

CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pharmacological Ways of Boosting CAR-T-Cell Therapy
2.1. Hematological Malignancies
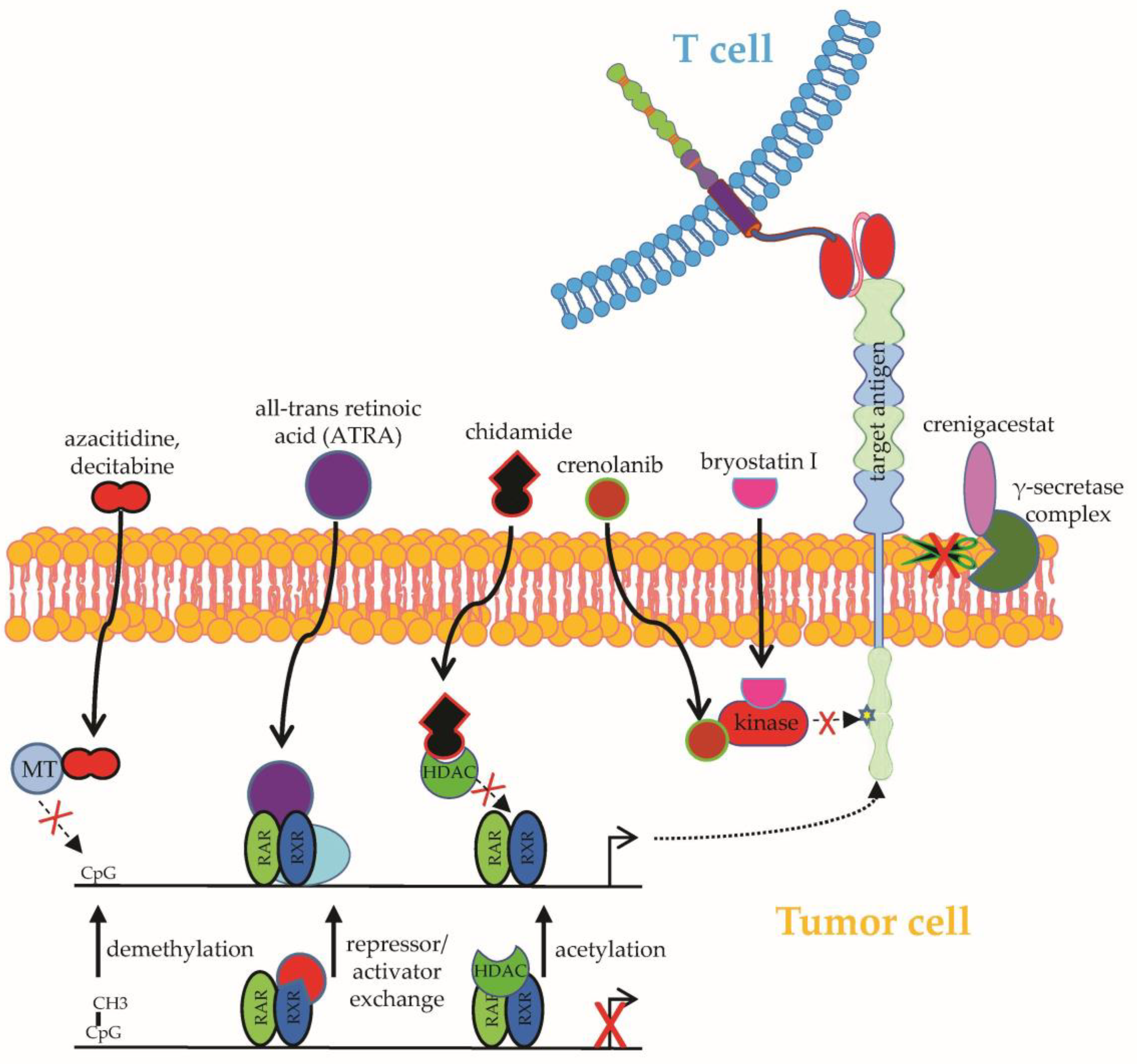
2.1.1. Upregulation of Target Antigens
2.1.2. Combinatorial Targeting
2.1.3. Improving CAR-T-Cell Functionality
2.2. Solid Tumors
2.2.1. Upregulation of Target Antigens
2.2.2. Combinatorial Targeting
2.2.3. Improving CAR-T-Cell Functionality
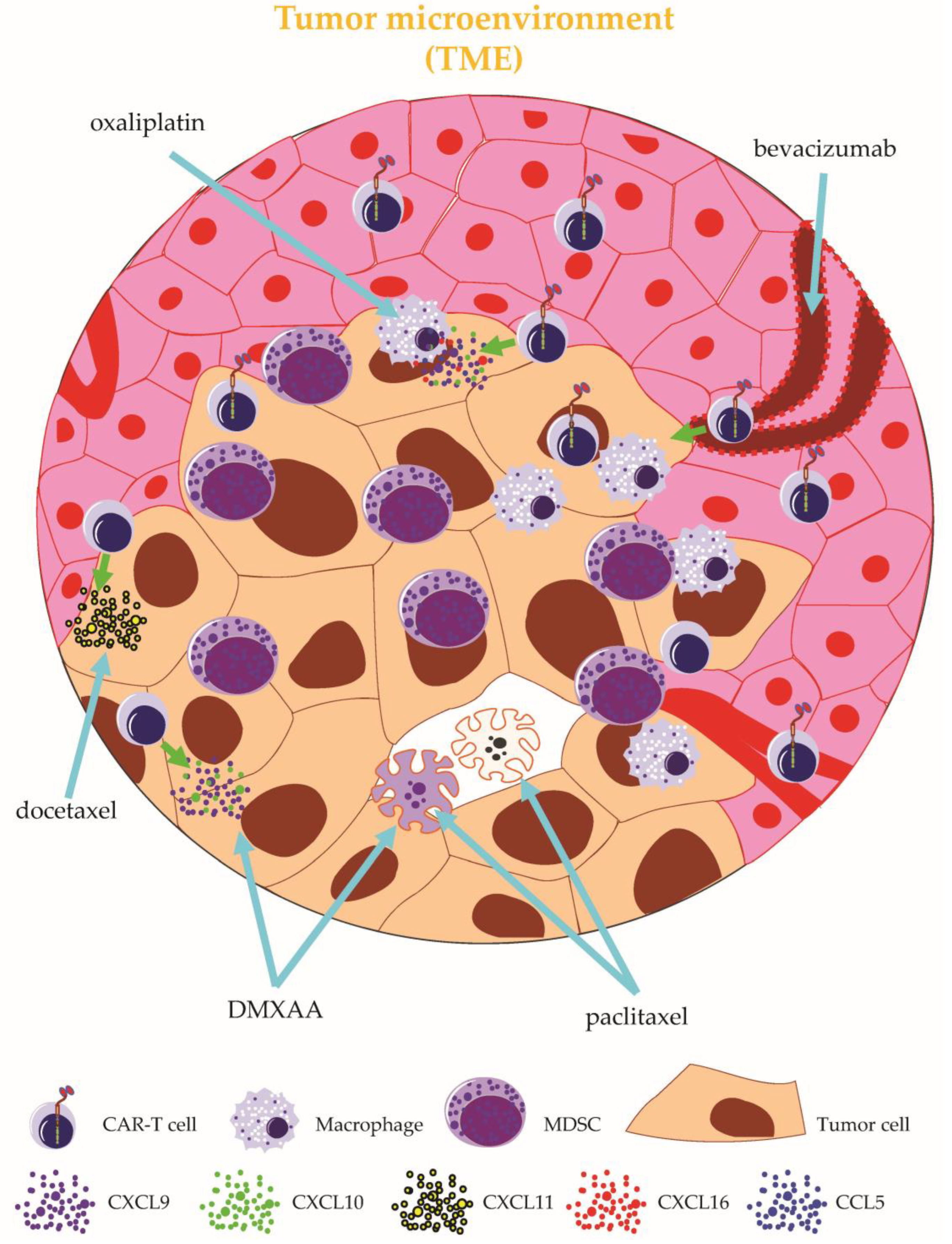
2.2.4. Targeting the Tumor Microenvironment
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Dickinson, M.; Munoz, J.; Ulrickson, M.L.; Thieblemont, C.; Oluwole, O.O.; Herrera, A.F.; Ujjani, C.S.; Lin, Y.; Riedell, P.A.; et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: The phase 2 ZUMA-12 trial. Nat. Med. 2022, 28, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Abken, H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology 2022, 107, 446–463. [Google Scholar] [CrossRef]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef]
- Ivica, N.A.; Young, C.M. Tracking the CAR-T Revolution: Analysis of Clinical Trials of CAR-T and TCR-T Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef]
- Chong, E.A.; Ruella, M.; Schuster, S.J. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N. Engl. J. Med. 2021, 384, 673–674. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving cars to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Achmad, H.; Bokov, D.; Abdelbasset, W.K.; Alsadoon, Z.; Chupradit, S.; Suksatan, W.; Shariatzadeh, S.; Hasanpoor, Z.; Yazdanifar, M.; et al. Hurdles to breakthrough in CAR T cell therapy of solid tumors. Stem Cell Res. Ther. 2022, 13, 140. [Google Scholar] [CrossRef]
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Ahmadi Najafabadi, M.; Yousefi, F.; Mirarefin, S.M.J.; Rahbarizadeh, F. Recent Advances in Solid Tumor CAR-T Cell Therapy: Driving Tumor Cells from Hero to Zero? Front. Immunol. 2022, 13, 795164. [Google Scholar] [CrossRef] [PubMed]
- Mansilla-Soto, J.; Eyquem, J.; Haubner, S.; Hamieh, M.; Feucht, J.; Paillon, N.; Zucchetti, A.E.; Li, Z.; Sjöstrand, M.; Lindenbergh, P.L.; et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med. 2022, 28, 345–352. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Brentjens, R.J. Tumors evading CARs-the chase is on. Nat. Med. 2018, 24, 1492–1493. [Google Scholar] [CrossRef]
- Leick, M.B.; Silva, H.; Scarfò, I.; Larson, R.; Choi, B.D.; Bouffard, A.A.; Gallagher, K.; Schmidts, A.; Bailey, S.R.; Kann, M.C.; et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell 2022, 40, 494–508. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Myburgh, R.; Matschulla, T.; Taromi, S.; Aumann, K.; Clarson, J.; Vinnakota, J.M.; Shoumariyeh, K.; et al. Demethylating therapy increases anti-CD123 CAR T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021, 12, 6436. [Google Scholar] [CrossRef]
- Li, S.; Xue, L.; Wang, M.; Qiang, P.; Xu, H.; Zhang, X.; Kang, W.; You, F.; Xu, H.; Wang, Y.; et al. Decitabine enhances cytotoxic effect of T cells with an anti-CD19 chimeric antigen receptor in treatment of lymphoma. Onco. Targets. Ther. 2019, 12, 5627–5638. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Zou, R.; Wang, P.; Zhu, Q.; Kang, L.; Ping, N.; Xia, F.; Liu, H.; Kong, D.; Yu, L.; et al. Decitabine-primed tandem CD19/CD22 CAR-T therapy in relapsed/refractory diffuse large B-cell lymphoma patients. Front. Immunol. 2022, 13, 969660. [Google Scholar] [CrossRef]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Hudecek, M.; Einsele, H. What is the future of immunotherapy in multiple myeloma? Blood 2020, 136, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- García-Guerrero, E.; Rodríguez-Lobato, L.G.; Sierro-Martínez, B.; Danhof, S.; Bates, S.; Frenz, S.; Haertle, L.; Götz, R.; Sauer, M.; Rasche, L.; et al. ATRA works synergistically with the γ-secretase inhibitor crenigacestat to augment BCMA on multiple myeloma and the efficacy of BCMA-CAR T-cells. Haematologica 2022. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.J.; Pont, M.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; Tuazon, S.A.; et al. Efficacy and Safety of Fully Human Bcma CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase Bcma Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2019, 134, 204. [Google Scholar] [CrossRef]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Q.; Xu, H.; Zhou, J. Upregulation of CD22 by Chidamide promotes CAR T cells functionality. Sci. Rep. 2021, 11, 20637. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Highfill, S.L.; Walsh, Z.; Nguyen, S.M.; Lei, H.; Shern, J.F.; Qin, H.; Kraft, I.L.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin. Cancer Res. 2019, 25, 5329–5341. [Google Scholar] [CrossRef] [Green Version]
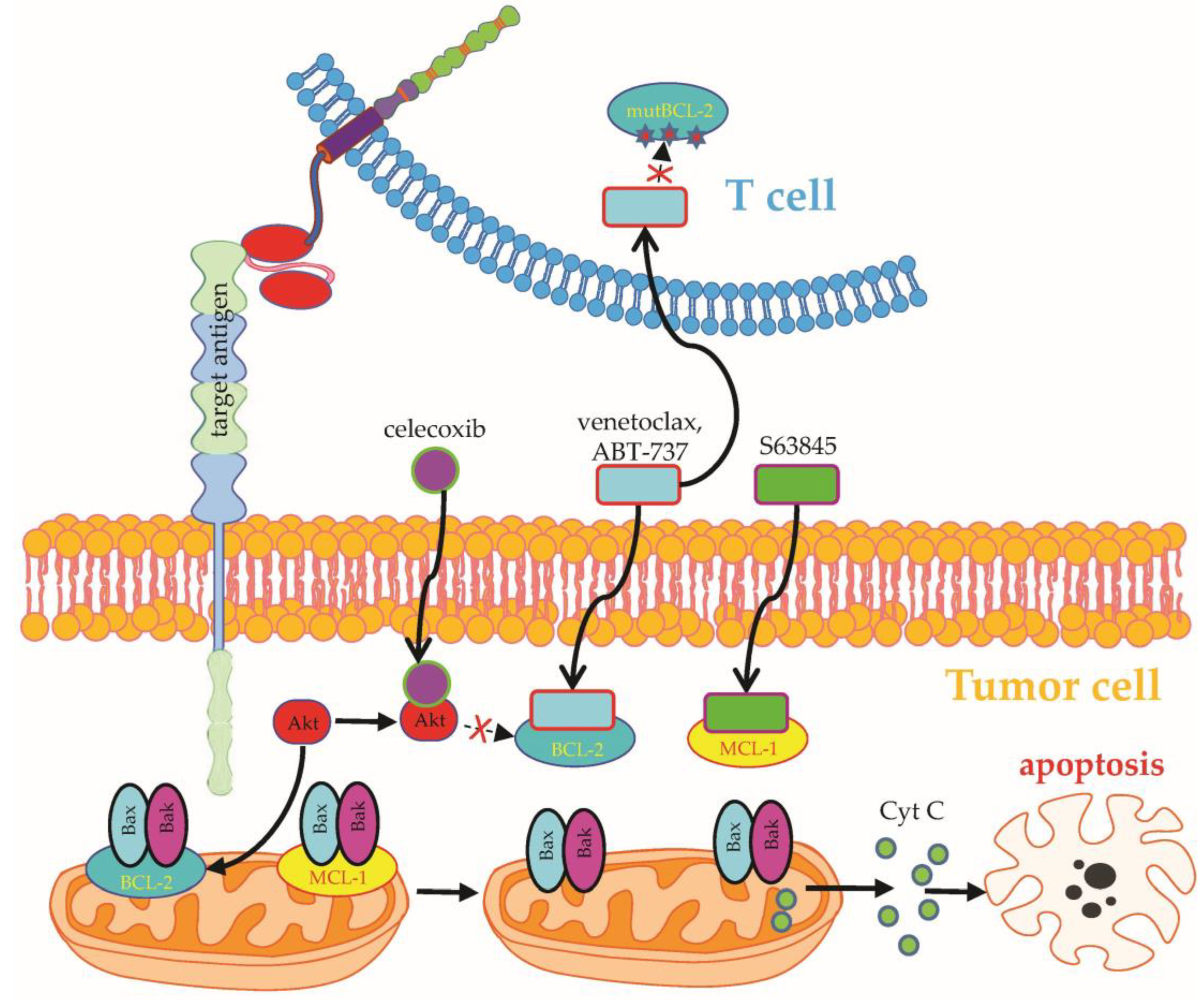
- Stolz, C.; Hess, G.; Hähnel, P.S.; Grabellus, F.; Hoffarth, S.; Schmid, K.W.; Schuler, M. Targeting Bcl-2 family proteins modulates the sensitivity of B-cell lymphoma to rituximab-induced apoptosis. Blood 2008, 112, 3312–3321. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Yu, L.; Yang, L. Mechanisms of venetoclax resistance and solutions. Front. Oncol. 2022, 12, 1005659. [Google Scholar] [CrossRef]
- Lee, Y.G.; Guruprasad, P.; Ghilardi, G.; Pajarillo, R.; Sauter, C.T.; Patel, R.; Ballard, H.J.; Hong, S.J.; Chun, I.; Yang, N.; et al. Modulation of BCL-2 in Both T Cells and Tumor Cells to Enhance Chimeric Antigen Receptor T-cell Immunotherapy against Cancer. Cancer Discov. 2022, 12, 2372–2391. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, L.; Ni, M.; Neuber, B.; Wang, S.; Gong, W.; Sauer, T.; Sellner, L.; Schubert, M.-L.; Hückelhoven-Krauss, A.; et al. Pre-sensitization of Malignant B Cells Through Venetoclax Significantly Improves the Cytotoxic Efficacy of CD19.CAR-T Cells. Front. Immunol. 2020, 11, 608167. [Google Scholar] [CrossRef] [PubMed]
- Mandeville, T.K.; Mavis, C.; Gu, J.; Olejniczak, S.; Paragh, G.; Dey, P.; Hernandez-Ilizaliturri, F.J. Contribution of Bcl-2 Inhibitor Venetoclax Toward Anti-CD19 CAR T Cell Efficacy in Relapsed/Refractory Diffuse Large B Cell Lymphoma. Blood 2021, 138, 1719. [Google Scholar] [CrossRef]
- Karlsson, H.; Lindqvist, A.C.; Fransson, M.; Paul-Wetterberg, G.; Nilsson, B.; Essand, M.; Nilsson, K.; Frisk, P.; Jernberg-Wiklund, H.; Loskog, A. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013, 20, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Freeman, C.L.; Sehn, L.H. A tale of two antibodies: Obinutuzumab versus rituximab. Br. J. Haematol. 2018, 182, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Torres-Collado, A.X.; Jazirehi, A.R. Overcoming Resistance of Human Non-Hodgkin’s Lymphoma to CD19-CAR CTL Therapy by Celecoxib and Histone Deacetylase Inhibitors. Cancers 2018, 10, 200. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Wang, L.; Ni, M.; Neuber, B.; Wang, S.; Gong, W.; Sauer, T.; Schubert, M.-L.; Hückelhoven-Krauss, A.; Xia, R.; et al. Dual Effects of Cyclooxygenase Inhibitors in Combination with CD19.CAR-T Cell Immunotherapy. Front. Immunol. 2021, 12, 670088. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S.; Shestova, O.; Fraietta, J.A.; Qayyum, S.; Zhang, Q.; Maus, M.V.; Liu, X.; Nunez-Cruz, S.; Klichinsky, M.; et al. The Addition of the BTK Inhibitor Ibrutinib to Anti-CD19 Chimeric Antigen Receptor T Cells (CART19) Improves Responses against Mantle Cell Lymphoma. Clin. Cancer Res. 2016, 22, 2684–2696. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.H.; Yeung, C.C.S.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020, 135, 1650–1660. [Google Scholar] [CrossRef]
- Gill, S.I.; Vides, V.; Frey, N.V.; Hexner, E.; Metzger, S.; O’Brien, M.; Hwang, W.-T.; Brogdon, J.L.; Davis, M.M.; Fraietta, J.A.; et al. Anti-CD19 CAR T Cells in Combination with Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia. Blood Adv. 2022, 6, 5774–5785. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Deng, H.; Mu, J.; Li, Q.; Pu, Y.; Jiang, Y.; Deng, Q.; Qian, Z. Ibrutinib improves the efficacy of anti-CD19-CAR T-cell therapy in patients with refractory non-Hodgkin lymphoma. Cancer Sci. 2021, 112, 2642–2651. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Gallagher, K.M.E.; Leick, M.B.; Kann, M.C.; Budka, J.; Sowrirajan, B.; Nguyen, K.; Shen, R.R.; Bot, A.; Maus, M.V. Effects of Prior Exposure to Tec Kinase(BTK/ITK) Inhibitors on Kte-X19 Products. Blood 2021, 138, 3849. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
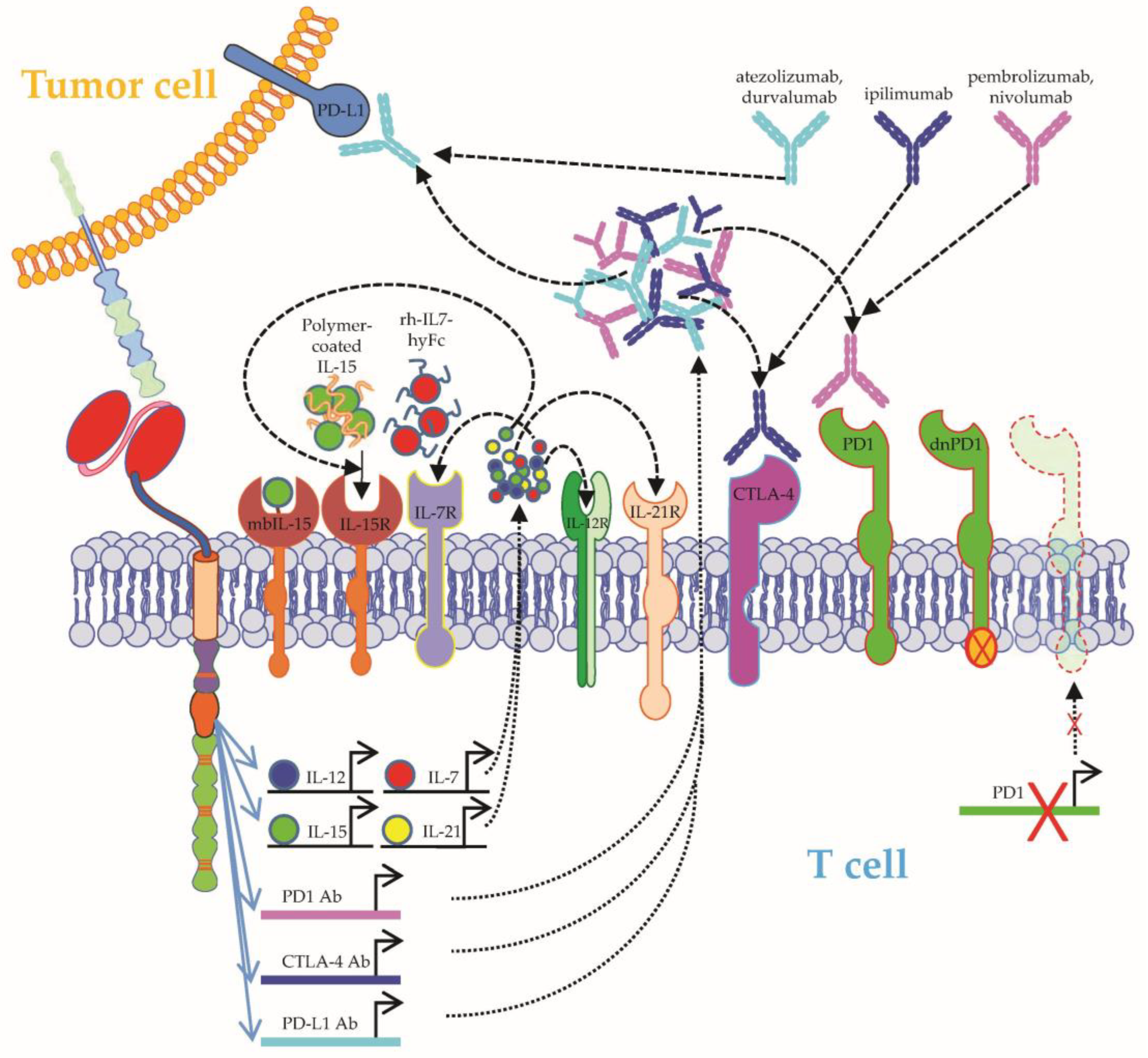
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Lanitis, E.; Rota, G.; Kosti, P.; Ronet, C.; Spill, A.; Seijo, B.; Romero, P.; Dangaj, D.; Coukos, G.; Irving, M. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J. Exp. Med. 2021, 218, e20192203. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.; Fraessle, S.; Steinmetz, R.; Hawkins, R.M.; Phi, T.-D.; Busch, D.; Miyazaki, T.; Marcondes, M.Q.; Riddell, S.R.; Turtle, C.J. Combination of NKTR-255, a Polymer Conjugated Human IL-15, with CD19 CAR T Cell Immunotherapy in a Preclinical Lymphoma Model. Blood 2019, 134, 2866. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jayasinghe, R.; Devenport, J.M.; Ritchey, J.K.; Rettig, M.P.; O’Neal, J.; Staser, K.W.; Kennerly, K.M.; Carter, A.J.; Gao, F.; et al. A long-acting interleukin-7, rhIL-7-hyFc, enhances CAR T cell expansion, persistence, and anti-tumor activity. Nat. Commun. 2022, 13, 3296. [Google Scholar] [CrossRef]
- Riley, J.L.; June, C.H. The road to recovery: Translating PD-1 biology into clinical benefit. Trends Immunol. 2007, 28, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, E.A.; Alanio, C.; Svoboda, J.; Nasta, S.D.; Landsburg, D.J.; Lacey, S.F.; Ruella, M.; Bhattacharyya, S.; Wherry, E.J.; Schuster, S.J. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 2022, 139, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Osborne, W.; Marzolini, M.; Tholouli, E.; Ramakrishnan, A.; Bachier, C.R.; McSweeney, P.A.; Irvine, D.; Zhang, M.; Al-Hajj, M.A.; Pule, M.; et al. Phase I Alexander study of AUTO3, the first CD19/22 dual targeting CAR T cell therapy, with pembrolizumab in patients with relapsed/refractory (r/r) DLBCL. J. Clin. Oncol. 2020, 38, 8001. [Google Scholar] [CrossRef]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination with Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Deng, Q.; Jiang, Y.-Y.; Zhang, R.; Zhu, H.-B.; Meng, J.-X.; Li, Y.-M. CAR-T 19 combined with reduced-dose PD-1 blockade therapy for treatment of refractory follicular lymphoma: A case report. Oncol. Lett. 2019, 18, 4415–4420. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, C.A.; Locke, F.L.; Miklos, D.B.; Herrera, A.F.; Westin, J.R.; Lee, J.; Rossi, J.M.; Zheng, L.; Avanzi, M.P.; Roberts, Z.J.; et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Blood 2018, 132, 4192. [Google Scholar] [CrossRef]
- Siddiqi, T.; Abramson, J.S.; Lee, H.J.; Schuster, S.; Hasskarl, J.; Montheard, S.; Dell Aringa, J.; Thompson, E.; Ananthakrishnan, R.; Lunning, M. Safety of Lisocabtagene Maraleucel Given with Durvalumab in Patients with Relapsed/Refractory Aggressive B-Cell Non Hodgkin Lymphoma: First Results from The Platform Study. Hematol. Oncol. 2019, 37, 171–172. [Google Scholar] [CrossRef] [Green Version]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Oh, J.-M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, Z.; Hong, C.; Schauner, R.; Dropulic, B.; Caimi, P.F.; de Lima, M.; Giraudo, M.F.; Gupta, K.; Reese, J.S.; Hwang, T.H.; et al. Sequential Single-Cell Transcriptional and Protein Marker Profiling Reveals TIGIT as a Marker of CD19 CAR-T Cell Dysfunction in Patients with Non-Hodgkin Lymphoma. Cancer Discov. 2022, 12, 1886–1903. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [Green Version]
- Works, M.; Soni, N.; Hauskins, C.; Sierra, C.; Baturevych, A.; Jones, J.C.; Curtis, W.; Carlson, P.; Johnstone, T.G.; Kugler, D.; et al. Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol. Cancer Ther. 2019, 18, 2246–2257. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.-C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Wei, R.; Feng, L.; Wu, Y.; He, F.; Xiao, M.; Cheng, Z. Lenalidomide enhances the efficacy of anti-BCMA CAR-T treatment in relapsed/refractory multiple myeloma: A case report and revies of the literature. Cancer Immunol. Immunother. 2022, 71, 39–44. [Google Scholar] [CrossRef]
- Shi, X.; Yan, L.; Shang, J.; Kang, L.; Yan, Z.; Jin, S.; Zhu, M.; Chang, H.; Gong, F.; Zhou, J.; et al. Anti-CD19 and anti-BCMA CAR T cell therapy followed by lenalidomide maintenance after autologous stem-cell transplantation for high-risk newly diagnosed multiple myeloma. Am. J. Hematol. 2022, 97, 537–547. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, Y.; Zou, Z.; Yang, Y.; Wang, X.; Xin, X.; Tu, S.; Li, Y. Combination strategies to optimize the efficacy of chimeric antigen receptor T cell therapy in haematological malignancies. Front. Immunol. 2022, 13, 954235. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, N.; Shi, H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget 2017, 8, 62730–62741. [Google Scholar] [CrossRef] [Green Version]
- Harrer, D.C.; Schenkel, C.; Berking, C.; Herr, W.; Abken, H.; Dörrie, J.; Schaft, N. Decitabine-Mediated Upregulation of CSPG4 in Ovarian Carcinoma Cells Enables Targeting by CSPG4-Specific CAR-T Cells. Cancers 2022, 14, 5033. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Dörrie, J.; Babalija, L.; Hoyer, S.; Gerer, K.F.; Schuler, G.; Heinzerling, L.; Schaft, N. BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int. J. Mol. Sci. 2018, 19, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Adusumilli, P.S.; Zauderer, M.G.; Rusch, V.W.; O’Cearbhaill, R.; Zhu, A.; Ngai, D.; McGee, E.; Chintala, N.; Messinger, J.; Cheema, W.; et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: Safety and preliminary efficacy in combination with anti-PD-1 agent. J. Clin. Oncol. 2019, 37, 2511. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Kumar, R.; Li, W.; Hoglund, V.; Wang, L.; Zhang, Y.; Shi, M.; Beak, D.; Cheuk, A.; Jensen, M.C.; et al. FGFR4-Targeted Chimeric Antigen Receptors Combined with Anti-Myeloid Polypharmacy Effectively Treat Orthotopic Rhabdomyosarcoma. Mol. Cancer Ther. 2022, 21, 1608–1621. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Li, W.; Yu, X.; Flores-Villanueva, P.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; Young, K.H.; Ma, X.; et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 2020, 9, 72. [Google Scholar] [CrossRef]
- Beenen, A.C.; Sauerer, T.; Schaft, N.; Dörrie, J. Beyond Cancer: Regulation and Function of PD-L1 in Health and Immune-Related Diseases. Int. J. Mol. Sci. 2022, 23, 8599. [Google Scholar] [CrossRef]
- Kilian, M.; Bunse, T.; Wick, W.; Platten, M.; Bunse, L. Genetically Modified Cellular Therapies for Malignant Gliomas. Int. J. Mol. Sci. 2021, 22, 12810. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; You, Y.; Shen, Z.; Shi, L. EGFRvIII-CAR-T Cells with PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol. Oncol. Res. 2020, 26, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.H.; Woroniecka, K.; Barbour, A.B.; Fecci, P.E.; Sanchez-Perez, L.; Sampson, J.H. CAR T cells and checkpoint inhibition for the treatment of glioblastoma. Expert Opin. Biol. Ther. 2020, 20, 579–591. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Roliński, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Mardiana, S.; John, L.B.; Henderson, M.A.; Slaney, C.Y.; von Scheidt, B.; Giuffrida, L.; Davenport, A.J.; Trapani, J.A.; Neeson, P.J.; Loi, S.; et al. A Multifunctional Role for Adjuvant Anti-4-1BB Therapy in Augmenting Antitumor Response by Chimeric Antigen Receptor T Cells. Cancer Res. 2017, 77, 1296–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
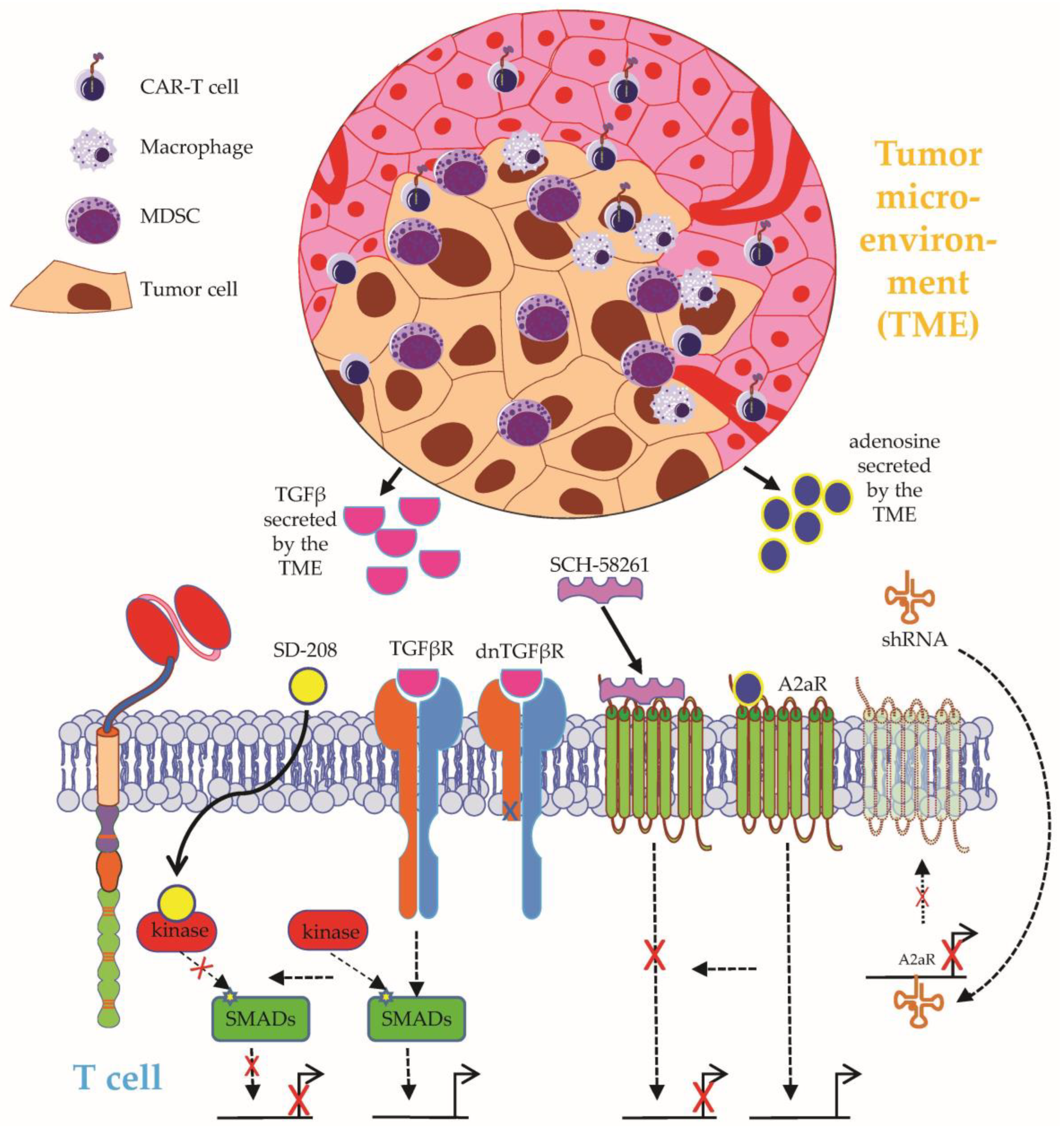
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-β-mediated repression in the adoptive T-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef] [PubMed]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.-Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.-T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Mølck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 49. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.G.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Investig. 2017, 127, 2176–2191. [Google Scholar] [CrossRef]
- Jung, M.; Yang, Y.; McCloskey, J.E.; Zaman, M.; Vedvyas, Y.; Zhang, X.; Stefanova, D.; Gray, K.D.; Min, I.M.; Zarnegar, R.; et al. Chimeric Antigen Receptor T Cell Therapy Targeting ICAM-1 in Gastric Cancer. Mol. Ther. Oncolytics 2020, 18, 587–601. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, S.; Chen, X.; Cheng, S.; Zhang, Z.; Li, F.; Huang, L.; Yang, Y.; Zhou, B.; Yue, D.; et al. Cancer-cell-secreted CXCL11 promoted CD8(+) T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J. Immunother. Cancer 2019, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Furlan, S.N.; Jaeger-Ruckstuhl, C.A.; Sarvothama, M.; Berger, C.; Smythe, K.S.; Garrison, S.M.; Specht, J.M.; Lee, S.M.; Amezquita, R.A.; et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy when Combined with Checkpoint Blockade. Cancer Cell 2021, 39, 193–208.e10. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; Leibowitz, M.S.; Sun, J.; Liousia, M.; Arguiri, E.; Albelda, S.M. Analysis and Augmentation of the Immunologic Bystander Effects of CAR T Cell Therapy in a Syngeneic Mouse Cancer Model. Mol. Ther. Oncolytics 2020, 18, 360–371. [Google Scholar] [CrossRef]
- Bocca, P.; Di Carlo, E.; Caruana, I.; Emionite, L.; Cilli, M.; de Angelis, B.; Quintarelli, C.; Pezzolo, A.; Raffaghello, L.; Morandi, F.; et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology 2017, 7, e1378843. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrer, D.C.; Dörrie, J.; Schaft, N. CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int. J. Mol. Sci. 2023, 24, 2342. https://doi.org/10.3390/ijms24032342
Harrer DC, Dörrie J, Schaft N. CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. International Journal of Molecular Sciences. 2023; 24(3):2342. https://doi.org/10.3390/ijms24032342
Chicago/Turabian StyleHarrer, Dennis Christoph, Jan Dörrie, and Niels Schaft. 2023. "CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy" International Journal of Molecular Sciences 24, no. 3: 2342. https://doi.org/10.3390/ijms24032342