
Microglial Cannabinoid CB2 Receptors in Pain Modulation


Abstract
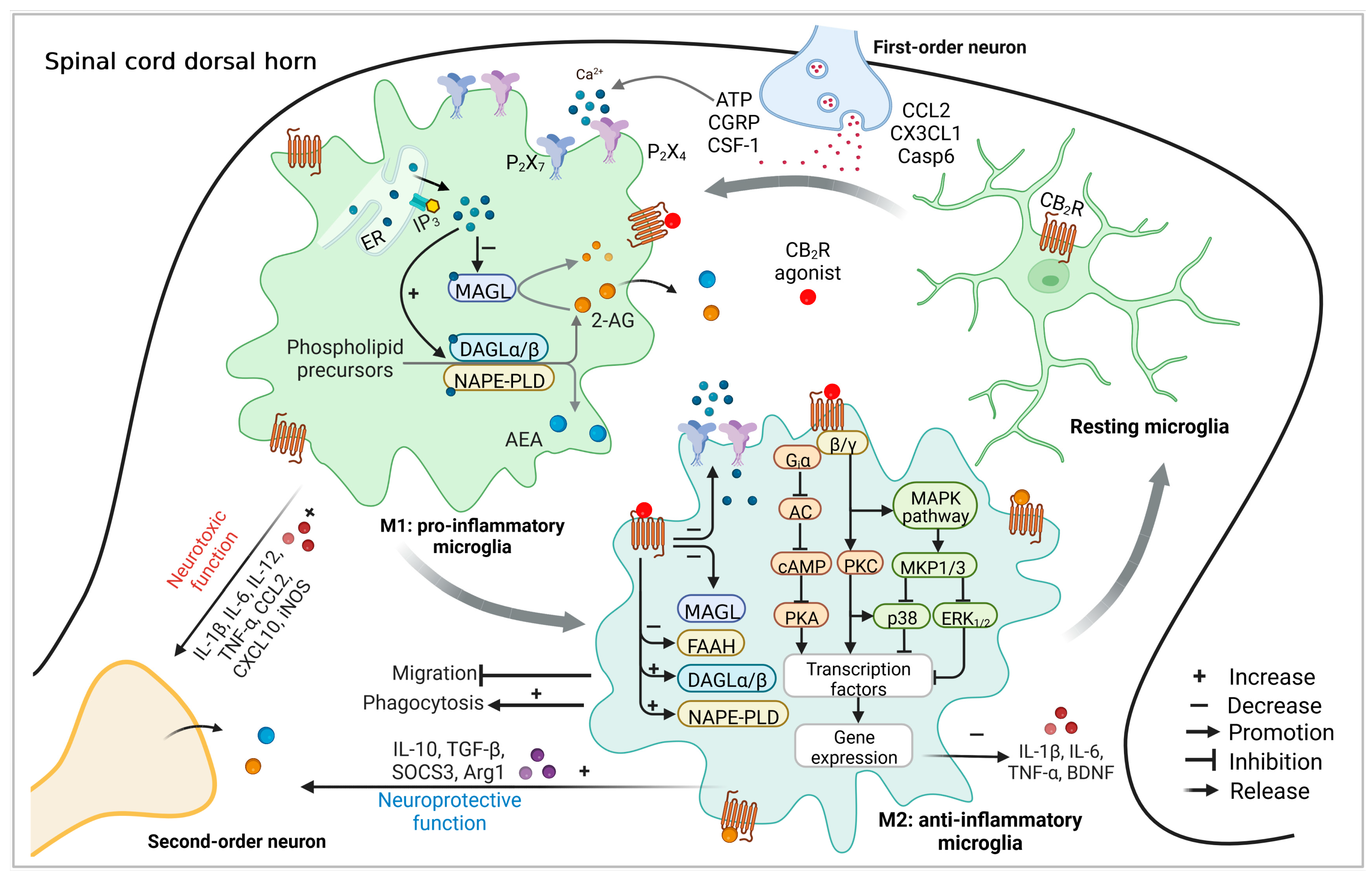
:1. Introduction
2. The Endocannabinoid System
3. Microglia Express the CB2R-Related Functional Endocannabinoid System
4. The Role of Microglial CB2R in Pathological Pain
4.1. Antinociceptive Effects of Well-Characterized CB2R Selective Agonists

{kind=link}
{kind=link}
| Agonist | In vitro binding profile | Pain Model | Route of Administration | Species | Efficacy | Reference | |
|---|---|---|---|---|---|---|---|
| CB1R | CB2R | ||||||
| HU308 | Ki > 10 μM Rat brain | Ki = 22.7 ± 3.9 nM Transfected cells | Formalin test | i.p., 50 mg/kg | Mouse | Antinociception | [148] |
| Arachidonic acid-induced ear Inflammation | i.p., 50 mg/kg | Mouse | Reduce ear swelling | [148] | |||
| Post-operative pain | i.p., 0.3–30 mg/kg | Rat | Antiallodynic effect | [149] | |||
| JWH-015 | Ki = 383 nM | Ki = 13.8 nM | Tail flick test | i.p., 1–100 mg/kg | Mouse | Antinociception | [21] |
| Tail immersion test/Paw pressure test | i.p., 5–20 mg/kg | Rat | Antinociception | [153] | |||
| Formalin test | i.p., 0.1–100 mg/kg | Mouse | Antinociception | [153] | |||
| Post-operative pain | i.t., 2–10 μg | Rat | Antiallodynic effect | [10,21,154] | |||
| i.p., 1–10 mg/kg | Rat | Antinociception | |||||
| Inflammatory pain (CFA) | i.p., 5–10 mg/kg | Rat | Antinociception | [153] | |||
| Neuropathic pain (SNI, SNL, L5NT, or bone cancer induced) | i.p., 1–10 mg/kg | Rat | Antinociception | [13,15,17,21,92,155] | |||
| i.t., 0.4–50 μg | Rat | Antiallodynic effect | |||||
| JWH-133 | Ki = 677 ± 132 nM Rat brain | Ki = 3.4 ± 1.0 nM Human embryonic kidney 293 cells | Formalin test | i.p., 0.1–10 mg/kg | Mouse | Antinociception | [162] |
| Inflammatory pain (Carrageenan or osteoarthritis induced) | s.c. 10 mg/kg | Rat | Increase weight bearing | [158,161] | |||
| i.pl., 5–15 μg | Rat | Inhibits mechanically evoked neuron responses | |||||
| Neuropathic pain (SNL or PSNL induced) | i.pl., 5–15 μg | Rat | Inhibits mechanically evoked neuron responses | [14,158,159,160] | |||
| i.t., 8–486 ng | Rat | Inhibits mechanically evoked neuron responses | |||||
| s.c. 1 mg/kg | Rat | Antiallodynic effect | |||||
| i.v., 0.15–0.3 mg/kg | Mouse | Antinociception | |||||
| GW405833 | Ki = 2043 ± 183 nM Cos-7 cells Ki = 273 ± 42.6 nM Rat brain | Ki = 14 ± 6 nM Cos-M6 cells Ki = 3.6 ± 1.1 nM Rat spleen | Hot plate test/Tail flick test | i.p., 100 mg/kg | Mouse | Antinociception | [166] |
| Formalin test | i.v., 3–10 mg/kg | Mouse | Antinociception | [9] | |||
| Post-operative pain | i.p., 0.3–30 mg/kg | Rat | Antiallodynic effect | [149,164] | |||
| Inflammatory pain (Carrageenan or CFA) | i.p., 3–30 mg/kg | Mouse | Antiallodynic effect | [164,166,167] | |||
| i.p., 0.1–100 mg/kg | Rat | Antinociception | |||||
| Neuropathic pain model (PSNL, L5NT, or CCI) | i.p., 3–30 mg/kg | Mouse | Antiallodynic effect | [9,164,166,168,169] | |||
| i.p., 0.01–30 mg/kg | Rat | Antiallodynic effect | |||||
| AM1241 | Ki = 280 ± 41 nM Rat brain | Ki = 3.4 ± 0.5 nM Mouse spleen | Hargreaves acute thermal stimulation | i.p., 0.3–3 mg/kg | Mouse | Antinociception | [22] |
| i.p., 0.033–0.33 mg/kg | Rat | Antinociception | [19] | ||||
| Hot plate test/Tail flick test | i.p., 0.3–10 mg/kg | Mouse | Antinociception | [22] | |||
| Formalin test | i.p., 3–10 mg/kg | Mouse | Antinociception | [9] | |||
| i.v., 0.3–3 mg/kg | Mouse | Antinociception | |||||
| Post-operative pain | i.p., 3–30 mg/kg | Rat | Antiallodynic effect | [149] | |||
| Inflammatory pain (Carrageenan, capsaicin, or CFA induced) | i.p., 0.033–1 mg/kg | Rat | Antinociception | [6,171,172,173] | |||
| i.pl., 0.033–4mg/kg | Rat | Antinociception/Reduce paw edema | |||||
| i.DRG, 100 nmol | Rat | Antinociception | |||||
| i.t., 100 nmol | Rat | Antinociception | |||||
| Neuropathic pain (SNL, bone cancer, vincristine-induced ) | i.t., 0.03–0.3 μg | Mouse | Antinociception | [93,176,177] [6,9,170,175] | |||
| i.p., 0.3–10 mg/kg | Mouse | Antiallodynic effect | |||||
| i.p., 0.1–3 mg/kg | Rat | Antinociception | |||||
| i.DRG, 100 nmol | Rat | Antinociception | |||||
| i.t., 0.01–10 μg | Rat | Antinociception | |||||
| i.v., 3–6 mg/kg | Rat | Antiallodynic effect | |||||
| MDA7 | hKi > 10,000 nM CHO-K1 cells rKi = 2565 nM CHO-K1 cells | hKi = 422 nM CHO-K1 cells rKi = 238 nM CHO-K1 cells | Hargreaves acute thermal stimulation | i.p., 1–10 mg/kg | Rat | Antinociception | [24] |
| Neuropathic pain (SNL or PTX) | i.p., 5–15 mg/kg | Rat | Antiallodynic effect | [24,25,26] | |||
4.2. Molecular Mechanisms Involved in the Action of Microglial CB2R in Pain Processing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| AEA | Arachidonoyl ethanolamide |
| ABDH6/12 | α/β domain hydrolases 6/12 |
| Arg-1 | Arginase 1 |
| ATP | Adenosine triphosphate |
| BDNF | Brain-derived neurotrophic factor |
| cAMP | cyclic Adenosine monophosphate |
| CB1R | Cannabinoid receptors 1 |
| CB2R | Cannabinoid receptors 2 |
| CCI | Chronic constriction injury |
| CFA | Complete Freund’s adjuvant |
| CGRP | Calcitonin gene-related peptide |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CSF-1 | Colony-stimulating factor-1 |
| DAGLα/β | Diacylglycerol lipases |
| DRG | Dorsal root ganglion |
| ECS | Endocannabinoid system |
| FAAH | Fatty acid amino hydrolase |
| IFN-γ | Interferon γ |
| IL-1β | Interleukin 1β |
| IL-10 | Interleukin 10 |
| LPI | Lysophosphatidylinositol |
| L5NT | Lumbar 5 nerve transection |
| MAPK | Mitogen-activated protein kinases |
| NO | Nitric oxide |
| NAPE | N-acyl phosphatidyl ethanolamine |
| NAPE-PLD | N-acyl-phosphatidyl ethanolamine-phospholipase D |
| NAT | N-acyltransferase |
| PC | Phosphatidylcholine |
| PE | Phosphatidylethanolamine |
| PGE2 | Prostaglandin E2 |
| PIP2 | Phosphatidyl inositol bis-phosphate |
| PLC | Phospholipase C |
| PNS | peripheral nervous systems |
| PSNL | Partial sciatic nerve ligation |
| PTX | Paclitaxel |
| SNL | Spinal nerve ligation |
| SOCS3 | Suppressor of cytokine signaling 3 |
| TGF-β | Transforming growth factor |
| TNF-α | Tumor necrosis factor α |
| 2-AG | 2-arachidonoyl-glycerol |
References
- Pratt, M.; Stevens, A.; Thuku, M.; Butler, C.; Skidmore, B.; Wieland, L.S.; Clemons, M.; Kanji, S.; Hutton, B. Benefits and harms of medical cannabis: A scoping review of systematic reviews. Syst. Rev. 2019, 8, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Sandoval, E.A.; Kolano, A.L.; Alvarado-Vazquez, P.A. Cannabis and Cannabinoids for Chronic Pain. Curr. Rheumatol. Rep. 2017, 19, 67. [Google Scholar] [CrossRef] [PubMed]
- Blair, R.E.; Deshpande, L.S.; Sombati, S.; Elphick, M.R.; Martin, B.R.; DeLorenzo, R.J. Prolonged exposure to WIN55,212-2 causes downregulation of the CB1 receptor and the development of tolerance to its anticonvulsant effects in the hippocampal neuronal culture model of acquired epilepsy. Neuropharmacology 2009, 57, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.P. Cannabinoids in pain management: CB1, CB2 and non-classic receptor ligands. Expert Opin. Investig. Drugs 2014, 23, 1123–1140. [Google Scholar] [CrossRef]
- Xu, B.; Xiao, J.; Xu, K.; Zhang, Q.; Chen, D.; Zhang, R.; Zhang, M.; Zhu, H.; Niu, J.; Zheng, T.; et al. VF-13, a chimeric peptide of VD-hemopressin(alpha) and neuropeptide VF, produces potent antinociception with reduced cannabinoid-related side effects. Neuropharmacology 2020, 175, 108178. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, G.C.; Pai, M.; Chandran, P.; Hooker, B.A.; Zhu, C.Z.; Salyers, A.K.; Wensink, E.J.; Zhan, C.; Carroll, W.A.; Dart, M.J.; et al. Central and peripheral sites of action for CB(2) receptor mediated analgesic activity in chronic inflammatory and neuropathic pain models in rats. Br. J. Pharmacol. 2011, 162, 428–440. [Google Scholar] [CrossRef] [Green Version]
- Guindon, J.; Hohmann, A.G. Cannabinoid CB2 receptors: A therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Klauke, A.L.; Racz, I.; Pradier, B.; Markert, A.; Zimmer, A.M.; Gertsch, J.; Zimmer, A. The cannabinoid CB(2) receptor-selective phytocannabinoid beta-caryophyllene exerts analgesic effects in mouse models of inflammatory and neuropathic pain. Eur. Neuropsychopharmacol. 2014, 24, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Beltramo, M.; Bernardini, N.; Bertorelli, R.; Campanella, M.; Nicolussi, E.; Fredduzzi, S.; Reggiani, A. CB2 receptor-mediated antihyperalgesia: Possible direct involvement of neural mechanisms. Eur. J. Neurosci. 2006, 23, 1530–1538. [Google Scholar] [CrossRef]
- Romero-Sandoval, A.; Eisenach, J.C. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology 2007, 106, 787–794. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Ikegami, M.; Kai, M.; Ohsawa, M.; Kamei, J. Activation of spinal cannabinoid CB2 receptors inhibits neuropathic pain in streptozotocin-induced diabetic mice. Neuroscience 2013, 250, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Gentry, K.R.; Dengler, E.C.; Wallace, J.A.; Kerwin, A.A.; Armijo, L.M.; Kuhn, M.N.; Thakur, G.A.; Makriyannis, A.; Milligan, E.D. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain 2012, 153, 1091–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, R.P.; Martinez, E.; DeLeo, J.A.; Romero-Sandoval, E.A. Spinal cannabinoid receptor type 2 agonist reduces mechanical allodynia and induces mitogen-activated protein kinase phosphatases in a rat model of neuropathic pain. J. Pain 2012, 13, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Cabanero, D.; Ramirez-Lopez, A.; Drews, E.; Schmole, A.; Otte, D.M.; Wawrzczak-Bargiela, A.; Huerga Encabo, H.; Kummer, S.; Ferrer-Montiel, A.; Przewlocki, R.; et al. Protective role of neuronal and lymphoid cannabinoid CB2 receptors in neuropathic pain. Elife 2020, 9, e55582. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Liu, Y.; Sun, B.; Sun, Y.; Hou, B.; Zhang, Y.; Ma, Z.; Gu, X. Intrathecal Injection of JWH-015 Attenuates Bone Cancer Pain Via Time-Dependent Modification of Pro-inflammatory Cytokines Expression and Astrocytes Activity in Spinal Cord. Inflammation 2015, 38, 1880–1890. [Google Scholar] [CrossRef]
- Wang, C.; Xu, K.; Wang, Y.; Mao, Y.; Huang, Y.; Liang, Y.; Liu, Y.; Hao, J.; Gu, X.; Ma, Z.; et al. Spinal cannabinoid receptor 2 activation reduces hypersensitivity associated with bone cancer pain and improves the integrity of the blood-spinal cord barrier. Reg. Anesth. Pain Med. 2020, 45, 783–791. [Google Scholar] [CrossRef]
- Romero-Sandoval, A.; Nutile-McMenemy, N.; DeLeo, J.A. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology 2008, 108, 722–734. [Google Scholar] [CrossRef] [Green Version]
- Rahn, E.J.; Thakur, G.A.; Wood, J.A.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Pharmacological characterization of AM1710, a putative cannabinoid CB2 agonist from the cannabilactone class: Antinociception without central nervous system side-effects. Pharmacol. Biochem. Behav. 2011, 98, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Malan, P.T., Jr.; Ibrahim, M.M.; Deng, H.; Liu, Q.; Mata, H.P.; Vanderah, T.; Porreca, F.; Makriyannis, A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain 2001, 93, 239–245. [Google Scholar] [CrossRef]
- Soliman, N.; Haroutounian, S.; Hohmann, A.G.; Krane, E.; Liao, J.; Macleod, M.; Segelcke, D.; Sena, C.; Thomas, J.; Vollert, J.; et al. Systematic review and meta-analysis of cannabinoids, cannabis-based medicines, and endocannabinoid system modulators tested for antinociceptive effects in animal models of injury-related or pathological persistent pain. Pain 2021, 162, S26–S44. [Google Scholar] [CrossRef] [PubMed]
- Grenald, S.A.; Young, M.A.; Wang, Y.; Ossipov, M.H.; Ibrahim, M.M.; Largent-Milnes, T.M.; Vanderah, T.W. Synergistic attenuation of chronic pain using mu opioid and cannabinoid receptor 2 agonists. Neuropharmacology 2017, 116, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.M.; Rude, M.L.; Stagg, N.J.; Mata, H.P.; Lai, J.; Vanderah, T.W.; Porreca, F.; Buckley, N.E.; Makriyannis, A.; Malan, T.P., Jr. CB2 cannabinoid receptor mediation of antinociception. Pain 2006, 122, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Astruc-Diaz, F.; McDaniel, S.W.; Xu, J.J.; Parola, S.; Brown, D.L.; Naguib, M.; Diaz, P. In vivo efficacy of enabling formulations based on hydroxypropyl-beta-cyclodextrins, micellar preparation, and liposomes for the lipophilic cannabinoid CB2 agonist, MDA7. J. Pharm. Sci. 2013, 102, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Naguib, M.; Diaz, P.; Xu, J.J.; Astruc-Diaz, F.; Craig, S.; Vivas-Mejia, P.; Brown, D.L. MDA7: A novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br. J. Pharmacol. 2008, 155, 1104–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Hocevar, M.; Bie, B.; Foss, J.F.; Naguib, M. Cannabinoid Type 2 Receptor System Modulates Paclitaxel-Induced Microglial Dysregulation and Central Sensitization in Rats. J. Pain 2019, 20, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Xu, J.J.; Diaz, P.; Brown, D.L.; Cogdell, D.; Bie, B.; Hu, J.; Craig, S.; Hittelman, W.N. Prevention of paclitaxel-induced neuropathy through activation of the central cannabinoid type 2 receptor system. Anesth. Analg. 2012, 114, 1104–1120. [Google Scholar] [CrossRef] [Green Version]
- Atwood, B.K.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hoffert, C.; Vu, H.K.; Groblewski, T.; Ahmad, S.; O’Donnell, D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur. J. Neurosci. 2003, 17, 2750–2754. [Google Scholar] [CrossRef]
- Racz, I.; Nadal, X.; Alferink, J.; Banos, J.E.; Rehnelt, J.; Martin, M.; Pintado, B.; Gutierrez-Adan, A.; Sanguino, E.; Manzanares, J.; et al. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J. Neurosci. 2008, 28, 12125–12135. [Google Scholar] [CrossRef] [Green Version]
- Brownjohn, P.W.; Ashton, J.C. Spinal cannabinoid CB2 receptors as a target for neuropathic pain: An investigation using chronic constriction injury. Neuroscience 2012, 203, 180–193. [Google Scholar] [CrossRef]
- Ashton, J.C.; Glass, M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiue, S.J.; Peng, H.Y.; Lin, C.R.; Wang, S.W.; Rau, R.H.; Cheng, J.K. Continuous Intrathecal Infusion of Cannabinoid Receptor Agonists Attenuates Nerve Ligation-Induced Pain in Rats. Reg. Anesth. Pain Med. 2017, 42, 499–506. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56 (Suppl. S1), 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef]
- Komorowska-Muller, J.A.; Schmole, A.C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef]
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.C.; Reddy, P.V.; Natarajan, V.; Schmid, H.H. Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J. Biol. Chem. 1983, 258, 9302–9306. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, D.J.; Gamble-George, J.C.; Marnett, L.J.; Patel, S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol. Sci. 2014, 35, 358–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.; Ribeiro, R.; Tanaka, M.; Zhang, Y. Activation of CB2 receptor is required for the therapeutic effect of ABHD6 inhibition in experimental autoimmune encephalomyelitis. Neuropharmacology 2015, 99, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Heimann, A.S.; Dale, C.S.; Guimaraes, F.S.; Reis, R.A.M.; Navon, A.; Shmuelov, M.A.; Rioli, V.; Gomes, I.; Devi, L.L.; Ferro, E.S. Hemopressin as a breakthrough for the cannabinoid field. Neuropharmacology 2021, 183, 108406. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.L.; Fang, Q.; Wang, Z.L.; Li, X.H.; Li, N.; Chang, X.M.; Pan, J.X.; Tang, H.Z.; Wang, R. Antinociceptive effects of central administration of the endogenous cannabinoid receptor type 1 agonist VDPVNFKLLSH-OH [(m)VD-hemopressin(alpha)], an N-terminally extended hemopressin peptide. J. Pharmacol. Exp. Ther. 2014, 348, 316–323. [Google Scholar] [CrossRef]
- Zheng, T.; Zhang, T.; Zhang, R.; Wang, Z.L.; Han, Z.L.; Li, N.; Li, X.H.; Zhang, M.N.; Xu, B.; Yang, X.L.; et al. Pharmacological characterization of rat VD-hemopressin(alpha), an alpha-hemoglobin-derived peptide exhibiting cannabinoid agonist-like effects in mice. Neuropeptides 2017, 63, 83–90. [Google Scholar] [CrossRef]
- Pan, J.X.; Wang, Z.L.; Li, N.; Han, Z.L.; Li, X.H.; Tang, H.H.; Wang, P.; Zheng, T.; Fang, Q.; Wang, R. Analgesic tolerance and cross-tolerance to the cannabinoid receptors ligands hemopressin, VD-hemopressin(alpha) and WIN55,212-2 at the supraspinal level in mice. Neurosci. Lett. 2014, 578, 187–191. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Gonsiorek, W.; Lunn, C.; Fan, X.; Narula, S.; Lundell, D.; Hipkin, R.W. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: Antagonism by anandamide. Mol. Pharmacol. 2000, 57, 1045–1050. [Google Scholar]
- Luk, T.; Jin, W.; Zvonok, A.; Lu, D.; Lin, X.Z.; Chavkin, C.; Makriyannis, A.; Mackie, K. Identification of a potent and highly efficacious, yet slowly desensitizing CB1 cannabinoid receptor agonist. Br. J. Pharmacol. 2004, 142, 495–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Showalter, V.M.; Compton, D.R.; Martin, B.R.; Abood, M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): Identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996, 278, 989–999. [Google Scholar] [PubMed]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Kotsikorou, E.; Madrigal, K.E.; Hurst, D.P.; Sharir, H.; Lynch, D.L.; Heynen-Genel, S.; Milan, L.B.; Chung, T.D.; Seltzman, H.H.; Bai, Y.; et al. Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 2011, 50, 5633–5647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.N.; Chambers, W.A.; Pertwee, R.G. Pharmacological actions and therapeutic uses of cannabis and cannabinoids. Anaesthesia 2001, 56, 1059–1068. [Google Scholar] [CrossRef]
- Pertwee, R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997, 74, 129–180. [Google Scholar] [CrossRef]
- La Manno, G.; Siletti, K.; Furlan, A.; Gyllborg, D.; Vinsland, E.; Mossi Albiach, A.; Mattsson Langseth, C.; Khven, I.; Lederer, A.R.; Dratva, L.M.; et al. Molecular architecture of the developing mouse brain. Nature 2021, 596, 92–96. [Google Scholar] [CrossRef]
- Zhang, Y.; Ke, J.; Zhou, Y.; Liu, X.; Huang, T.; Wang, F. Sex-specific characteristics of cells expressing the cannabinoid 1 receptor in the dorsal horn of the lumbar spinal cord. J. Comp. Neurol. 2022, 530, 2451–2473. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, N.E.; Abood, M.E.; Kessler, F.K.; Martin, B.R.; Schatz, A.R. Identification of a functionally relevant cannabinoid receptor on mouse spleen cells that is involved in cannabinoid-mediated immune modulation. Mol. Pharmacol. 1992, 42, 736–742. [Google Scholar] [PubMed]
- Bouaboula, M.; Rinaldi, M.; Carayon, P.; Carillon, C.; Delpech, B.; Shire, D.; Le Fur, G.; Casellas, P. Cannabinoid-receptor expression in human leukocytes. Eur. J. Biochem. 1993, 214, 173–180. [Google Scholar] [CrossRef]
- Gerard, C.M.; Mollereau, C.; Vassart, G.; Parmentier, M. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochem. J. 1991, 279 Pt 1, 129–134. [Google Scholar] [CrossRef]
- Stander, S.; Schmelz, M.; Metze, D.; Luger, T.; Rukwied, R. Distribution of cannabinoid receptor 1 (CB1) and 2 (CB2) on sensory nerve fibers and adnexal structures in human skin. J. Dermatol. Sci. 2005, 38, 177–188. [Google Scholar] [CrossRef]
- Hill, K.P.; Palastro, M.D.; Johnson, B.; Ditre, J.W. Cannabis and Pain: A Clinical Review. Cannabis Cannabinoid Res. 2017, 2, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Mitrirattanakul, S.; Ramakul, N.; Guerrero, A.V.; Matsuka, Y.; Ono, T.; Iwase, H.; Mackie, K.; Faull, K.F.; Spigelman, I. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain 2006, 126, 102–114. [Google Scholar] [CrossRef] [Green Version]
- Svizenska, I.; Dubovy, P.; Sulcova, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—A short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliu, A.; Mestre, L.; Guaza, C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol. Ther. 2016, 166, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, D.N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Holgado, E.; Vela, J.M.; Arevalo-Martin, A.; Almazan, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Turcotte, D.; Le Dorze, J.A.; Esfahani, F.; Frost, E.; Gomori, A.; Namaka, M. Examining the roles of cannabinoids in pain and other therapeutic indications: A review. Expert. Opin. Pharmacother. 2010, 11, 17–31. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Hasko, J.; Skuba, A.; Fan, S.; Dykstra, H.; McCormick, R.; Reichenbach, N.; Krizbai, I.; Mahadevan, A.; Zhang, M.; et al. Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood-brain barrier dysfunction under inflammatory conditions. J. Neurosci. 2012, 32, 4004–4016. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, S.J.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G.A. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; Van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef]
- Juan-Pico, P.; Fuentes, E.; Bermudez-Silva, F.J.; Javier Diaz-Molina, F.; Ripoll, C.; Rodriguez de Fonseca, F.; Nadal, A. Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium 2006, 39, 155–162. [Google Scholar] [CrossRef]
- Idris, A.I.; van ‘t Hof, R.J.; Greig, I.R.; Ridge, S.A.; Baker, D.; Ross, R.A.; Ralston, S.H. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat. Med. 2005, 11, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Abrams, D.I.; Guzman, M. Cannabis in cancer care. Clin. Pharmacol. Ther. 2015, 97, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. Cannabis, cannabinoid receptors, and endocannabinoid system: Yesterday, today, and tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.J.; Xi, Z.X. Progress in brain cannabinoid CB2 receptor research: From genes to behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef] [Green Version]
- Viscomi, M.T.; Oddi, S.; Latini, L.; Pasquariello, N.; Florenzano, F.; Bernardi, G.; Molinari, M.; Maccarrone, M. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J. Neurosci. 2009, 29, 4564–4570. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, H.; Horiuchi, Y.; Ishikawa, M.; Koga, M.; Imai, K.; Suzuki, Y.; Morikawa, M.; Inada, T.; Watanabe, Y.; Takahashi, M.; et al. Brain cannabinoid CB2 receptor in schizophrenia. Biol. Psychiatry 2010, 67, 974–982. [Google Scholar] [CrossRef]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shi, L.; Sun, B.; Zhang, Y.; Hou, B.; Sun, Y.; Ma, Z.; Gu, X. A Single Intrathecal or Intraperitoneal Injection of CB2 Receptor Agonist Attenuates Bone Cancer Pain and Induces a Time-Dependent Modification of GRK2. Cell. Mol. Neurobiol. 2017, 37, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Curto-Reyes, V.; Llames, S.; Hidalgo, A.; Menendez, L.; Baamonde, A. Spinal and peripheral analgesic effects of the CB2 cannabinoid receptor agonist AM1241 in two models of bone cancer-induced pain. Br. J. Pharmacol. 2010, 160, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.M.; Stella, N. CB2 receptor-mediated migration of immune cells: It can go either way. Br. J. Pharmacol. 2008, 153, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, K.; Shirasaka, R.; Yoshihara, K.; Mikuriya, S.; Tanaka, K.; Takanami, K.; Inoue, K.; Sakamoto, H.; Ohkawa, Y.; Masuda, T.; et al. A spinal microglia population involved in remitting and relapsing neuropathic pain. Science 2022, 376, 86–90. [Google Scholar] [CrossRef]
- Tansley, S.; Gu, N.; Guzman, A.U.; Cai, W.; Wong, C.; Lister, K.C.; Munoz-Pino, E.; Yousefpour, N.; Roome, R.B.; Heal, J.; et al. Microglia-mediated degradation of perineuronal nets promotes pain. Science 2022, 377, 80–86. [Google Scholar] [CrossRef]
- Gao, Y.J.; Ji, R.R. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Kuhn, J.A.; Wang, X.; Colquitt, B.; Solorzano, C.; Vaman, S.; Guan, A.K.; Evans-Reinsch, Z.; Braz, J.; Devor, M.; et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 2016, 19, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Berta, T.; Park, C.K.; Xu, Z.Z.; Xie, R.G.; Liu, T.; Lu, N.; Liu, Y.C.; Ji, R.R. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J. Clin. Investig. 2014, 124, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. S1), S10–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecha, M.; Feliu, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutierrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Fenn, A.M.; Hall, J.C.; Gensel, J.C.; Popovich, P.G.; Godbout, J.P. IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: Consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J. Neurosci. 2014, 34, 8904–8917. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, C.; Nent, E.; Bilkei-Gorzo, A.; Zimmer, A. Involvement of leptin signaling in the development of cannabinoid CB2 receptor-dependent mirror image pain. Sci. Rep. 2018, 8, 10827. [Google Scholar] [CrossRef] [Green Version]
- Lisboa, S.F.; Gomes, F.V.; Guimaraes, F.S.; Campos, A.C. Microglial Cells as a Link between Cannabinoids and the Immune Hypothesis of Psychiatric Disorders. Front. Neurol. 2016, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- van den Hoogen, N.J.; Harding, E.K.; Davidson, C.E.D.; Trang, T. Cannabinoids in Chronic Pain: Therapeutic Potential Through Microglia Modulation. Front. Neural Circuits 2021, 15, 816747. [Google Scholar] [CrossRef]
- Klegeris, A.; Bissonnette, C.J.; McGeer, P.L. Reduction of human monocytic cell neurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 receptor. Br. J. Pharmacol. 2003, 139, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, F.; Del Giudice, E.; Furegato, S.; Passarotto, M.; Leon, A. Cannabinoids ablate release of TNFalpha in rat microglial cells stimulated with lypopolysaccharide. Glia 2003, 41, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Ren, P.; Wang, Q.; Jiang, S.K.; Zhang, M.; Li, J.Y.; Wang, L.L.; Guan, D.W. Cannabinoid 2 receptor attenuates inflammation during skin wound healing by inhibiting M1 macrophages rather than activating M2 macrophages. J. Inflamm. 2018, 15, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, Y.; Olson, J.M.; Carlisle, S.J.; Cabral, G.A. The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J. Pharmacol. Exp. Ther. 1999, 288, 1357–1366. [Google Scholar]
- Moreno-Garcia, A.; Bernal-Chico, A.; Colomer, T.; Rodriguez-Antiguedad, A.; Matute, C.; Mato, S. Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination. Biomolecules 2020, 10, 1228. [Google Scholar] [CrossRef]
- De Meij, J.; Alfanek, Z.; Morel, L.; Decoeur, F.; Leyrolle, Q.; Picard, K.; Carrier, M.; Aubert, A.; Sere, A.; Lucas, C.; et al. Microglial Cannabinoid Type 1 Receptor Regulates Brain Inflammation in a Sex-Specific Manner. Cannabis Cannabinoid Res. 2021, 6, 488–507. [Google Scholar] [CrossRef]
- Pietr, M.; Kozela, E.; Levy, R.; Rimmerman, N.; Lin, Y.H.; Stella, N.; Vogel, Z.; Juknat, A. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009, 583, 2071–2076. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Witting, A.; Walter, L.; Wacker, J.; Moller, T.; Stella, N. P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 3214–3219. [Google Scholar] [CrossRef] [Green Version]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [Green Version]
- Witting, A.; Chen, L.; Cudaback, E.; Straiker, A.; Walter, L.; Rickman, B.; Moller, T.; Brosnan, C.; Stella, N. Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proc. Natl. Acad. Sci. USA 2006, 103, 6362–6367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Araujo, D.J.; Tjoa, K.; Saijo, K. The Endocannabinoid System as a Window Into Microglial Biology and Its Relationship to Autism. Front. Cell Neurosci. 2019, 13, 424. [Google Scholar] [CrossRef]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Rossi, S.; Bari, M.; De Chiara, V.; Fezza, F.; Musella, A.; Gasperi, V.; Prosperetti, C.; Bernardi, G.; Finazzi-Agro, A.; et al. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat. Neurosci. 2008, 11, 152–159. [Google Scholar] [CrossRef]
- Garcia-Ovejero, D.; Arevalo-Martin, A.; Petrosino, S.; Docagne, F.; Hagen, C.; Bisogno, T.; Watanabe, M.; Guaza, C.; Di Marzo, V.; Molina-Holgado, E. The endocannabinoid system is modulated in response to spinal cord injury in rats. Neurobiol. Dis. 2009, 33, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Eljaschewitsch, E.; Witting, A.; Mawrin, C.; Lee, T.; Schmidt, P.M.; Wolf, S.; Hoertnagl, H.; Raine, C.S.; Schneider-Stock, R.; Nitsch, R.; et al. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 2006, 49, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Maroof, N.; Pardon, M.C.; Kendall, D.A. Endocannabinoid signalling in Alzheimer’s disease. Biochem. Soc. Trans. 2013, 41, 1583–1587. [Google Scholar] [CrossRef] [Green Version]
- Arevalo-Martin, A.; Garcia-Ovejero, D.; Gomez, O.; Rubio-Araiz, A.; Navarro-Galve, B.; Guaza, C.; Molina-Holgado, E.; Molina-Holgado, F. CB2 cannabinoid receptors as an emerging target for demyelinating diseases: From neuroimmune interactions to cell replacement strategies. Br. J. Pharmacol. 2008, 153, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Cowley, T.R.; Blau, C.W.; Dempsey, C.N.; Noonan, J.; Gowran, A.; Tanveer, R.; Olango, W.M.; Finn, D.P.; Campbell, V.A.; et al. The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippocampus of aged rats and restores an age-related deficit in long-term potentiation. J. Neuroinflamm. 2012, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.K.; Devi, L.A. The highs and lows of cannabinoid receptor expression in disease: Mechanisms and their therapeutic implications. Pharmacol. Rev. 2011, 63, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.C.; Mackie, K. Review of the Endocannabinoid System. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Correa, F.; Docagne, F.; Mestre, L.; Clemente, D.; Hernangomez, M.; Loria, F.; Guaza, C. A role for CB2 receptors in anandamide signalling pathways involved in the regulation of IL-12 and IL-23 in microglial cells. Biochem. Pharmacol. 2009, 77, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, R.S.; Sorgi, C.A.; Peti, A.P.F.; Veras, F.P.; Faccioli, L.H.; Galdino, G. Involvement of Spinal Cannabinoid CB2 Receptors in Exercise-Induced Antinociception. Neuroscience 2019, 418, 177–188. [Google Scholar] [CrossRef]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Tolon, R.M.; Nunez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martinez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef]
- Malek, N.; Popiolek-Barczyk, K.; Mika, J.; Przewlocka, B.; Starowicz, K. Anandamide, Acting via CB2 Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plast. 2015, 2015, 130639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spittau, B.; Wullkopf, L.; Zhou, X.; Rilka, J.; Pfeifer, D.; Krieglstein, K. Endogenous transforming growth factor-beta promotes quiescence of primary microglia in vitro. Glia 2013, 61, 287–300. [Google Scholar] [CrossRef]
- Rom, S.; Persidsky, Y. Cannabinoid receptor 2: Potential role in immunomodulation and neuroinflammation. J. Neuroimmune Pharmacol. 2013, 8, 608–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, J.; Desroches, J.; Beaulieu, P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br. J. Pharmacol. 2007, 150, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Guindon, J.; Guijarro, A.; Piomelli, D.; Hohmann, A.G. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br. J. Pharmacol. 2011, 163, 1464–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokal, D.M.; Elmes, S.J.; Kendall, D.A.; Chapman, V. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurones via activation of CB2 receptors in anaesthetised rats. Neuropharmacology 2003, 45, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Jaggar, S.I.; Hasnie, F.S.; Sellaturay, S.; Rice, A.S. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 1998, 76, 189–199. [Google Scholar] [CrossRef]
- Hanus, L.; Breuer, A.; Tchilibon, S.; Shiloah, S.; Goldenberg, D.; Horowitz, M.; Pertwee, R.G.; Ross, R.A.; Mechoulam, R.; Fride, E. HU-308: A specific agonist for CB(2), a peripheral cannabinoid receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 14228–14233. [Google Scholar] [CrossRef] [Green Version]
- LaBuda, C.J.; Koblish, M.; Little, P.J. Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postoperative pain. Eur. J. Pharmacol. 2005, 527, 172–174. [Google Scholar] [CrossRef]
- Thapa, D.; Cairns, E.A.; Szczesniak, A.M.; Toguri, J.T.; Caldwell, M.D.; Kelly, M.E.M. The Cannabinoids Delta(8)THC, CBD, and HU-308 Act via Distinct Receptors to Reduce Corneal Pain and Inflammation. Cannabis Cannabinoid Res. 2018, 3, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Huffman, J.W.; Padgett, L.W. Recent developments in the medicinal chemistry of cannabimimetic indoles, pyrroles and indenes. Curr. Med. Chem. 2005, 12, 1395–1411. [Google Scholar] [CrossRef]
- Huffman, J.W.; Zengin, G.; Wu, M.J.; Lu, J.; Hynd, G.; Bushell, K.; Thompson, A.L.; Bushell, S.; Tartal, C.; Hurst, D.P.; et al. Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB(1) and CB(2) receptors: Steric and electronic effects of naphthoyl substituents. New highly selective CB(2) receptor agonists. Bioorg. Med. Chem. 2005, 13, 89–112. [Google Scholar] [CrossRef]
- Craft, R.M.; Greene, N.Z.; Wakley, A.A. Antinociceptive effects of JWH015 in female and male rats. Behav. Pharmacol. 2018, 29, 280–289. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, W.; Liu, Y.; Liu, X.; Ma, Z.; Gu, X. Intrathecal injection of JWH015 attenuates remifentanil-induced postoperative hyperalgesia by inhibiting activation of spinal glia in a rat model. Anesth. Analg. 2014, 118, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Huang, Y.; Zhang, Y.; Wang, C.; Wu, H.; Tian, X.; Liu, Y.; Hou, B.; Liang, Y.; Rong, H.; et al. Cannabinoid receptor 2selective agonist JWH015 attenuates bone cancer pain through the amelioration of impaired autophagy flux induced by inflammatory mediators in the spinal cord. Mol. Med. Rep. 2019, 20, 5100–5110. [Google Scholar] [CrossRef] [Green Version]
- Huffman, J.W.; Liddle, J.; Yu, S.; Aung, M.M.; Abood, M.E.; Wiley, J.L.; Martin, B.R. 3-(1′,1′-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: Synthesis of selective ligands for the CB2 receptor. Bioorg. Med. Chem. 1999, 7, 2905–2914. [Google Scholar] [CrossRef]
- Jonsson, K.O.; Persson, E.; Fowler, C.J. The cannabinoid CB2 receptor selective agonist JWH133 reduces mast cell oedema in response to compound 48/80 in vivo but not the release of beta-hexosaminidase from skin slices in vitro. Life Sci. 2006, 78, 598–606. [Google Scholar] [CrossRef]
- Elmes, S.J.; Jhaveri, M.D.; Smart, D.; Kendall, D.A.; Chapman, V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naive rats and in rat models of inflammatory and neuropathic pain. Eur. J. Neurosci. 2004, 20, 2311–2320. [Google Scholar] [CrossRef]
- Sagar, D.R.; Kelly, S.; Millns, P.J.; O’Shaughnessey, C.T.; Kendall, D.A.; Chapman, V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur. J. Neurosci. 2005, 22, 371–379. [Google Scholar] [CrossRef]
- Burston, J.J.; Sagar, D.R.; Shao, P.; Bai, M.; King, E.; Brailsford, L.; Turner, J.M.; Hathway, G.J.; Bennett, A.J.; Walsh, D.A.; et al. Cannabinoid CB2 receptors regulate central sensitization and pain responses associated with osteoarthritis of the knee joint. PLoS ONE 2013, 8, e80440. [Google Scholar] [CrossRef] [Green Version]
- Elmes, S.J.R.; Winyard, L.A.; Medhurst, S.J.; Clayton, N.M.; Wilson, A.W.; Kendall, D.A.; Chapman, V. Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain 2005, 118, 327–335. [Google Scholar] [CrossRef]
- Yuill, M.B.; Hale, D.E.; Guindon, J.; Morgan, D.J. Anti-nociceptive interactions between opioids and a cannabinoid receptor 2 agonist in inflammatory pain. Mol. Pain 2017, 13, 1744806917728227. [Google Scholar] [CrossRef] [Green Version]
- Huffman, J.W. The search for selective ligands for the CB2 receptor. Curr. Pharm. Des. 2000, 6, 1323–1337. [Google Scholar] [CrossRef]
- Valenzano, K.J.; Tafesse, L.; Lee, G.; Harrison, J.E.; Boulet, J.M.; Gottshall, S.L.; Mark, L.; Pearson, M.S.; Miller, W.; Shan, S.; et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 2005, 48, 658–672. [Google Scholar] [CrossRef]
- Thakur, G.A.; Tichkule, R.; Bajaj, S.; Makriyannis, A. Latest advances in cannabinoid receptor agonists. Expert Opin. Ther. Pat. 2009, 19, 1647–1673. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, G.T.; Gottshall, S.L.; Boulet, J.M.; Chaffer, S.M.; Harrison, J.E.; Pearson, M.S.; Turchin, P.I.; Mark, L.; Garrison, A.E.; Valenzano, K.J. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur. J. Pharmacol. 2005, 528, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Clayton, N.; Marshall, F.H.; Bountra, C.; O’Shaughnessy, C.T. CB1 and CB2 cannabinoid receptors are implicated in inflammatory pain. Pain 2002, 96, 253–260. [Google Scholar] [CrossRef]
- Hu, B.; Doods, H.; Treede, R.D.; Ceci, A. Depression-like behaviour in rats with mononeuropathy is reduced by the CB2-selective agonist GW405833. Pain 2009, 143, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Leichsenring, A.; Andriske, M.; Backer, I.; Stichel, C.C.; Lubbert, H. Analgesic and antiinflammatory effects of cannabinoid receptor agonists in a rat model of neuropathic pain. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 379, 627–636. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Deng, H.; Zvonok, A.; Cockayne, D.A.; Kwan, J.; Mata, H.P.; Vanderah, T.W.; Lai, J.; Porreca, F.; Makriyannis, A.; et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: Pain inhibition by receptors not present in the CNS. Proc. Natl. Acad. Sci. USA 2003, 100, 10529–10533. [Google Scholar] [CrossRef] [Green Version]
- Nackley, A.G.; Makriyannis, A.; Hohmann, A.G. Selective activation of cannabinoid CB(2) receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 2003, 119, 747–757. [Google Scholar] [CrossRef]
- Quartilho, A.; Mata, H.P.; Ibrahim, M.M.; Vanderah, T.W.; Porreca, F.; Makriyannis, A.; Malan, T.P., Jr. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology 2003, 99, 955–960. [Google Scholar] [CrossRef]
- Gutierrez, T.; Farthing, J.N.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: A comparative analysis. Br. J. Pharmacol. 2007, 150, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Nackley, A.G.; Zvonok, A.M.; Makriyannis, A.; Hohmann, A.G. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. J. Neurophysiol. 2004, 92, 3562–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, J.L.; Gentry, K.R.; Dengler, E.C.; Wallace, J.A.; Kerwin, A.A.; Kuhn, M.N.; Zvonok, A.M.; Thakur, G.A.; Makriyannis, A.; Milligan, E.D. Immunofluorescent spectral analysis reveals the intrathecal cannabinoid agonist, AM1241, produces spinal anti-inflammatory cytokine responses in neuropathic rats exhibiting relief from allodynia. Brain Behav. 2012, 2, 155–177. [Google Scholar] [CrossRef] [PubMed]
- Rahn, E.J.; Makriyannis, A.; Hohmann, A.G. Activation of cannabinoid CB1 and CB2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. Br. J. Pharmacol. 2007, 152, 765–777. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Ondoua, A.N.; Wright, C.; Vardanyan, A.; King, T.; Largent-Milnes, T.M.; Nelson, M.; Jimenez-Andrade, J.M.; Mantyh, P.W.; Vanderah, T.W. A cannabinoid 2 receptor agonist attenuates bone cancer-induced pain and bone loss. Life Sci. 2010, 86, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, P.; Phatak, S.S.; Xu, J.; Fronczek, F.R.; Astruc-Diaz, F.; Thompson, C.M.; Cavasotto, C.N.; Naguib, M. 2,3-Dihydro-1-benzofuran derivatives as a series of potent selective cannabinoid receptor 2 agonists: Design, synthesis, and binding mode prediction through ligand-steered modeling. ChemMedChem 2009, 4, 1615–1629. [Google Scholar] [CrossRef] [Green Version]
- Arena Pharmaceuticals. Olorinab (APD371) [Arena Pharmaceuticals Pipeline]. Arena Pharmaceuticals. Available online: https://www.arenapharm.com/pipeline/apd371/ (accessed on 18 February 2021).
- Soethoudt, M.; Grether, U.; Fingerle, J.; Grim, T.W.; Fezza, F.; de Petrocellis, L.; Ullmer, C.; Rothenhausler, B.; Perret, C.; van Gils, N.; et al. Cannabinoid CB(2) receptor ligand profiling reveals biased signalling and off-target activity. Nat. Commun. 2017, 8, 13958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiera, R.; Hummers, L.; Chung, L.; Frech, T.M.; Domsic, R.; Hsu, V.; Furst, D.E.; Gordon, J.; Mayes, M.; Simms, R.; et al. Safety and Efficacy of Lenabasum in a Phase II, Randomized, Placebo-Controlled Trial in Adults With Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1350–1360. [Google Scholar] [CrossRef]
- Spiera, R.; Khanna, D.; Kuwana, M.; Furst, D.E.; Frech, T.M.; Hummers, L.; Stevens, W.; Matucci-Cerinic, M.; Baron, M.; Distler, O.; et al. A randomised, double-blind, placebo-controlled phase 3 study of lenabasum in diffuse cutaneous systemic sclerosis: RESOLVE-1 design and rationale. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S131), 124–133. [Google Scholar] [CrossRef]
- Kinsey, S.G.; Mahadevan, A.; Zhao, B.; Sun, H.; Naidu, P.S.; Razdan, R.K.; Selley, D.E.; Imad Damaj, M.; Lichtman, A.H. The CB2 cannabinoid receptor-selective agonist O-3223 reduces pain and inflammation without apparent cannabinoid behavioral effects. Neuropharmacology 2011, 60, 244–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, L.; Palazzo, E.; Tambaro, S.; Giordano, C.; Gatta, L.; Scafuro, M.A.; Rossi, F.S.; Lazzari, P.; Pani, L.; de Novellis, V.; et al. 1-(2′,4′-dichlorophenyl)-6-methyl-N-cyclohexylamine-1,4-dihydroindeno[1,2-c]pyraz ole-3-carboxamide, a novel CB2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiol. Dis. 2010, 37, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tang, Y.; Xie, M.; Bie, B.; Wu, J.; Yang, H.; Foss, J.F.; Yang, B.; Rosenquist, R.W.; Naguib, M. Activation of cannabinoid receptor 2 attenuates mechanical allodynia and neuroinflammatory responses in a chronic post-ischemic pain model of complex regional pain syndrome type I in rats. Eur. J. Neurosci. 2016, 44, 3046–3055. [Google Scholar] [CrossRef]
- Xu, J.J.; Diaz, P.; Bie, B.; Astruc-Diaz, F.; Wu, J.; Yang, H.; Brown, D.L.; Naguib, M. Spinal gene expression profiling and pathways analysis of a CB2 agonist (MDA7)-targeted prevention of paclitaxel-induced neuropathy. Neuroscience 2014, 260, 185–194. [Google Scholar] [CrossRef]
- Gomez-Canas, M.; Morales, P.; Garcia-Toscano, L.; Navarrete, C.; Munoz, E.; Jagerovic, N.; Fernandez-Ruiz, J.; Garcia-Arencibia, M.; Pazos, M.R. Biological characterization of PM226, a chromenoisoxazole, as a selective CB2 receptor agonist with neuroprotective profile. Pharmacol. Res. 2016, 110, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Nent, E.; Nozaki, C.; Schmole, A.C.; Otte, D.; Zimmer, A. CB2 receptor deletion on myeloid cells enhanced mechanical allodynia in a mouse model of neuropathic pain. Sci. Rep. 2019, 9, 7468. [Google Scholar] [CrossRef] [Green Version]
- La Porta, C.; Bura, S.A.; Aracil-Fernandez, A.; Manzanares, J.; Maldonado, R. Role of CB1 and CB2 cannabinoid receptors in the development of joint pain induced by monosodium iodoacetate. Pain 2013, 154, 160–174. [Google Scholar] [CrossRef]
- Racz, I.; Nadal, X.; Alferink, J.; Banos, J.E.; Rehnelt, J.; Martin, M.; Pintado, B.; Gutierrez-Adan, A.; Sanguino, E.; Bellora, N.; et al. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J. Neurosci. 2008, 28, 12136–12145. [Google Scholar] [CrossRef] [Green Version]
- Slipetz, D.M.; O’Neill, G.P.; Favreau, L.; Dufresne, C.; Gallant, M.; Gareau, Y.; Guay, D.; Labelle, M.; Metters, K.M. Activation of the human peripheral cannabinoid receptor results in inhibition of adenylyl cyclase. Mol. Pharmacol. 1995, 48, 352–361. [Google Scholar]
- Bouaboula, M.; Poinot-Chazel, C.; Marchand, J.; Canat, X.; Bourrie, B.; Rinaldi-Carmona, M.; Calandra, B.; Le Fur, G.; Casellas, P. Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur. J. Biochem. 1996, 237, 704–711. [Google Scholar] [CrossRef]
- Niu, J.; Huang, D.; Zhou, R.; Yue, M.; Xu, T.; Yang, J.; He, L.; Tian, H.; Liu, X.; Zeng, J. Activation of dorsal horn cannabinoid CB2 receptor suppresses the expression of P2Y12 and P2Y13 receptors in neuropathic pain rats. J. Neuroinflamm. 2017, 14, 185. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Suter, M.R. p38 MAPK, microglial signaling, and neuropathic pain. Mol. Pain 2007, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.Y.; Gerner, P.; Woolf, C.J.; Ji, R.R. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005, 114, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Zhu, N.; Grist, J.; Ma, Z.; Loeb, J.A.; Bennett, D.L. Following nerve injury neuregulin-1 drives microglial proliferation and neuropathic pain via the MEK/ERK pathway. Glia 2011, 59, 554–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, S.; Nath, N.; Smith, B.; Viollet, B.; Singh, A.K.; Singh, I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: A possible role of AMP-activated protein kinase. J. Neurosci. 2004, 24, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Burgos, E.; Gomez-Nicola, D.; Pascual, D.; Martin, M.I.; Nieto-Sampedro, M.; Goicoechea, C. Cannabinoid agonist WIN 55,212-2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. Eur. J. Pharmacol. 2012, 682, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, F.; Hernangomez, M.; Mestre, L.; Loria, F.; Spagnolo, A.; Docagne, F.; Di Marzo, V.; Guaza, C. Anandamide enhances IL-10 production in activated microglia by targeting CB(2) receptors: Roles of ERK1/2, JNK, and NF-kappaB. Glia 2010, 58, 135–147. [Google Scholar] [CrossRef]
- Hernangomez, M.; Mestre, L.; Correa, F.G.; Loria, F.; Mecha, M.; Inigo, P.M.; Docagne, F.; Williams, R.O.; Borrell, J.; Guaza, C. CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 2012, 60, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef]
- Romero-Sandoval, E.A.; Horvath, R.; Landry, R.P.; DeLeo, J.A. Cannabinoid receptor type 2 activation induces a microglial anti-inflammatory phenotype and reduces migration via MKP induction and ERK dephosphorylation. Mol. Pain 2009, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.W.; Shao, Q.H.; Wang, X.T.; Ma, K.L.; Chen, N.H.; Yuan, Y.H. CB2 receptor activation inhibits the phagocytic function of microglia through activating ERK/AKT-Nurr1 signal pathways. Acta Pharmacol. Sin. 2022, 43, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, L.; Jiang, B.; Feng, Z.; Yang, L.; Tang, J.; Chen, Q.; Zhang, J.; Tan, Q.; Feng, H.; et al. Cannabinoid receptor-2 stimulation suppresses neuroinflammation by regulating microglial M1/M2 polarization through the cAMP/PKA pathway in an experimental GMH rat model. Brain Behav. Immun. 2016, 58, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, M.; Liu, L.; Zhang, P.; Liu, D.; Zheng, X.; Zhong, X.; Wang, G. Low-dose cannabinoid receptor 2 agonist induces microglial activation in a cancer pain-morphine tolerance rat model. Life Sci. 2021, 264, 118635. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, K.; Wu, Y.; Tian, Z.; Xu, Y.; Wu, C.; Wang, Z. Microglial Cannabinoid CB2 Receptors in Pain Modulation. Int. J. Mol. Sci. 2023, 24, 2348. https://doi.org/10.3390/ijms24032348
Xu K, Wu Y, Tian Z, Xu Y, Wu C, Wang Z. Microglial Cannabinoid CB2 Receptors in Pain Modulation. International Journal of Molecular Sciences. 2023; 24(3):2348. https://doi.org/10.3390/ijms24032348
Chicago/Turabian StyleXu, Kangtai, Yifei Wu, Zhuangzhuang Tian, Yuanfan Xu, Chaoran Wu, and Zilong Wang. 2023. "Microglial Cannabinoid CB2 Receptors in Pain Modulation" International Journal of Molecular Sciences 24, no. 3: 2348. https://doi.org/10.3390/ijms24032348
APA StyleXu, K., Wu, Y., Tian, Z., Xu, Y., Wu, C., & Wang, Z. (2023). Microglial Cannabinoid CB2 Receptors in Pain Modulation. International Journal of Molecular Sciences, 24(3), 2348. https://doi.org/10.3390/ijms24032348