.4 | 27 | 31 |
| GG-p(CAR20B80) | 34.35 | - | 158.5 | 20:80 | 190.4 | 27 | 31 |
| Systems | Critical Strain (%) | G′ (Pa) | tan (δ) | ||||
|---|---|---|---|---|---|---|---|
| 0.02 Hz | 1.00 Hz | 10.0 Hz | 0.02 Hz | 1.00 Hz | 10.0 Hz | ||
| GG-p(CAR50B50) | 0.012 ± 0.003 | 183 ± 34 | 681 ± 51 | 1012 ± 22 | 0.81 ± 0.09 | 0.35 ± 0.02 | 0.23 ± 0.01 |
| GG-p(TAR50B50) | 0.012 ± 0.003 | 133 ± 24 | 574 ± 48 | 886 ± 21 | 0.88 ± 0.06 | 0.39 ± 0.03 | 0.25 ± 0.01 |
| GG-pB | 0.086 ± 0.020 | 120 ± 20 | 495 ± 40 | 765 ± 20 | 0.85 ± 0.05 | 0.38 ± 0.03 | 0.24 ± 0.01 |
| Entry | Type of IPN | Drug | POLYMER 1 | POLYMER 2 | Polymerization Method | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| (A) Polysaccharides (B) p(Meth)acryl(ate/amide)s (C) Others | (A) Xrlinked? (Y/N) (B) if Yes, Xrlinker | (A) Polysaccharides (B) p(Meth)acryl(ate/amide)s (C) Others | (A) Xrlinked? (Y/N) (B) if Yes, Xrlinker | |||||
| 1 | 2nd-Generation Semi-IPN | Diclofenac sodium | (A) Sodium Alginate | Y; Ca2+ ions (from CaCl2) | (A) Guar Gum, Xanthan gum, Gellan gum (C) PVA | N; --- | --- Physical mixture and then ionic gelation method | [28] |
| 2 | 2nd-Generation Full-IPN | Amoxicillin | (A) Sterculia foetida | Y; Glutaraldehyde | (A) Pullulan | Y; Glutaraldehyde | Physical mixture and then Xrlinked together | [29] |
| 3 | 2nd-Generation Full-IPN | Esomeprazole | (A) CTS | Y; Glutaraldehyde | (C) PVA | Y; Glutaraldehyde | Physical mixture and then Xrlinked together | [30] |
| 4 | 3rd-Generation Semi-IPN | Clarithromycin (H. Pylori) | (A) CTS and (C) PVP, MMT | N; --- | (B) PAAm-co-PAA | Y; MBA | Polymer 2: Free radical polymerization (TEMED, APS) | [31] |
| 5 | 3rd-Generation Semi-IPN | 5-Fluorouracil | (A) Gelatin and (C) PVP | N; --- | (B) PAA | Y; EGDMA | Polymer 2: Free radical polymerization (APS, NaHSO3) | [32] |
| 6 | 3rd-Generation Semi-IPN | Curcumine | (B) PDMAEMA | N; --- | (B) PAAm | Y; MBA | Polymer 1: Free radical polymerization (AIBN, toluene) Polymer 2: Free radical polymerization (TEMED, APS) | [33] |
| 7 | 3rd-Generation Full-IPN | Resveratrol | (A) Sodium Alginate and Gelatin | Y; Ester formation between COOH and OH groups from polymers (coupling agent: EDC/NHS) | (B) PHEMA | Y; PEGDMA | Polymer 2: Free radical polymerization (PPS, TEMED) | [34] |
| 8 | 3rd-Generation Full-IPN | --- | (B) PDEAAm-co-PIAM | Y; Xrlinked with monomer IAM | (B) PDEAAm | Y; MBA | Polymer 1: Free radical polymerization (TEMED, APS) Polymer 2: Free radical polymerization (TEMED, APS) | [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grosso, R.; Benito, E.; Carbajo-Gordillo, A.I.; García-Martín, M.G.; Perez-Puyana, V.; Sánchez-Cid, P.; de-Paz, M.-V. Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept. Int. J. Mol. Sci. 2023, 24, 2281. https://doi.org/10.3390/ijms24032281
Grosso R, Benito E, Carbajo-Gordillo AI, García-Martín MG, Perez-Puyana V, Sánchez-Cid P, de-Paz M-V. Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept. International Journal of Molecular Sciences. 2023; 24(3):2281. https://doi.org/10.3390/ijms24032281
Chicago/Turabian StyleGrosso, Roberto, Elena Benito, Ana I. Carbajo-Gordillo, M. Gracia García-Martín, Víctor Perez-Puyana, Pablo Sánchez-Cid, and M.-Violante de-Paz. 2023. "Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept" International Journal of Molecular Sciences 24, no. 3: 2281. https://doi.org/10.3390/ijms24032281
APA StyleGrosso, R., Benito, E., Carbajo-Gordillo, A. I., García-Martín, M. G., Perez-Puyana, V., Sánchez-Cid, P., & de-Paz, M.-V. (2023). Biodegradable Guar-Gum-Based Super-Porous Matrices for Gastroretentive Controlled Drug Release in the Treatment of Helicobacter pylori: A Proof of Concept. International Journal of Molecular Sciences, 24(3), 2281. https://doi.org/10.3390/ijms24032281