Abstract
Variants in desmoplakin gene (DSP MIM *125647) have been usually associated with Arrhythmogenic Cardiomyopathy (ACM), or Dilated Cardiomyopathy (DCM) inherited in an autosomal dominant manner. A cohort of 18 probands, characterized as heterozygotes for DSP variants by a target Next Generation Sequencing (NGS) cardiomyopathy panel, was analyzed. Cardiological, genetic data, and imaging features were retrospectively collected. A total of 16 DSP heterozygous pathogenic or likely pathogenic variants were identified, 75% (n = 12) truncating variants, n = 2 missense variants, n = 1 splicing variant, and n = 1 duplication variant. The mean age at diagnosis was 40.61 years (IQR 31–47.25), 61% of patients being asymptomatic (n = 11, New York Heart Association (NYHA) class I) and 39% mildly symptomatic (n = 7, NYHA class II). Notably, 39% of patients (n = 7) presented with a clinical history of presumed myocarditis episodes, characterized by chest pain, myocardial enzyme release, 12-lead electrocardiogram abnormalities with normal coronary arteries, which were recurrent in 57% of cases (n = 4). About half of the patients (55%, n = 10) presented with a varied degree of left ventricular enlargement (LVE), four showing biventricular involvement. Eleven patients (61%) underwent implantable cardioverter defibrillator (ICD) implantation, with a mean age of 46.81 years (IQR 36.00–64.00). Cardiac magnetic resonance imaging (CMRI) identified in all 18 patients a delayed enhancement (DE) area consistent with left ventricular (LV) myocardial fibrosis, with a larger localization and extent in patients presenting with recurrent episodes of myocardial injury. These clinical and genetic data confirm that DSP-related cardiomyopathy may represent a distinct clinical entity characterized by a high arrhythmic burden, variable degrees of LVE, Late Gadolinium Enhancement (LGE) with subepicardial distribution and episodes of myocarditis-like picture.
1. Introduction
Arrhythmogenic Cardiomyopathy (ACM) is an inherited disorder characterized by the progressive replacement of the ventricular myocardium by fibrofatty tissue, which predispose to develop ventricular arrhythmias. In contrast to what was previously thought, the disorder does not involve only the right ventricle (RV), but it may also affect the left ventricle (LV), or both, as reported in several studies [1,2,3,4,5,6]. Recently, an international expert consensus document proposed new criteria for the diagnosis of ACM (the “Padua criteria”), to cover all the phenotypic expressions of the disease [7]. Affected patients usually present arrhythmic symptoms, such as palpitations, chest tightness, and syncopes, and may present variable signs of heart failure (HF). Notably, the ACM phenotype can also overlap with other cardiomyopathies (CMPs), particularly dilated cardiomyopathy (DCM), in which arrhythmia may be present along with other distinctive phenotypic features such as ventricular dilation and depressed myocardial performance. In fact, according to the European Society of Cardiology (ESC), DCM is currently defined by the presence of left ventricular or biventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions (hypertension, valve disease) or coronary artery disease, sufficient to cause global systolic impairment [8]. It is important to underline that both conditions are characterized by a remarkable phenotypic variability, which make complex the classification in clinically distinct entities [9,10]. Furthermore, biological studies of pathogenic mechanisms underlying the development of inherited cardiomyopathies have led to an awareness of final common protein pathways [11]. Although DCM is predominantly caused by variants in Titin (TTN) or Lamin A/C (LMNA) genes, genes encoding for desmosomal proteins are known to be involved, too [10,12]. Mutations in desmosomal genes are indeed the most common genetic cause of ACM [13,14,15].
Desmosomes are intercellular junctions, which provide strong adhesion between cells and link intracellularly to the intermediate filament of cytoskeleton, giving mechanical strength to tissues. They are mostly represented in tissues subjected to continuous mechanical stress, such as epidermidis and myocardium [16,17]. Desmoplakin, encoded by the DSP gene (MIM *125647), is the most prevalent component of desmosome. There are three different isoforms, produced by alternative splicing of the DSP gene, which differ from each other by the length of rod domain: a long isoform (DSP-I), with a whole rod domain; an intermediate isoform (DSP-Ia), including a half of rod domain; and a short isoform (DSP-II), which misses the majority of rod domain. DSP-I is the most prevalent cardiac isoform, which connects the cardiac desmosome to intermediate filaments, and it is crucial for force transmission in the myocardium [18].
Thus, variants in the DSP gene have been associated with both DCM and ACM phenotypes, especially left-dominant ventricular cardiomyopathy (ALVC), inherited in autosomal manner [19,20]. Variants causing a desmoplakin loss of function lead to the failure of intercellular adhesion and have been associated with histological findings of inflammatory infiltrates and fibrosis of the left ventricular myocardium [21,22]. Moreover, small DSP case series reported in these patients the occurrence of recurrent myocarditis, that may be the initial presentation of cardiomyopathy, preceding systolic dysfunction [23,24,25]. Considering these findings, which appear to differentiate DSP-related cardiomyopathy from classic right ventricular arrhythmogenic cardiomyopathy (ARVC) and DCM, some authors have suggested the possibility of a new distinct clinical entity characterized by a prevalent fibrotic and inflammatory component requiring its own specific clinic management [22,26].
With these assumptions in the present study, we retrospectively evaluated the clinical and instrumental characteristics of a selected cohort of patients carrying heterozygous DSP pathogenic (P), or likely pathogenic (LP) variants, to identify distinctive phenotypic features and rule out a possible genotype-phenotype correlation.
2. Results
2.1. Genetics Variants
A total of 16 heterozygous variants in the DSP gene were identified in 18 probands (Table 1) since the same variants were detected in two couples of two unrelated patients (rs869025395 in patients ID 5 and 17; rs1581819043 in patients ID 7 and 9). Of them, 75% (n = 12) were truncating variants, two were missense, one a splicing variant, and one a duplication variant. The majority of variants were located in “coiled coil domain” (n = 8), followed by “rod domain” (n = 4), and “spectrin domain” (n = 3) and “pectin repeat domain” (n = 1). More than half of the identified variants are located in the head of the protein. Of 16 variants, 10 were not reported in scientific literature nor in ClinVar [27]; five variants were cited in ClinVar [27]; seven were reported in the Single Nucleotide Polymorphism Database (dbSNP) [28]; seven were classified as “pathogenic” (P); and nine as “likely pathogenic” (LP) according to American College of Medical Genetics (ACMG) criteria [29]. Two patients (ID 9 and 16) also carried a heterozygous variant in the DSC2 gene. Specifically, patient ID 9 carried the variant c.2381C>T, p.(Ser794Leu) and patient ID 16 carried the variant c.304G>A, p.(Glu102Lys). Both of these examples are cited in ClinVar as a variant of uncertain significance (VoUS)/likely benign variant and reported in Genomic Aggregation Database (gnomAD), with an allele frequency of 0.004 and 0.000777, respectively [27,30].

Table 1.
Variants identified in desmoplakin (DSP NM_004415) gene by next generation sequencing (NGS) analysis in probands.
2.2. Demographics and Clinical Presentation of Probands
The demographics and genetics data of 18 unrelated individuals who carried LP or P variants in the DSP gene were collected (Table 2). The mean age at diagnosis was 40.61 years (IQR 31–47.25); the youngest patient was a girl who presented recurrent myocarditis-like episodes, as did the other three patients, by the age of 18 years. At the time of first genetic counseling, none of the probands have presented a major arrhythmic event (MAE), defined as fatal or life-threatening arrhythmic events, except two female patients (ID 1 and 16) who presented a sustained ventricular tachycardia (SVT) pharmacologically cardioverted. The majority of patients were asymptomatic (61%, n = 11, NYHA I) or mildly symptomatic (39%, n = 7, NYHA II). Palpitations were the most common reported symptoms (78%, n = 14), followed by chest pain (44%, n = 8), with 39% of patients (n = 7) showing both symptoms. Seven patients (39%) suffered from myocarditis-like episodes, in four cases (57%) they were recurrent. Finally, two patients (ID 5 and 13) presented a syncopal episode. Nine patients (50%) presented a family history of sudden cardiac death (SCD), eight patients (44%) presented a family history of DCM and/or ACM and four patients (22%) presented family history for both conditions. Notably, more than half of the patients (61%, n = 11) underwent ICD implantation.
Table 2.
Clinical characteristics of probands carrying likely pathogenic or pathogenic variants in desmoplakin (DSP) gene.
2.3. Cardiac Imaging
All probands underwent a full cardiologic evaluation before they were referred to genetic counseling. Data from echocardiography and/or cardiac magnetic resonance imaging (CMRI) at first clinical assessment were retrospectively collected (Table 3). The majority of patients (61%, n = 11) presented with a mild decreased or at lower limits systolic function, frequently associated with cardiac wall abnormalities involving predominantly the left ventricle. At the time of diagnosis, slightly more than half of the patients (55%, n = 10) showed left ventricular enlargement (LVE) with a variable degree of ventricular dilation and four of them (40%) showed biventricular enlargement. Of note in all patients, CMRI lead to the identification of an area of delayed enhancement (DE) consistent with LV myocardial fibrosis, even if ventricular ejection fraction (VEF) was preserved. The most frequent localization of DE was subepicardial with left ventricular walls (LVW) and intraventricular septum involvement with a non-ischemic pattern. Particularly, in patient ID 1, CMRI showed morpho-functional modifications consistent with ACM with biventricular phenotype, while in probands ID 3 and 4, CMRI identified subepicardial DE distributed along postero-lateral and infero-lateral LVW, consistent with ALVC. In patients ID 7, 8, 9, and 10 affected by recurrent episodes of myocardial injury, CMRI showed a more extensive DE distribution with subepicardial or mid-subepicardial localization, as expected in a post-inflammatory cardiomyopathy (Supplementary Figures S1A, S2A, S3A and S4). In patient ID 8 myocardial fibrosis was also confirmed by the histological exam of endomyocardial bioptic tissue, which showed several areas of polychronic fibrosis, surrounded by myocytes with clear and significant involutional aspects with the rarefaction and disaggregation of the contractile material, nuclear clearing or complete nuclear involution.
Table 3.
Cardiac imaging of probands at first clinical assessment.
2.4. Electrocardiography, Arrythmias and ICD
At electrocardiography (ECG) baseline examination all probands showed an atrioventricular and intraventricular conduction substantially within normal limits (Table 4). Most individuals (67%, n = 12) presented QRS abnormalities, such as low voltage in peripheral leads (33%, n = 6), the fragmentation of QRS complex (fQRS) (28%, n = 5) and poor R wave progression (PRWP) (n = 1). Non-specific abnormalities (NSA) in ventricular repolarization (VR) were the most common anomalies reported (61%, n = 11). In two patients, ECG showed T wave inversion (TWI), respectively, in anterior and infero-lateral leads in patient ID 1 and inferior leads in patient ID 4. Holter ECG monitoring of 24 h is available in all patients, showing a high burden of ventricular ectopy (VE) with frequent or very frequent premature ventricular contractions (PVCs) of at least two distinct morphologies in 72% of individuals (n = 13). Of them, in eight cases (62%) they were organized in repetitive fashion, both couples and salvos. In two patients Holter ECG monitoring of 24 h revealed a low burden of VE with occasional polymorphic PVCs. Specifically, Holter ECG performed in patient ID 10, during treatment with metoprolol, showed some PVCs evenly distributed throughout the 24 h, mostly singles, some organized in couples, and two polymorphic triplets. An ECG stress test, later performed, showed some PVCs of different morphology at the peak of exercise and some monomorphic in the recovery phase. In patient ID 17, an ECG Holter showed some PVCs, mostly singles and monomorphic, some of different morphology and organized in couples. Of note, in patient ID 16, Holter ECG monitoring showed frequent polymorphic VE with some episodes of SVT, prior to define clinical and genetic diagnosis. ECG stress test data were available for four of these patients; none of them resulted positive for inducible ischemia. Particularly, an ECG stress test of patient ID 4 highlighted several PVCs of infundibular origin, both singles and organized in couples, predominantly in recovery phase; patient ID 5’s test revealed numerous PVCs, during exercise and in recovery phase, with left bundle branch and right bundle branch block morphology, some organized in couples, absents at peak exercise. In patient ID 8, an ECG stress test performed at first clinical assessment showed several, singles, polymorphic PVCs and three couples at the beginning of the exercise, which disappeared right after. Finally, in patient ID 17, and ECG stress test identified a symptomatic triplet at the peak of the exercise. A further test performed after 3 years in patient ID 8 showed in rest phase frequent, singles, polymorphic PVCs, with two couples and two triplets during exercise, persistent in recovery phase.
Table 4.
Electrocardiographic findings and arrhythmia burden of our cohort.
As previously reported in Table 2, 11 patients (61%) underwent ICD implantation; in nine of them for primary prevention, and in ID 1 and ID 16 for secondary prevention after SVT. Mean age at ICD implantation was 46.81 years (IQR 36.00–64.00), after an average interval from diagnosis of 3.1 years (IQR 0.00–3.00). Nine patients underwent implantation of an intracardiac device, six bicameral (Dual-Chamber; D-C) and three monocameral (Single-Chamber; S-C), respectively, while the other two were implanted with subcutaneous ICD (S-ICD). At the time of ICD implantation, Left Ventricular Ejection Fraction (LVEF) mean value was 43.00% (IQR 35.00–50.00) (Table 5).
Table 5.
Data related to implantable cardioverter defibrillator (ICD) implantation.
2.5. Segregation Data Analysis
After the identification of LP or P variants, genetic screening was offered to first degree relatives of affected probands. Data from the segregation analysis of three families (ID 2, ID 6, and ID 7) are available.
In the family of patient ID 2, genetic testing was performed in both his two sons and in the only nephew, whose father, the probands’ brother, suddenly died at the age of 51 years (Figure 1). Segregation analysis was not performed on the probands’ parents and brother because they had already died (Figure 1). His first child (IV:1) resulted as a carrier for heterozygous pathogenic variant Q833X in the DSP gene. At the time of the genetic counseling, he was 22 years old and apparently asymptomatic, with no history of syncope, chest pain and/or palpitations. Of note, a recent cardiologic evaluation was not available for the patient, thus it is not possible to exclude an unknown underlying condition. The second child (IV: 2) and the nephew (IV: 3) resulted negative at genetic testing.
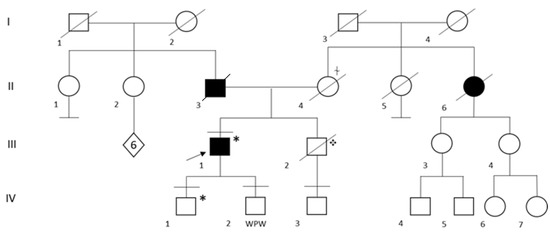
Figure 1.
Family Pedigree of Patient ID 2. Black circle and squares indicate individuals with a clinical diagnosis of dilated cardiomyopathy. Black arrow indicates the proband. Black lines at the top indicate individuals who underwent to genetic testing. Black lines at the bottom indicate individuals without offspring. WPW: Wolf Parkinson White syndrome, ✥ Sudden Cardiac Death (SCD) at the age of 51 years. * Q833X heterozygous carrier, ⍭ Aortic dissection.
In the family of patient ID 6, genetic testing was performed on her two daughters, which resulted in both being carriers of E1068Vfs*1 in the DSP gene (Figure 2). Her first daughter (IV:1), at 38 years old, received a full cardiologic evaluation, which resulted within the limits. Her second daughter (IV:2) was 28 years old at the time of genetic counselling and she did not undergo cardiological examination in the previous two years; neither of them reported any symptoms.
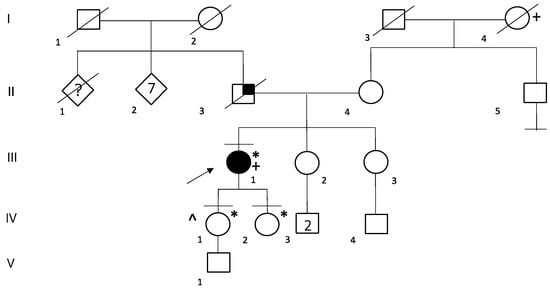
Figure 2.
Family Pedigree of Patient ID 6. Black circle and squares indicate individuals with Arrhythmogenic cardiomyopathy. Black arrow indicates the proband. Black lines at the top indicate individuals who underwent to genetic testing. Black lines at the bottom indicate individuals without offspring. Little black square indicates lung cancer, * E1068Vfs*19 heterozygous carrier, + Breast cancer, ^ Bilateral hypoacusia, probably due to a meningitis infection on her 6th month of life.
In the family of patient ID 7, genetic testing was performed on both probands’ parents and led to the identification of maternal segregation of P variant G1737Dfs*16 (Figure 3). The probands’ mother has a personal history of frequent ventricular extrasystoles in the absence of any signs of morpho-functional abnormalities at echocardiography performed during the previous year.
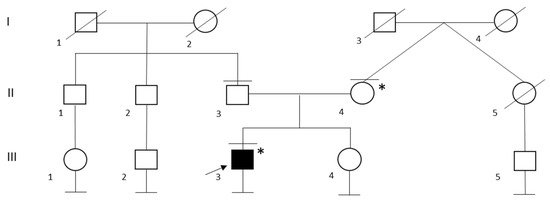
Figure 3.
Family Pedigree of Patient ID 7. Black circle and squares indicate individuals with a clinical diagnosis of dilated cardiomyopathy. Black arrow indicates the proband. Black lines at the top indicate individuals who underwent to genetic testing. Black lines at the bottom indicates individuals without offspring. * G1737Dfs*16 heterozygous carrier.
3. Discussion
DSP variants have been recently reported to be causative of a form of cardiomyopathy characterized by recurrent myocardial injury episodes and left ventricular fibrosis [22]. In this cohort, seven patients reported a previous history of myocardial injury episodes and all of them presented DE at CMRI (Table 2 and Table 3). Extensive infectious and immunological screenings were performed in patients who suffered from presumed myocarditis episodes and both resulted negative. In one case (patient ID 8), a cardiac biopsy was performed, showing histological findings consistent with inflammatory cardiomyopathy, and resulting negative for viral markers. In this patient, myocardial injury was the first clinical presentation of disease along with frequent polymorphic PVCs. A short time after the first episodes, the patient presented a drastic decrease in systolic function, previously reported as normal or at the lower limits (Table 3), with a LVEF reduced by up to 25% and only partially recovered after appropriate clinical management, leading to S-ICD implantation at the age of 34 years (Table 5). Notably, even if they were not detected at the first clinical cardiological assessment (Table 4), performed when he was 31 years old, TWI in V5-V6 and low voltage QRS complex, the latter finding already known, were observed at the last ECG baseline, carried out at the age of 36 years (Supplementary Figure S2B). After 5 months from the first genetic counseling, the patient presented two episodes of SVT: one was treated with direct current (DC) shock, and the other resolved spontaneously. At CMRI, circumferential DE was observed. Similarly, patient ID 9 was referred for medical attention at the age of 18 years due to recurrent episodes of myocardial injury associated with chest pain and palpitations. Holter ECG registration detected frequent PVCs with two morphologies isolated and organized in 10 couples and one triplet (Supplementary Figure S3C). Differently from patient ID 8, LVEF was preserved at the time of genetic counselling, when she was 23 years old. CMRI detected an extensive and persistent mid- and subepicardial DE, involving 30% of myocardial mass (Supplementary Figure S3A). In contrast, in patient ID 7, no history of arrhythmias or others electrocardiographic anomalies was reported, nor they were detected at subsequent examinations, except for fQRS at ECG baseline (Supplementary Figure S1B). CMRI revealed an extensive subepicardial DE of inferior LVW in a non-ischemic pattern (Supplementary Figure S1A). Patient ID 10 sought medical attention due to three episodes of chest pain and palpitations with myocardial enzyme release, initially diagnosed as a presumed viral myocarditis. CMRI performed after the first episode showed an extensive, nearly circumferential, LGE of LVW (Supplementary Figure S4) with spotty intramiocardial distribution in the inferior segment of IVS and in the mid-lateral and apical wall. This latter finding was no longer reported in the following exams. Of note, CMRI performed in occasion of these episodes failed in identifying any signs of myocardial edema. Additionally, in patients ID 15 and 18, the first clinical presentation was chest pain with myocardial enzyme release, and ECG abnormalities with normal coronary arteries. Finally, patient ID 5, unlike other patients who acceded to medical attention primarily for their myocarditis-like history, was firstly evaluated at the age of 31 years due to frequent polymorphic PVCs, absent at the peak of exercise. Both echocardiography and CMRI findings confirmed a diagnosis of DCM with left ventricular non compaction (LVNC). Particularly, CMRI revealed a moderate LVE with depressed biventricular systolic function and both intramural and subepicardial DE, respectively, of IVS and inferior LVW, midapical segments.
Myocarditis has been previously proposed as a possible clinical presentation of ACM [31,32] and more specifically DSP-related cardiomyopathies [22,23,24]. Myocarditis diagnosis is often difficult due to variable clinical presentation and etiology. In most cases, due to the lack of histological data, the diagnosis is frequently based on clinical presentation, CMRI, and laboratory data, since endomyocardial biopsy is not routinary performed [33]. Therefore, it is not uncommon that myocarditis diagnosis is presumed and not proven. This applies also to myocarditis episodes reported in DSP-related cardiomyopathies. Moreover, some of the cases reported in scientific literature did not meet the current clinical criteria of myocarditis diagnosis.
In 2005, Bauce et al. first described clinical myocarditis in two siblings affected by familial ARVC due to a missense variant in the DSP gene, already known to be associated with the disease. In both cases, chest pain and an elevation of myocardial enzymes in the absence of any abnormalities at coronary angiography were detected and reported as the first clinical symptoms [34]. Notably, in the same year, Sen-Chowdhry et al. in a cohort study of 42 patients affected by ALVC, reported that four of them presented with chest pain with enzyme release and had previously been diagnosed as affected by viral myocarditis, suggesting the possibility of myocarditis-like episodes as clinical presentation in ALVC patients [2]. The authors suggested that inflammatory myocarditis may be part of the natural history of ACM, due to genetic causes rather than infective ones. In fact, variants in the desmosomal gene, such as the DSP gene, may affect intercellular adhesions and/or intermediate filament function, leading to myocyte loss and inflammatory reaction followed by fibrous or fibrofatty tissue replacement. This process may present in an episodic fashion, triggered by unknown stimuli, which activate cell loss and inflammation in myocardial tissue, with a clinical presentation that has been defined ‘hot phase’ [35,36]. More recently, Smith et al. reported myocardial inflammatory episodes in 15% of their cohort among DSP patients. In addition, in 90% of patients with available CMRI data, the DE of LV was observed [22]. Similarly, Wang et al., in 2021, in a large cohort study of DSP carriers, reported a documented myocardial injury in 22% of patients, and in 90% of them CMRI showed a LGE distribution suggestive of myocardial fibrosis. The authors observed that myocardial injury also preceded the appearance of arrhythmias and heart failure [26]. Finally, in 2020, Piriou et al. selected six families, among those with a potential inherited cardiomyopathy phenotype, with at least one individual with a documented episode of acute myocarditis, and at least one individual with a cardiomyopathy or a history of SCD. Genetic testing identified in five of them a heterozygous pathogenic variant in DSP, and clinical evaluation confirmed a ALVC diagnosis. Histological data available in some of the DSP carriers showed a coexistence of slight inflammatory infiltrates, interstitial fibrosis, and the presence of viral genomes without overt systemic viral infection.
The authors concluded that acute myocarditis should be considered as a further criterion for ACM diagnosis, and genetic testing should be performed in patients who experience acute myocarditis and have a family history of cardiomyopathy, or sudden death [37].
Our findings confirm myocardial episodic injuries as a clinical manifestation of DSP-cardiomyopathy, which may be the first clinical sign of disease, as occurred in seven patients with an history of presumed myocarditis.
Remarkably, all patients in this cohort presented LV DE at CMRI, even if systolic function was preserved. Notably, a larger extent of DE was observed in those patients with myocarditis-like history and, most commonly, it involved subepicardial layers of LV. Many studies demonstrated that the larger the extent of DE, the higher is the risk of SCD [38,39]. However, some studies do not attribute additional prediction value to the extent of LGE [40]. Additionally, data about the role of DE distribution remains unclear. Specifically, septal midwall LGE have been associated with a greater risk of SCD [41] and it has even been proposed as an exclusive predictor for SCD or ICD implantation [42]. On the contrary, other authors proposed the subepicardial distribution of LGE as an independent predictor for MAE [43]. Finally, some studies have not demonstrated an association between DE location and additional SCD risk [44]. Recently, Cipriani et al. clinically characterized a population of 87 ACM patients and 153 DCM patients who underwent CMRI with quantitative tissue characterization. All ACM patients presented LGE predominantly localized in the subepicardial LVW. A greater amount of LGE was observed in those patients with a decreased LVEF [45]. The authors suggested that, due to the extensive amount of LV fibrosis identified in ACM patients, the prophylactic implantation of ICD may be considered even if LVEF is not severely depressed [45].
In our cohort, 72% of patients presented a high burden of VE with frequent or very frequent PVCs of at least two distinct morphologies, consistent with an arrhythmogenic substrate. Notably, they were variably observed during exercise, rest, or recovery phase. Three patients suffered from MAE and three presented TWI in peripheral leads, with a lower frequency compared to literature data [22,26]. This could be related to the relatively small size of our cohort. Remarkably, 61% of patients (n = 11) underwent ICD implantation, nine of them for primary prevention and two for secondary prevention. This finding appears to be substantially in line with data which emerged from other larger cohort studies, which reported a little higher percentage of ICD implantation in DSP carriers [46,47].
A total of 16 LP or P variants were identified, 75% (n = 12) were truncating variants, two were missense, one a splicing variant, and one a duplication variant (Figure 3). Thirteen variants were not previously reported in literature and of them, 10 were not cited by ClinVar [27]. The variants of ID 3 [48], ID 5 and 17 [49], ID 15 [26,50], and ID 18 [51,52] were previously reported in literature (Table 1). Since most variants were truncating ones, it was not possible to establish an accurate genotype-phenotype correlation based on our data. However, based on molecular predicted consequences, it is possible to hypothesize that variants causing a loss of function of DSP are more likely to be implicated in a severe form of cardiomyopathy [21]. Generally, we observed that a significant number of variants (31%, n = 5) were located in exon 23, followed by exon 24 (19%, n = 3). Of note, two variants, one missense and one duplication, respectively, were identified in exon 7 (Figure 4).
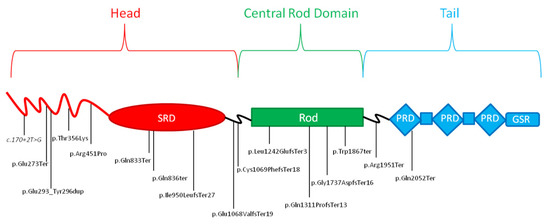
Figure 4.
Schematic representation of the main structural domains of the desmoplakin protein and relative position of the genetic variants reported in this study. Splicing variant reported in italics. SRD:Spectrin repeat domain; PRD:plakin repeat domains; GSR:glycine/serine/arginine-rich domain.
It has been previously reported in literature that not truncating variants in exon 7 may alter DSP protein function by interfering with physiological binding with plakoglobin protein, leading to the loss of desmosomal cadherin-JUP complexes [53]. Notably, two patients (ID 7 and 9) were heterozygous carriers of the same novel truncating variant G1737Dfs*16. Both of them presented a history of myocardial injury with a substantially impaired LVEF and an extensive subepicardial DE at CMRI; patient ID 9 also presented frequent polymorphic EV. Another two patients (ID 5 and 17) resulted to be carriers of the same truncating variant R1951X, previously described in literature [49]. However, in this case, the patients shared few clinical features, such as polymorphic VE, reported as frequent in patient ID 5 and occasional in patient ID 17. This finding may be due to the known clinical variability among individuals carrying the same variants reported for many inherited heart disorders, and/or to the age gap between the two patients, which does not allow to exclude the possibility of a future manifestation of further shared clinical signs or symptoms.
According to the recent Padua Criteria [7], only four patients presented sufficient requirements for the diagnosis of ALVC and two patients of ACM with biventricular involvement. Five patients were diagnosed as affected by DCM. These data suggest that in the left ventricular dominant form of ACM, genetic testing might be determinant to achieve a right diagnosis.
4. Materials and Methods
4.1. Subject Selection
We selected a cohort of 15 probands among those patients referred to Medical Genetics Unit of Policlinico Tor Vergata Hospital of Rome from 2018 to 2022, with a suspected or confirmed diagnosis of ACM or DCM, carriers of heterozygous variants in DSP gene. We checked whether or not selected DSP variants should be reclassified according to current ACMG standards and guidelines [29]. We excluded five patients carrying DSP variants classified as of uncertain significance due to the lack of data about their actual pathogenicity and the impossibility to rightfully establish a genotype-phenotype correlation. We included 10 probands heterozygous carriers of DSP variants classified as likely pathogenic (LP) or pathogenic (P). Genomic database ClinVar, gnomAD (v2.1.1) and dbSNP [27,28,30] were consulted as well as scientific literature and data were reported. Segregation data analysis of probands’ relatives were collected when available. Further 8 probands carrying likely pathogenic (LP) or pathogenic (P) variants were collected after clinical and genetic diagnosis in other reference centers, for a total of 18 probands included in the study.
4.2. Data Collection
Demographic and clinical data of DSP patients were retrospectively collected, including sex, age at diagnosis, personal history of major arrhythmias, family history of SCD and/or non-ischemic cardiomyopathies, and data from cardiologic evaluation of symptoms and imaging features. Indeed, patients were referred to our centre after a full cardiologic evaluation at Policlinico Casilino Hospital or San Giovanni Calibita Hospital, Fatebenefratelli, Isola Tiberina, comprehensive at least of baseline ECG, Holter ECG monitoring, transthoracic echocardiography (TTE) and/or CMRI. Coronary artery disease and/or others cardiovascular conditions potentially causative of pathologic myocardial modifications were excluded in all patients. Six probands had received their cardiac evaluation and genetic testing at the ASST Papa Giovanni XXIII Hospital of Bergamo. Data were collected in the same way as reported above. All cardiologic screenings were performed according to standards clinical protocols.
4.3. Genetic Analysis
The DNA was extracted with the EZ1 DNA Blood kit (Qiagen, Hilden, Germany) following manifacture’s protocol. DNA was analyzed by Nanodrop (Thermofisher, Waltham, MA, USA) to evaluate the quality by measurement of the levels of absorbance at different wavelengths. At the same time, the samples were quantified with the Qubit Fluorometer 2.0 through the use of the Qubit High Sensitivity Assay Kit (Thermofisher, Waltham, MA, USA) for determining the concentration.
An on-demand custom panel was designed using Ion AmpliSeq Designer software 7.48 version. We selected 32 genes from literature belong to Cardiomyopathies and Channelopathies (Supplementary Table S1). To generate the libraries were necessary 15 ng of genomic DNA (gDNA), for each samples using Ion Chef System (Thermofisher), following the manufacturer’s instructions. The resultant template of 16 samples was sequenced on the Ion Torrent S5 platform (Thermofisher) using the 530 chip. Alignment and variant calling were carried out by the Torrent Suite (Thermofisher) using as human reference genome the GRCh37/hg19 version. Data were analyzed using the IonReporter software and the pathogenicity of the identified variants was evaluated according to the ACMG standards and guidelines [46]. The variants identified as VoUS, LP or P were selected for Sanger confirmation, performed using specific oligonulceotides whose sequence is shown in Table S2 (Supplementary Materials). At ASST Papa Giovanni XXIII Hospital in Bergamo, genetic testing has been performed by bioinformatic selection of the cardiac gene panel (Supplementary Table S1) from whole exome data (Agilent, Santa Clara, CA, USA, Clinical Research Exome v.2) sequenced on the Illumina NovaSeq 6000 platform.
5. Conclusions
Despite the small size of this cohort, clinical and genetic data suggest that DSP-related cardiomyopathy could be considered a distinct clinical entity characterized by a high arrhythmic burden, variable LVE, and LV scar. Myocarditis-like episodes may represent the first clinical presentation of the disease. These results are in line with recent literature data and contribute to a better delineation of this emerging specific phenotype.
We propose to consider genetic testing, according to the following red-flags: (i) young adult subjects with family history of ACM/DCM and/or SCD; (ii) myocarditis-like picture characterized by chest pain, myocardial enzyme release, 12-lead electrocardiogram abnormalities with normal coronary arteries; and (iii) the presence of LGE with subepicardial distribution. Current criteria for risk stratification based mainly on depressed systolic function may not be sufficient for patients affected by DSP-related cardiomyopathy. The presence of extensive myocardial fibrosis could set up an arrhythmic substrate, which predispose to the development of malignant arrhythmias. Primary ICD implantation may be considered even in the absence of impaired LVEF. Further studies on a larger cohort are needed to identify more accurately distinctive phenotypic features and rule out a likely genotype-phenotype correlation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24032490/s1.
Author Contributions
Conceptualization, E.M., F.D.L., R.M. and F.S.; methodology, V.F., A.L. and L.P.; software, V.F. and A.L.; investigation, F.R., A.M., L.C., S.B. and L.P.; data curation, F.R., A.M., L.C., S.B., F.D.L., E.M. and A.I.; writing—original draft preparation, F.D.L. and E.M.; writing—review and editing, F.D.L., E.M., A.I., M.I., R.M., F.S. and G.N. All authors have read and agreed to the published version of the manuscript.
Funding
Italian Ministry of Health, GENERA project to G.N. (CUP E83C22004130005—Piano Sviluppo e Coesione Salute—FSC 2014-2020—Traiettoria 3).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Policlinico Tor Vergata, Rome, Italy; approval code: protocol code 254.20; approval date: December 2020, and all patients involved in the study signed an informed consent accordingly to ethical guidelines. Patients were collected in a path that belongs to common clinical practice.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Some of the variants have been submitted to ClinVar (SUB12648635 clivar).
Acknowledgments
Thanks to “Heal Italia” PE0000019, PE PNRR, M4-C2-1.3—Funded by NextGenerationEU. We are grateful to patients and family members for participating in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tavora, F.; Zhang, M.; Franco, M.; Oliveira, J.B.; Li, L.; Fowler, D.; Zhao, Z.; Cresswell, N.; Burke, A. Distribution of biventricular disease in arrhythmogenic cardiomyopathy: An autopsy study. Hum. Pathol. 2012, 43, 592–596. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef]
- Bennett, R.G.; Haqqani, H.M.; Berruezo, A.; Della Bella, P.; Marchlinski, F.E.; Hsu, C.J.; Kumar, S. Arrhythmogenic Cardiomyopathy in 2018–2019: ARVC/ALVC or Both? Heart Lung Circ. 2019, 28, 164–177. [Google Scholar] [CrossRef]
- Corrado, D.; Zorzi, A. Natural history of arrhythmogenic cardiomyopathy: Redefining the age range of clinical presentation. Heart Rhythm 2017, 14, 892–893. [Google Scholar] [CrossRef]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Ram-pazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef]
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Rivera Sorensen, C.; Sheppard, M.N.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- McNally, E.; MacLeod, H.; Dellefave-Castillo, L. Arrhythmogenic Right Ventricular Cardiomyopathy. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Insights into Genetics and Pathophysiology of Arrhythmogenic Cardiomyopathy. Curr. Heart Fail. Rep. 2021, 18, 378–390. [Google Scholar] [CrossRef]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Cheng, L.T.; Wang, Z.F.; Wu, Y.Q. Desmoplakin and clinical manifestations of desmoplakin cardiomyopathy. Chin. Med. J. 2021, 134, 1771–1779. [Google Scholar] [CrossRef]
- Bariani, R.; Cason, M.; Rigato, I.; Cipriani, A.; Celeghin, R.; De Gaspari, M.; Bueno Marinas, M.; Mattesi, G.; Pergola, V.; Rizzo, S.; et al. Clinical profile and long-term follow-up of a cohort of patients with desmoplakin cardiomyopathy. Heart Rhythm 2022, 19, 1315–1324. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C. Arrhythmogenic left ventricular cardiomyopathy. Heart 2022, 108, 733–743. [Google Scholar] [CrossRef]
- López-Ayala, J.M.; Gómez-Milanés, I.; Sánchez Muñoz, J.J.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdés, M.; et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Reichl, K.; Kreykes, S.E.; Martin, C.M.; Shenoy, C. Desmoplakin Variant-Associated Arrhythmogenic Cardiomyopathy Presenting as Acute Myocarditis. Circ. Genom. Precis. Med. 2018, 11, e002373. [Google Scholar] [CrossRef]
- Ghawanmeh, M.; Simon Frances, B.; Kerai, A.; Patel, P.; Du, J.; Kumar, P. Management of Recurrent Myocarditis Due to Desmoplakin Cardiomyopathy: Diagnostic and Therapeutic Challenges. JACC Case Rep. 2022, 4, 59–62. [Google Scholar] [CrossRef]
- Rezaei Bookani, K.; Minga, I.; Wodskow, J.; Harris, J.; Gordon, R.; Sarswat, N.; Pursnani, A. A case series of desmoplakin cardiomyopathy: A mimic of viral myocarditis. Eur. Heart J. Case Rep. 2022, 6, ytac341. [Google Scholar] [CrossRef]
- Wang, W.; Murray, B.; Tichnell, C.; Gilotra, N.A.; Zimmerman, S.L.; Gasperetti, A.; Scheel, P.; Tandri, H.; Calkins, H.; James, C.A. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. Europace 2022, 24, 268–277. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443, Correction in Nature 2021, 590, E53; Correction in Nature 2021, 597, E3–E4. [Google Scholar] [CrossRef]
- Scheel, P.J., 3rd; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am. J. Cardiol. 2021, 145, 128–134. [Google Scholar] [CrossRef]
- Scheel, P.J., 3rd; Murray, B.; Tichnell, C.; James, C.A.; Tandri, H.; Calkins, H.; Chelko, S.P.; Gilotra, N.A. Myocarditis: An Important Presentation of Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC). J. Card. Fail. 2020, 26, S37–S38. [Google Scholar] [CrossRef]
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648d. [Google Scholar] [CrossRef]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2007, 50, 1813–1821. [Google Scholar] [CrossRef]
- Bariani, R.; Cipriani, A.; Rizzo, S.; Celeghin, R.; Bueno Marinas, M.; Giorgi, B.; De Gaspari, M.; Rigato, I.; Leoni, L.; Zorzi, A.; et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. Europace 2021, 23, 907–917. [Google Scholar] [CrossRef]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneau, T.; Conan, E.; et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef]
- Aquaro, G.D.; Grigoratos, C.; Bracco, A.; Proclemer, A.; Todiere, G.; Martini, N.; Habtemicael, Y.G.; Carerj, S.; Sinagra, G.; Di Bella, G. Late Gadolinium Enhancement-Dispersion Mapping: A New Magnetic Resonance Imaging Technique to Assess Prognosis in Patients with Hypertrophic Cardiomyopathy and Low-Intermediate 5-Year Risk of Sudden Death. Circ. Cardiovasc. Imaging 2020, 13, e010489. [Google Scholar] [CrossRef]
- Aquaro, G.D.; De Luca, A.; Cappelletto, C.; Raimondi, F.; Bianco, F.; Botto, N.; Lesizza, P.; Grigoratos, C.; Minati, M.; Dell’Omodarme, M.; et al. Prognostic Value of Magnetic Resonance Phenotype in Patients with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2753–2765. [Google Scholar] [CrossRef]
- Halliday, B.P.; Gulati, A.; Ali, A.; Guha, K.; Newsome, S.; Arzanauskaite, M.; Vassiliou, V.S.; Lota, A.; Izgi, C.; Tayal, U.; et al. Association between Midwall Late Gadolinium Enhancement and Sudden Cardiac Death in Patients with Dilated Cardiomyopathy and Mild and Moderate Left Ventricular Systolic Dysfunction. Circulation 2017, 135, 2106–2115. [Google Scholar] [CrossRef]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.; Ismail, N.A.; Dweck, M.R.; et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013, 309, 896–908, Correction in JAMA 2013, 310, 99. [Google Scholar] [CrossRef]
- Almehmadi, F.; Joncas, S.X.; Nevis, I.; Zahrani, M.; Bokhari, M.; Stirrat, J.; Fine, N.M.; Yee, R.; White, J.A. Prevalence of myocardial fibrosis patterns in patients with systolic dysfunction: Prognostic significance for the prediction of sudden cardiac arrest or appropriate implantable cardiac defibrillator therapy. Circ. Cardiovasc. Imaging 2014, 7, 593–600. [Google Scholar] [CrossRef]
- Shin, D.G.; Lee, H.-J.; Park, J.; Uhm, J.-S.; Pak, H.-N.; Lee, M.-H.; Kim, Y.J.; Joung, B. Pattern of late gadolinium enhancement predicts arrhythmic events in patients with non-ischemic cardiomyopathy. Int. J. Cardiol. 2016, 222, 9–15. [Google Scholar] [CrossRef]
- Keil, L.; Chevalier, C.; Kirchhof, P.; Blankenberg, S.; Lund, G.; Müllerleile, K.; Magnussen, C. CMR-Based Risk Stratification of Sudden Cardiac Death and Use of Implantable Cardioverter-Defibrillator in Non-Ischemic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 7115. [Google Scholar] [CrossRef]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis with Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef]
- Reza, N.; de Feria, A.; Chowns, J.L.; Hoffman-Andrews, L.; Vann, L.; Kim, J.; Marzolf, A.; Owens, A.T. Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP). Cardiogenetics 2022, 12, 24–36. [Google Scholar] [CrossRef]
- Bariani, R.; Rigato, I.; Cason, M.; Marinas, M.B.; Celeghin, R.; Pilichou, K.; Bauce, B. Genetic Background and Clinical Features in Arrhythmogenic Left Ventricular Cardiomyopathy: A Systematic Review. J. Clin. Med. 2022, 11, 4313. [Google Scholar] [CrossRef]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef]
- Cuenca, S.; Ruiz-Cano, M.J.; Gimeno-Blanes, J.R.; Jurado, A.; Salas, C.; Gomez-Diaz, I.; Padron-Barthe, L.; Grillo, J.J.; Vilches, C.; Segovia, J.; et al. Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. J. Heart Lung Transplant. 2016, 35, 625–635. [Google Scholar] [CrossRef]
- Pigors, M.; Schwieger-Briel, A.; Cosgarea, R.; Diaconeasa, A.; Bruckner-Tuderman, L.; Fleck, T.; Has, C. Desmoplakin mutations with palmoplantar keratoderma, woolly hair and cardiomyopathy. Acta Derm. Venereol. 2015, 95, 337–340. [Google Scholar] [CrossRef]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef]
- Ye, J.Z.; Delmar, M.; Lundby, A.; Olesen, M.S. Reevaluation of genetic variants previously associated with arrhythmogenic right ventricular cardiomyopathy integrating population-based cohorts and proteomics data. Clin. Genet. 2019, 96, 506–514. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).