
Short-Term Starvation Weakens the Efficacy of Cell Cycle Specific Chemotherapy Drugs through G1 Arrest


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
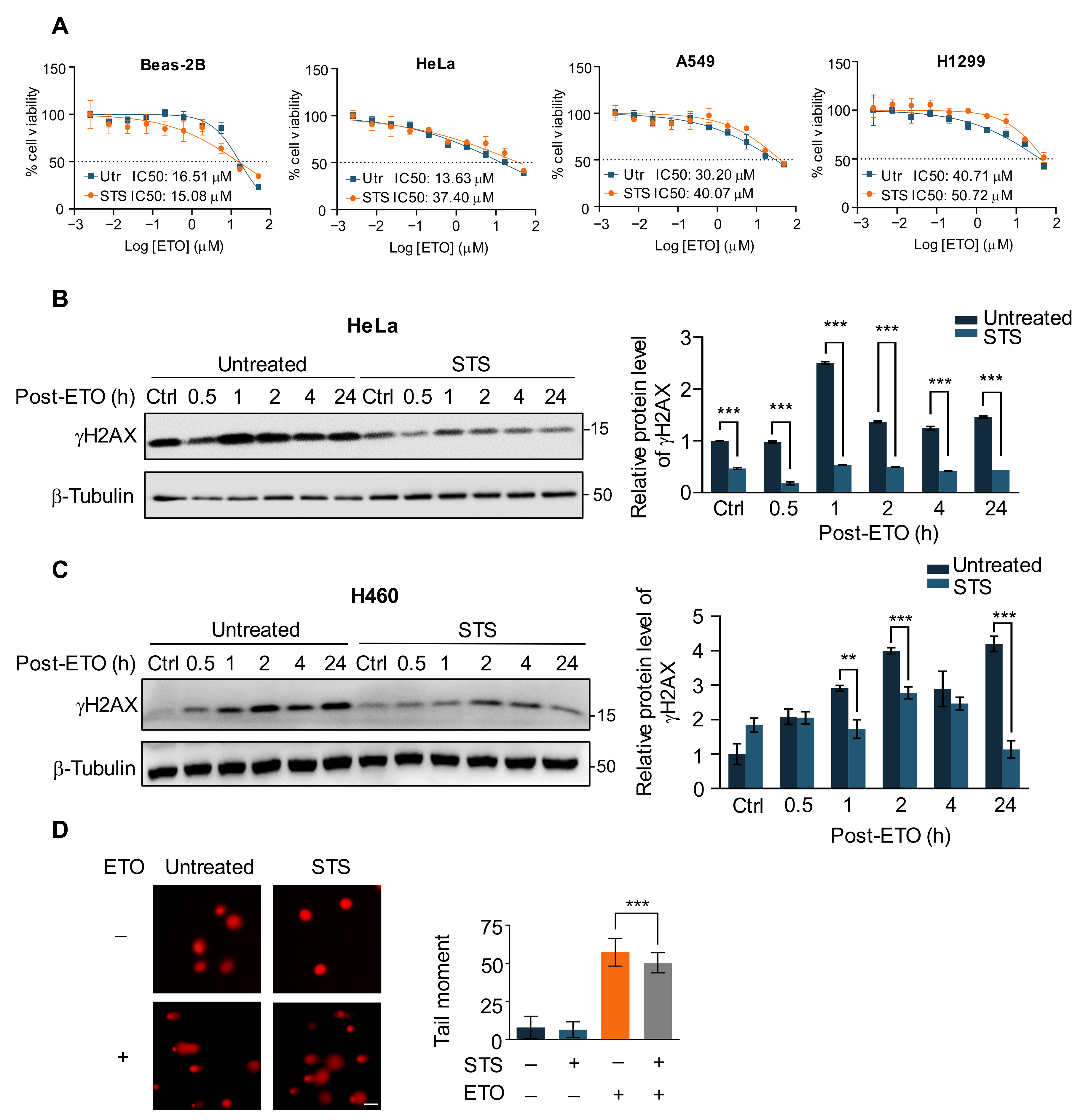
2.1. Short-Term Starvation Protects Cancer Cells against Chemotherapy Drug ETO
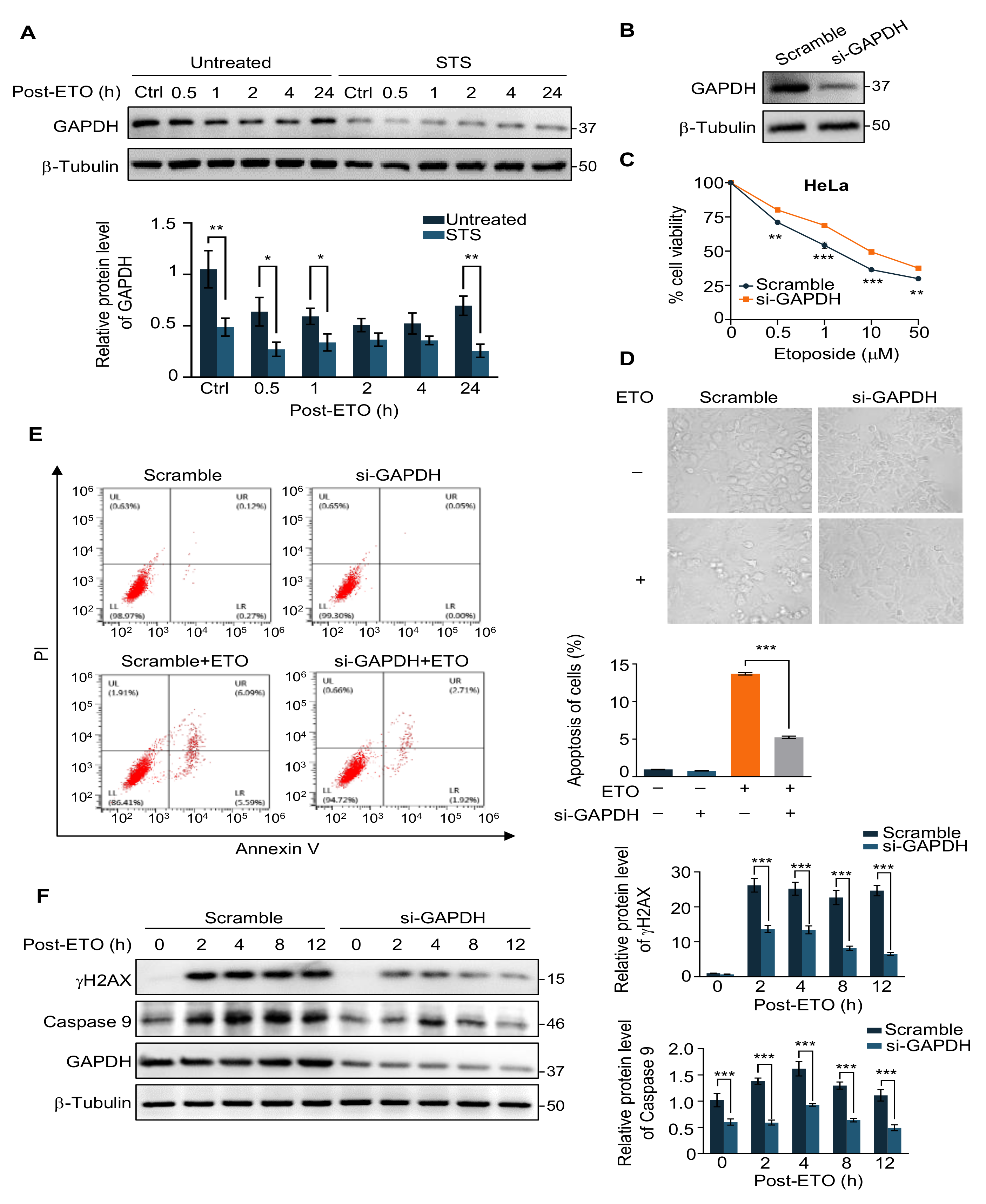
2.2. GAPDH Is Involved in the Protection of Cells from ETO-Induced Cell Apoptosis by STS
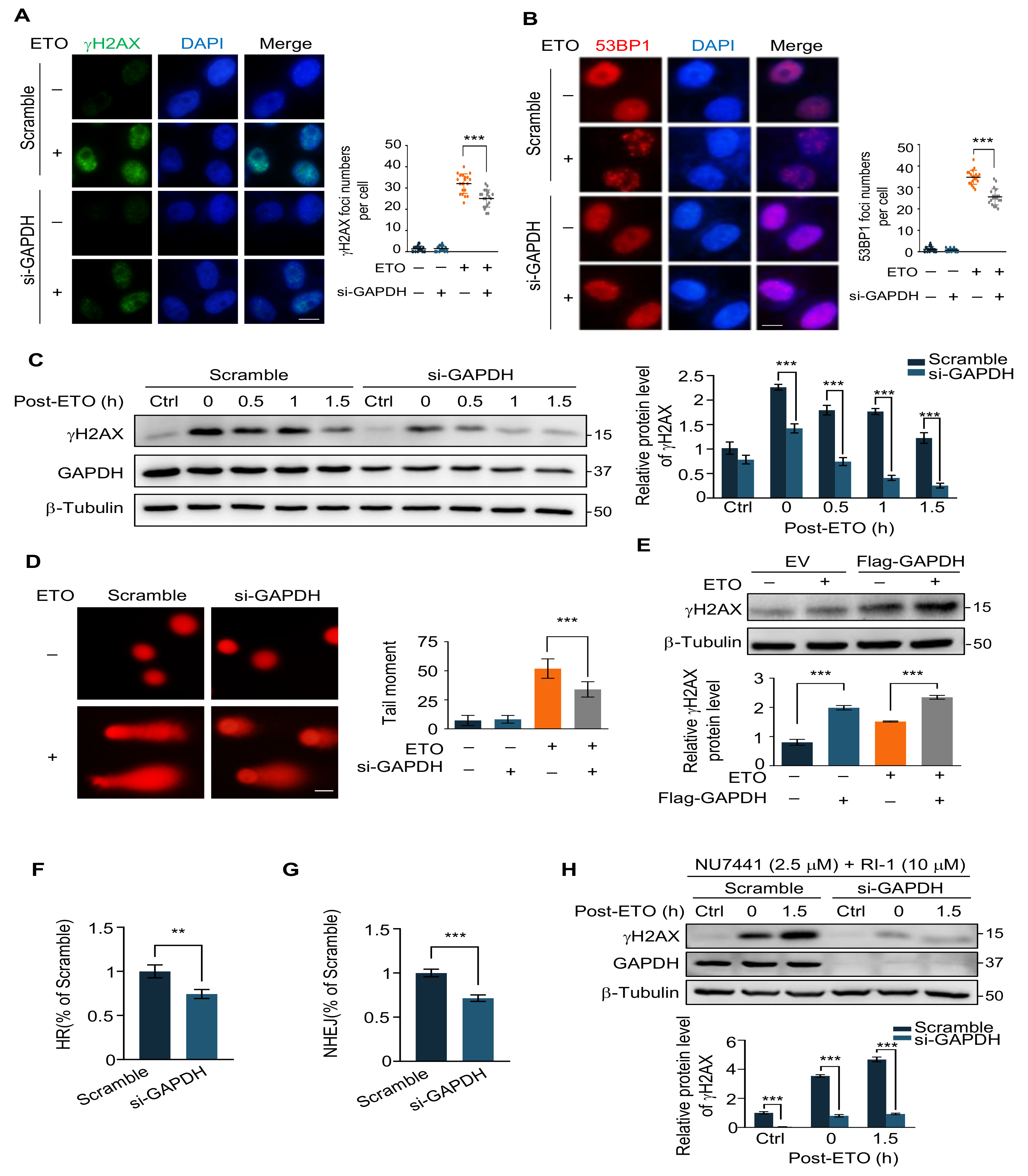
2.3. GAPDH Knockdown Decreases ETO-Induced DNA Damage
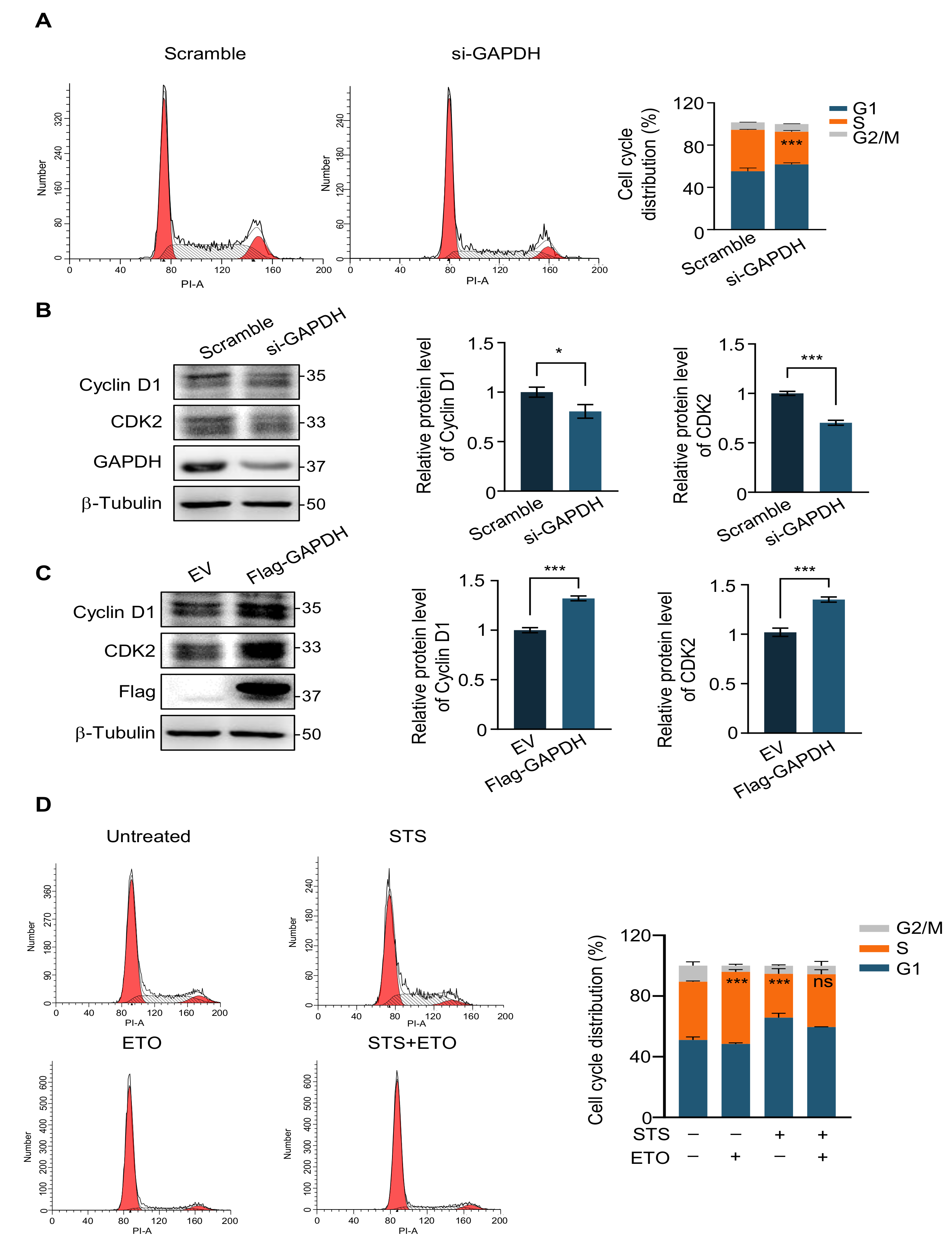
2.4. G1 Phase Arrest Weakens the Efficacy of Chemotherapy Drugs
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Regent
4.2. Antibodies
4.3. Western Blot
4.4. Cell Viability Assay
4.5. siRNA Sequences and Transfection
4.6. Immunofluorescence
4.7. Apoptosis Assay
4.8. Comet Assay
4.9. HR and NHEJ Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Longo, V.D. Starvation, detoxification, and multidrug resistance in cancer therapy. Drug Resist. Updat. 2012, 15, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive reciprocity of lipid and glucose metabolism in human short-term starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Salvadori, G.; Zanardi, F.; Iannelli, F.; Lobefaro, R.; Vernieri, C.; Longo, V.D. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 2021, 33, 2247–2259. [Google Scholar] [CrossRef]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef]
- Chang, M.; Wang, M.; Wang, M.; Shu, M.; Ding, B.; Li, C.; Pang, M.; Cui, S.; Hou, Z.; Lin, J. A Multifunctional Cascade Bioreactor Based on Hollow-Structured Cu2 MoS4 for Synergetic Cancer Chemo-Dynamic Therapy/Starvation Therapy/Phototherapy/Immunotherapy with Remarkably Enhanced Efficacy. Adv. Mater. 2019, 31, e1905271. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Jia, X.; Du, S.; Yin, Y.; Zhang, X. Cascade catalytic nanoplatform for enhanced starvation and sonodynamic therapy. J. Drug Target. 2020, 28, 195–203. [Google Scholar] [CrossRef]
- Yang, B.; Ding, L.; Chen, Y.; Shi, J. Augmenting Tumor-Starvation Therapy by Cancer Cell Autophagy Inhibition. Adv. Sci. 2020, 7, 1902847. [Google Scholar] [CrossRef]
- Zhang, M.K.; Li, C.X.; Wang, S.B.; Liu, T.; Song, X.L.; Yang, X.Q.; Feng, J.; Zhang, X.Z. Tumor Starvation Induced Spatiotemporal Control over Chemotherapy for Synergistic Therapy. Small 2018, 14, e1803602. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, Z.; Zeng, X.; Chen, X.; Gu, Z. Advances in nanomedicine for cancer starvation therapy. Theranostics 2019, 9, 8026–8047. [Google Scholar] [CrossRef] [PubMed]
- Hande, K.R. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Anand, J.; Sun, Y.; Zhao, Y.; Nitiss, K.C.; Nitiss, J.L. Detection of Topoisomerase Covalent Complexes in Eukaryotic Cells. Methods Mol. Biol. 2018, 1703, 283–299. [Google Scholar] [CrossRef]
- Menendez, D.; Anand, J.R.; Murphy, C.C.; Bell, W.J.; Fu, J.; Slepushkina, N.; Buehler, E.; Martin, S.E.; Lal-Nag, M.; Nitiss, J.L.; et al. Etoposide-induced DNA damage is increased in p53 mutants: Identification of ATR and other genes that influence effects of p53 mutations on Top2-induced cytotoxicity. Oncotarget 2022, 13, 332–346. [Google Scholar] [CrossRef]
- Sun, Y.; Miller Jenkins, L.M.; Su, Y.P.; Nitiss, K.C.; Nitiss, J.L.; Pommier, Y. A conserved SUMO pathway repairs topoisomerase DNA-protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Sci. Adv. 2020, 6, eaba6290. [Google Scholar] [CrossRef]
- Sun, Y.; Saha, S.; Wang, W.; Saha, L.K.; Huang, S.N.; Pommier, Y. Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair 2020, 89, 102837. [Google Scholar] [CrossRef]
- Lee, K.C.; Padget, K.; Curtis, H.; Cowell, I.G.; Moiani, D.; Sondka, Z.; Morris, N.J.; Jackson, G.H.; Cockell, S.J.; Tainer, J.A.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef]
- Xiao, H.; Mao, Y.; Desai, S.D.; Zhou, N.; Ting, C.Y.; Hwang, J.; Liu, L.F. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 3239–3244. [Google Scholar] [CrossRef]
- Loehrer, P.J., Sr. Etoposide therapy for testicular cancer. Cancer 1991, 67 (Suppl. S1), 220–224. [Google Scholar] [CrossRef]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Nayak, M.S.; Yang, J.M.; Hait, W.N. Effect of a single nucleotide polymorphism in the murine double minute 2 promoter (SNP309) on the sensitivity to topoisomerase II-targeting drugs. Cancer Res. 2007, 67, 5831–5839. [Google Scholar] [CrossRef] [PubMed]
- Tinkum, K.L.; Stemler, K.M.; White, L.S.; Loza, A.J.; Jeter-Jones, S.; Michalski, B.M.; Kuzmicki, C.; Pless, R.; Stappenbeck, T.S.; Piwnica-Worms, D.; et al. Fasting protects mice from lethal DNA damage by promoting small intestinal epithelial stem cell survival. Proc. Natl. Acad. Sci. USA 2015, 112, E7148–E7154. [Google Scholar] [CrossRef]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Glaser, P.E.; Gross, R.W. Rapid plasmenylethanolamine-selective fusion of membrane bilayers catalyzed by an isoform of glyceraldehyde-3-phosphate dehydrogenase: Discrimination between glycolytic and fusogenic roles of individual isoforms. Biochemistry 1995, 34, 12193–12203. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Hsu, J.W.; Park, S.Y.; Li, J.; Oldham, W.M.; Beznoussenko, G.V.; Mironov, A.A.; Loscalzo, J.; Hsu, V.W. GAPDH inhibits intracellular pathways during starvation for cellular energy homeostasis. Nature 2018, 561, 263–267. [Google Scholar] [CrossRef]
- Ci, S.; Xia, W.; Liang, W.; Qin, L.; Zhang, Y.; Dianov, G.L.; Wang, M.; Zhao, X.; Wu, C.; Alagamuthu, K.K.; et al. Src-mediated phosphorylation of GAPDH regulates its nuclear localization and cellular response to DNA damage. FASEB J. 2020, 34, 10443–10461. [Google Scholar] [CrossRef]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra127. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Mohd Omar, M.F.; Soong, R. The Warburg effect and drug resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef]
- Zhong, X.Y.; Yuan, X.M.; Xu, Y.Y.; Yin, M.; Yan, W.W.; Zou, S.W.; Wei, L.M.; Lu, H.J.; Wang, Y.P.; Lei, Q.Y. CARM1 Methylates GAPDH to Regulate Glucose Metabolism and Is Suppressed in Liver Cancer. Cell Rep. 2018, 24, 3207–3223. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef]
- Li, W.; Liu, C.; Huang, Z.; Shi, L.; Zhong, C.; Zhou, W.; Meng, P.; Li, Z.; Wang, S.; Luo, F.; et al. AKR1B10 negatively regulates autophagy through reducing GAPDH upon glucose starvation in colon cancer. J. Cell Sci. 2021, 134, jcs255273. [Google Scholar] [CrossRef]
- Kluska, M.; Wozniak, K. Natural Polyphenols as Modulators of Etoposide Anti-Cancer Activity. Int. J. Mol. Sci. 2021, 22, 6602. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N.O.; Tafani, M.; Rothman, R.J.; Russo, M.A.; Farber, J.L. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochromec. J. Biol. Chem. 2002, 277, 16547–16552. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Tammaro, M.; Barr, P.; Ricci, B.; Yan, H. Replication-dependent and transcription-dependent mechanisms of DNA double-strand break induction by the topoisomerase 2-targeting drug etoposide. PLoS ONE 2013, 8, e79202. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Ban, Y.; Lyu, Y.L.; Liu, L.F. Proteasome-dependent processing of topoisomerase I-DNA adducts into DNA double strand breaks at arrested replication forks. J. Biol. Chem. 2009, 284, 28084–28092. [Google Scholar] [CrossRef] [PubMed]
- Nam, C.; Yamauchi, H.; Nakayama, H.; Doi, K. Etoposide induces apoptosis and cell cycle arrest of neuroepithelial cells in a p53-related manner. Neurotoxicol. Teratol. 2006, 28, 664–672. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, G.A.; Visser, G.W.; Vrouenraets, M.B. Photosensitizer-antibody conjugates for detection and therapy of cancer. Adv. Drug Deliv. Rev. 2004, 56, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Patel, A. Benign vs Malignant Tumors. JAMA Oncol. 2020, 6, 1488. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updat. 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Kozal, K.; Jozwiak, P.; Krzeslak, A. Contemporary Perspectives on the Warburg Effect Inhibition in Cancer Therapy. Cancer Control. 2021, 28, 10732748211041243. [Google Scholar] [CrossRef]
- Weng, M.L.; Chen, W.K.; Chen, X.Y.; Lu, H.; Sun, Z.R.; Yu, Q.; Sun, P.F.; Xu, Y.J.; Zhu, M.M.; Jiang, N.; et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1alpha pathway suppression. Nat. Commun. 2020, 11, 1869. [Google Scholar] [CrossRef] [PubMed]
- Ehl, S.; Astigarraga, I.; von Bahr Greenwood, T.; Hines, M.; Horne, A.; Ishii, E.; Janka, G.; Jordan, M.B.; La Rosee, P.; Lehmberg, K.; et al. Recommendations for the Use of Etoposide-Based Therapy and Bone Marrow Transplantation for the Treatment of HLH: Consensus Statements by the HLH Steering Committee of the Histiocyte Society. J. Allergy Clin. Immunol. Pract. 2018, 6, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Ragavan, M.; Das, M. Systemic Therapy of Extensive Stage Small Cell Lung Cancer in the Era of Immunotherapy. Curr. Treat. Options Oncol. 2020, 21, 64. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, M.; Hou, J.; Shao, S.; Liang, W.; Wang, S.; Yang, Y.; Guo, Z.; Pan, F. Short-Term Starvation Weakens the Efficacy of Cell Cycle Specific Chemotherapy Drugs through G1 Arrest. Int. J. Mol. Sci. 2023, 24, 2498. https://doi.org/10.3390/ijms24032498
Shi M, Hou J, Shao S, Liang W, Wang S, Yang Y, Guo Z, Pan F. Short-Term Starvation Weakens the Efficacy of Cell Cycle Specific Chemotherapy Drugs through G1 Arrest. International Journal of Molecular Sciences. 2023; 24(3):2498. https://doi.org/10.3390/ijms24032498
Chicago/Turabian StyleShi, Munan, Jiajia Hou, Shan Shao, Weichu Liang, Shiwei Wang, Yuzhou Yang, Zhigang Guo, and Feiyan Pan. 2023. "Short-Term Starvation Weakens the Efficacy of Cell Cycle Specific Chemotherapy Drugs through G1 Arrest" International Journal of Molecular Sciences 24, no. 3: 2498. https://doi.org/10.3390/ijms24032498
APA StyleShi, M., Hou, J., Shao, S., Liang, W., Wang, S., Yang, Y., Guo, Z., & Pan, F. (2023). Short-Term Starvation Weakens the Efficacy of Cell Cycle Specific Chemotherapy Drugs through G1 Arrest. International Journal of Molecular Sciences, 24(3), 2498. https://doi.org/10.3390/ijms24032498