
Ion Channels in Gliomas—From Molecular Basis to Treatment



Abstract
:1. Introduction—Classification and Etiopathogenesis of Brain Tumors
2. Proliferation
2.1. K+ Channels
2.1.1. hERG and EAG
2.1.2. KATP Channels
2.1.3. Ca2+-Activated K+ Channels
2.1.4. Kir4.1
2.2. TRP Channels
2.2.1. TRPV1 and TRPV2
2.2.2. TRPM2 and TRPM3
2.2.3. TRPML1
2.2.4. TRPC1 and TRPC6
2.2.5. TRPM7 and TRPM8
2.2.6. TRPML2
2.3. Chloride Channels
2.3.1. CIC
2.3.2. VRAC
2.3.3. TMEM16A
2.3.4. CLIC
2.4. NHE
2.5. ASIC
2.6. PIEZO
2.7. Voltage-Gated Na+ Channels
2.8. AMPAR
3. Infiltration and Cellular Migration
3.1. Ion Channels and Exchangers Enriched in Glioma Cell Leading Edge
3.1.1. NKCC1
3.1.2. BK Channels
3.1.3. TRPC
3.2. Ion Channels Enriched in Trailing Edge
3.2.1. KCa3.1 (IK)
3.2.2. CLC-3
3.3. Additional Modifiers of Migration Invasion
3.3.1. TRPM
3.3.2. TRPV4
3.3.3. ASIC1
3.3.4. PIEZO
3.3.5. NHE
3.4. Kir4.1—Inhibitor of Cellular Migration
3.5. Tumor Microenvironment (TME) influence on Migration and Invasion
4. Glioma Metabolism
4.1. VDAC in Metabolic Homeostasis of Gliomas
4.2. K+ Channels in Glioma Metabolism
4.3. Role of pH in Glioma Metabolism
4.4. TRPC Channels in Hypoxia-Induced Metabolic Shifts in Gliomas
5. Blood–Brain Barrier, Microenvironment, and Angiogenesis
5.1. Blood–Brain Barrier in Brain Tumors
5.2. Overview of BBB Ion Channels and Transporters
5.3. Aquaporins
5.4. Microenvironment in Brain Tumors
5.5. TME in Tumor-Associated Epilepsy (TAE)
5.5.1. TME in Tumor-Associated Brain Edema (TABE)
5.5.2. TME, Tumor pH, and Metabolic Environment
5.6. Angiogenesis and Blood Vessel Co-Option in Brain Tumors
5.6.1. Angiogenesis in Brain Tumors
5.6.2. Co-Option in Brain Tumors
5.6.3. TRPC6 in Angiogenesis of Glioma
5.6.4. Other TRP Channels in Tumor Angiogenesis
6. Therapeutic Outlook for Ion Channels in Brain Cancer
6.1. Repurposing of Drugs
6.2. Potential Pharmacological and Nonpharmacological Targets
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partap, S.; Monje, M. Pediatric Brain Tumors. Continuum 2018, 26, 1553–1583. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, K.; Abul, K.A.; Jon, C.A.; associate, e.; Jerrold, R.T. Robbins & Cotran Pathologic Basis of Disease, 10th ed.; With Illustrations by James, A.P.; Elsevier: Philadelphia, PA, USA, 2021; pp. 1241–1304. [Google Scholar]
- Pekmezci, M.; Villanueva-Meyer, J.E.; Goode, B.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Bastian, B.C.; Chamyan, G.; Maher, O.M.; Khatib, Z.; et al. The genetic landscape of ganglioglioma. Acta Neuropathol. Commun. 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Menyhárt, O.; Győrffy, B. Principles of tumorigenesis and emerging molecular drivers of SHH-activated medulloblastomas. Ann. Clin. Transl. Neurol. 2019, 6, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Stuhmer, W.; Martin, S.; Schell, J.; Reichmann, A.; Rohde, V.; Pardo, L. Analysis of the expression of Kv10.1 potassium channel in patients with brain metastases and glioblastoma multiforme: Impact on survival. BMC Cancer 2015, 15, 839. [Google Scholar] [CrossRef] [Green Version]
- Pointer, K.B.; Clark, P.A.; Eliceiri, K.W.; Salamat, M.S.; Robertson, G.A.; Kuo, J.S. Administration of Non-Torsadogenic human Ether-a-go-go-Related Gene Inhibitors Is Associated with Better Survival for High hERG-Expressing Glioblastoma Patients. Clin. Cancer Res. 2017, 23, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.K.; Bomben, V.C.; Sontheimer, H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006, 54, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R.; et al. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramia, M.; Sforna, L.; Franciolini, F.; Catacuzzeno, L. The Volume-Regulated Anion Channel in Glioblastoma. Cancers 2019, 11, 307. [Google Scholar] [CrossRef] [Green Version]
- Catacuzzeno, L.; Fioretti, B.; Franciolini, F. Expression and Role of the Intermediate-Conductance Calcium-Activated Potassium Channel KCa3.1 in Glioblastoma. J. Signal Transduct. 2012, 2012, 421564. [Google Scholar] [CrossRef] [Green Version]
- Catacuzzeno, L.; Sforna, L.; Esposito, V.; Limatola, C.; Franciolini, F. Ion Channels in Glioma Malignancy. Rev. Physiol. Biochem. Pharmacol. 2021, 181, 223–267. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Ren, Y.; Kang, L.; Zhang, L. Transmembrane protein with unknown function 16A overexpression promotes glioma formation through the nuclear factor-κB signaling pathway. Mol. Med. Rep. 2014, 9, 1068–1074. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, J.Y.; Jung, C.W.; Lee, Y.S.; An, J.Y.; Kim, E.H.; Kim, K.H.; Lee, S.P.; Park, J.Y.; Park, M.J. ANO1 regulates the maintenance of stemness in glioblastoma stem cells by stabilizing EGFRvIII. Oncogene 2021, 40, 1490–1502. [Google Scholar] [CrossRef]
- Setti, M.; Savalli, N.; Osti, D.; Richichi, C.; Angelini, M.; Brescia, P.; Fornasari, L.; Carro, M.S.; Mazzanti, M.; Pelicci, G. Functional role of CLIC1 ion channel in glioblastoma-derived stem/progenitor cells. J. Natl. Cancer Inst. 2013, 105, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Kurata, T.; Rajendran, V.; Fan, S.; Ohta, T.; Numata, M.; Fushida, S. NHE5 regulates growth factor signaling, integrin trafficking, and degradation in glioma cells. Clin. Exp. Metastasis 2019, 36, 527–538. [Google Scholar] [CrossRef]
- King, P.; Wan, J.; Guo, A.A.; Guo, S.; Jiang, Y.; Liu, M. Regulation of gliomagenesis and stemness through acid sensor ASIC1a. Int. J. Oncol. 2021, 59, 82. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.-K.; Hu, M.; Draghici, D.; et al. A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Liu, X.; van Wijnbergen, J.W.M.; Yuan, L.; Liu, Y.; Zhang, C.; Jia, W. Identification of PIEZO1 as a potential prognostic marker in gliomas. Sci. Rep. 2020, 10, 16121. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Schiapparelli, P.; Ap Rhys, C.; Guerrero-Cazares, H.; Smith, C.; Kim, D.H.; Kone, L.; Farber, H.; Lee, D.Y.; An, S.S.; et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012, 10, e1001320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiapparelli, P.; Guerrero-Cazares, H.; Magaña-Maldonado, R.; Hamilla, S.M.; Ganaha, S.; Goulin Lippi Fernandes, E.; Huang, C.H.; Aranda-Espinoza, H.; Devreotes, P.; Quinones-Hinojosa, A. NKCC1 Regulates Migration Ability of Glioblastoma Cells by Modulation of Actin Dynamics and Interacting with Cofilin. EBioMedicine 2017, 21, 94–103. [Google Scholar] [CrossRef] [Green Version]
- Stegen, B.; Butz, L.; Klumpp, L.; Zips, D.; Dittmann, K.; Ruth, P.; Huber, S.M. Ca2+-Activated IK K+ Channel Blockade Radiosensitizes Glioblastoma Cells. Mol. Cancer Res. 2015, 13, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Luo, L.; Begum, G.; Kohanbash, G.; Song, Q.; Rao, A.; Amankulor, N.; Sun, B.; Sun, D.; Jia, W. Elevated Na/H exchanger 1 (SLC9A1) emerges as a marker for tumorigenesis and prognosis in gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 255. [Google Scholar] [CrossRef] [Green Version]
- Cong, Z.X.; Wang, H.D.; Zhou, Y.; Wang, J.W.; Pan, H.; Zhang, D.D.; Zhang, L.; Zhu, L. Temozolomide and irradiation combined treatment-induced Nrf2 activation increases chemoradiation sensitivity in human glioblastoma cells. J. Neurooncol. 2014, 116, 41–48. [Google Scholar] [CrossRef]
- McLean, L.A.; Roscoe, J.; Jorgensen, N.K.; Gorin, F.A.; Cala, P.M. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am. J. Physiol. Cell Physiol. 2000, 278, C676–C688. [Google Scholar] [CrossRef] [Green Version]
- Olsen, M.L.; Sontheimer, H. Mislocalization of Kir channels in malignant glia. Glia 2004, 46, 63–73. [Google Scholar] [CrossRef]
- Thuringer, D.; Chanteloup, G.; Boucher, J.; Pernet, N.; Boudesco, C.; Jego, G.; Chatelier, A.; Bois, P.; Gobbo, J.; Cronier, L.; et al. Modulation of the inwardly rectifying potassium channel Kir4.1 by the pro-invasive miR-5096 in glioblastoma cells. Oncotarget 2017, 8, 37681–37693. [Google Scholar] [CrossRef] [Green Version]
- Debska-Vielhaber, G.; Godlewski, M.M.; Kicinska, A.; Skalska, J.; Kulawiak, B.; Piwonska, M.; Zablocki, K.; Kunz, W.S.; Szewczyk, A.; Motyl, T. Large-conductance K+ channel openers induce death of human glioma cells. J. Physiol. Pharmacol. 2009, 60, 27–36. [Google Scholar]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Bell, B.A.; Krishna, S. Increased aquaporin 1 water channel expression in human brain tumours. Br. J. Cancer 2002, 87, 621–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshio, K.; Binder, D.K.; Liang, Y.; Bollen, A.; Feuerstein, B.; Berger, M.S.; Manley, G.T. Expression of the aquaporin-1 water channel in human glial tumors. Neurosurgery 2005, 56, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hindy, N.; Bankfalvi, A.; Herring, A.; Adamzik, M.; Lambertz, N.; Zhu, Y.; Siffert, W.; Sure, U.; Sandalcioglu, I.E. Correlation of Aquaporin-1 Water Channel Protein Expression with Tumor Angiogenesis in Human Astrocytoma. Anticancer Res. 2013, 33, 609–613. [Google Scholar] [PubMed]
- Warth, A.; Kröger, S.; Wolburg, H. Redistribution of aquaporin-4 in human glioblastoma correlates with loss of agrin immunoreactivity from brain capillary basal laminae. Acta Neuropathol. 2004, 107, 311–318. [Google Scholar] [CrossRef]
- Noell, S.; Wolburg-Buchholz, K.; Mack, A.F.; Ritz, R.; Tatagiba, M.; Beschorner, R.; Wolburg, H.; Fallier-Becker, P. Dynamics of expression patterns of AQP4, dystroglycan, agrin and matrix metalloproteinases in human glioblastoma. Cell Tissue Res. 2012, 347, 429–441. [Google Scholar] [CrossRef]
- Nakanishi, M.; Morita, Y.; Hata, K.; Muragaki, Y. Acidic microenvironments induce lymphangiogenesis and IL-8 production via TRPV1 activation in human lymphatic endothelial cells. Exp. Cell Res. 2016, 345, 180–189. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Taylor, K.R.; Monje, M. Invasive glioma cells: The malignant pioneers that follow the current. Cell 2022, 185, 2846–2848. [Google Scholar] [CrossRef]
- Venkataramani, V.; Yang, Y.; Schubert, M.C.; Reyhan, E.; Tetzlaff, S.K.; Wißmann, N.; Botz, M.; Soyka, S.J.; Beretta, C.A.; Pramatarov, R.L.; et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 2022, 185, 2899–2917.e31. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: Ion channels, membrane voltage and the cell cycle. Cell Cycle 2014, 8, 3527–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cone, C.D., Jr. Unified theory on the basic mechanism of normal mitotic control and oncogenesis. J. Theor. Biol. 1971, 30, 151–181. [Google Scholar] [CrossRef]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stuhmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [Green Version]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Higashimori, H.; Sontheimer, H. Role of Kir4.1 channels in growth control of glia. Glia 2007, 55, 1668–1679. [Google Scholar] [CrossRef] [Green Version]
- Funck, V.R.; Ribeiro, L.R.; Pereira, L.M.; de Oliveira, C.V.; Grigoletto, J.; Della-Pace, I.D.; Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Larrick, J.W.; et al. Contrasting effects of Na+, K+-ATPase activation on seizure activity in acute versus chronic models. Neuroscience 2015, 298, 171–179. [Google Scholar] [CrossRef]
- Foroutan, S.; Brillault, J.; Forbush, B.; O’Donnell, M.E. Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl− cotransporter. Am. J. Physiol.-Cell Physiol. 2005, 289, C1492–C1501. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Hicks, K.; O’Neil, R.G.; Dubinsky, W.S.; Brown, R.C. TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am. J. Physiol. Cell Physiol. 2010, 298, C1583–C1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balbuena, P.; Li, W.; Rzigalinski, B.A.; Ehrich, M. Malathion/oxon and lead acetate increase gene expression and protein levels of transient receptor potential canonical channel subunits TRPC1 and TRPC4 in rat endothelial cells of the blood-brain barrier. Int. J. Toxicol. 2012, 31, 238–249. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykocki, N.R.; Brayden, J.E.; Hill-Eubanks, D.; Nelson, M.T. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, I.D.; Wang, S.; Brown, P.D.; Barrand, M.A.; Hladky, S.B. Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflüg. Arch.—Eur. J. Physiol. 2008, 456, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, H.; Suzuki, Y.; Yamamura, H.; Asai, K.; Imaizumi, Y. Hypoxic stress up-regulates Kir2.1 expression and facilitates cell proliferation in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2016, 476, 386–392. [Google Scholar] [CrossRef]
- Staudacher, I.; Jehle, J.; Staudacher, K.; Pledl, H.W.; Lemke, D.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PLoS ONE 2014, 9, e88164. [Google Scholar] [CrossRef] [Green Version]
- Shugg, T.; Dave, N.; Amarh, E.; Assiri, A.A.; Pollok, K.E.; Overholser, B.R. Letrozole targets the human ether-a-go-go-related gene potassium current in glioblastoma. Basic Clin. Pharmacol. Toxicol. 2021, 128, 357–365. [Google Scholar] [CrossRef]
- Huang, L.; Li, B.; Li, W.; Guo, H.; Zou, F. ATP-sensitive potassium channels control glioma cells proliferation by regulating ERK activity. Carcinogenesis 2009, 30, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. J. Neurosci. Res. 2004, 78, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Takayasu, T.; Kurisu, K.; Esquenazi, Y.; Ballester, L.Y. Ion Channels and Their Role in the Pathophysiology of Gliomas. Mol. Cancer Ther. 2020, 19, 1959–1969. [Google Scholar] [CrossRef]
- Simon, O.J.; Müntefering, T.; Grauer, O.M.; Meuth, S.G. The role of ion channels in malignant brain tumors. J. Neuro-Oncol. 2015, 125, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Edalat, L.; Stegen, B.; Klumpp, L.; Haehl, E.; Schilbach, K.; Lukowski, R.; Kühnle, M.; Bernhardt, G.; Buschauer, A.; Zips, D.; et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016, 7, 14259–14278. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.; Sforna, L.; Carlomagno, S.; Mangino, G.; Miscusi, M.; Pessia, M.; Franciolini, F.; Calogero, A.; Catacuzzeno, L. Overexpression of Large-Conductance Calcium-Activated Potassium Channels in Human Glioblastoma Stem-Like Cells and Their Role in Cell Migration. J. Cell. Physiol. 2017, 232, 2478–2488. [Google Scholar] [CrossRef] [PubMed]
- Hoa, N.T.; Ge, L.; Martini, F.; Chau, V.; Ahluwalia, A.; Kruse, C.A.; Jadus, M.R. Temozolomide induces the expression of the glioma Big Potassium (gBK) ion channel, while inhibiting fascin-1 expression: Possible targets for glioma therapy. Expert Opin. Ther. Targets 2016, 20, 1155–1167. [Google Scholar] [CrossRef]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K(+) channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Ion channels and transporters [corrected] in cancer. 2. Ion channels and the control of cancer cell migration. Am. J. Physiol. Cell Physiol. 2011, 301, C541–C549. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Turner, K.L.; Seifert, S.; Sontheimer, H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J. Neurosci. 2013, 33, 1427–1440. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.L.; Honasoge, A.; Robert, S.M.; McFerrin, M.M.; Sontheimer, H. A proinvasive role for the Ca(2+) -activated K(+) channel KCa3.1 in malignant glioma. Glia 2014, 62, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.L.; Sontheimer, H. Cl− and K+ channels and their role in primary brain tumour biology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130095. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Bock, J.; Grassmé, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, C.P.; O’Keefe, E.; Wallace, E.; Loftus, T.; Keaney, J.; Kealy, J.; Humphries, M.M.; Molloy, M.G.; Meaney, J.F.; Farrell, M.; et al. Blood-Brain Barrier Dysfunction as a Hallmark Pathology in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Bondjers, C.; He, L.; Takemoto, M.; Norlin, J.; Asker, N.; Hellström, M.; Lindahl, P.; Betsholtz, C. Microarray analysis of blood microvessels from PDGF-B and PDGF-Rbeta mutant mice identifies novel markers for brain pericytes. FASEB J. 2006, 20, 1703–1705. [Google Scholar] [CrossRef]
- He, L.; Vanlandewijck, M.; Raschperger, E.; Andaloussi Mäe, M.; Jung, B.; Lebouvier, T.; Ando, K.; Hofmann, J.; Keller, A.; Betsholtz, C. Analysis of the brain mural cell transcriptome. Sci. Rep. 2016, 6, 35108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, A.; Muñoz-Manchado, A.B.; Codeluppi, S.; Lönnerberg, P.; La Manno, G.; Juréus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Peretti, M.; Raciti, F.M.; Carlini, V.; Verduci, I.; Sertic, S.; Barozzi, S.; Garré, M.; Pattarozzi, A.; Daga, A.; Barbieri, F.; et al. Mutual Influence of ROS, pH, and CLIC1 Membrane Protein in the Regulation of G(1)-S Phase Progression in Human Glioblastoma Stem Cells. Mol. Cancer Ther. 2018, 17, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- Habela, C.W.; Ernest, N.J.; Swindall, A.F.; Sontheimer, H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J. Neurophysiol. 2009, 101, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Habela, C.W.; Olsen, M.L.; Sontheimer, H. ClC3 is a critical regulator of the cell cycle in normal and malignant glial cells. J. Neurosci. 2008, 28, 9205–9217. [Google Scholar] [CrossRef]
- Lui, V.C.; Lung, S.S.; Pu, J.K.; Hung, K.N.; Leung, G.K. Invasion of human glioma cells is regulated by multiple chloride channels including ClC-3. Anticancer Res. 2010, 30, 4515–4524. [Google Scholar]
- Wang, B.; Xie, J.; He, H.Y.; Huang, E.W.; Cao, Q.H.; Luo, L.; Liao, Y.S.; Guo, Y. Suppression of CLC-3 chloride channel reduces the aggressiveness of glioma through inhibiting nuclear factor-κB pathway. Oncotarget 2017, 8, 63788–63798. [Google Scholar] [CrossRef] [PubMed]
- Sforna, L.; Cenciarini, M.; Belia, S.; Michelucci, A.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. Hypoxia Modulates the Swelling-Activated Cl Current in Human Glioblastoma Cells: Role in Volume Regulation and Cell Survival. J. Cell. Physiol. 2017, 232, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bomben, V.C.; Sontheimer, H. Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia 2010, 58, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Bomben, V.C.; Turner, K.L.; Barclay, T.T.; Sontheimer, H. Transient receptor potential canonical channels are essential for chemotactic migration of human malignant gliomas. J. Cell. Physiol. 2011, 226, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Bomben, V.C.; Sontheimer, H.W. Inhibition of transient receptor potential canonical channels impairs cytokinesis in human malignant gliomas. Cell Prolif. 2008, 41, 98–121. [Google Scholar] [CrossRef]
- Ding, X.; He, Z.; Zhou, K.; Cheng, J.; Yao, H.; Lu, D.; Cai, R.; Jin, Y.; Dong, B.; Xu, Y.; et al. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. [Google Scholar] [CrossRef] [Green Version]
- Ge, R.; Tai, Y.; Sun, Y.; Zhou, K.; Yang, S.; Cheng, T.; Zou, Q.; Shen, F.; Wang, Y. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 2009, 283, 43–51. [Google Scholar] [CrossRef]
- Chinigò, G.; Castel, H.; Chever, O.; Gkika, D. TRP Channels in Brain Tumors. Front. Cell Dev. Biol. 2021, 9, 617801. [Google Scholar] [CrossRef] [PubMed]
- Ertilav, K.; Nazıroğlu, M.; Ataizi, Z.S.; Braidy, N. Selenium Enhances the Apoptotic Efficacy of Docetaxel Through Activation of TRPM2 Channel in DBTRG Glioblastoma Cells. Neurotox. Res. 2019, 35, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Oyama, A.; Hagiwara, T.; Miyazaki, A.; Mori, Y.; Kiuchi, Y.; Shimizu, S. Facilitation of H2O2-induced A172 human glioblastoma cell death by insertion of oxidative stress-sensitive TRPM2 channels. Anticancer Res. 2007, 27, 3987–3992. [Google Scholar] [PubMed]
- Wan, J.; Guo, A.A.; Chowdhury, I.; Guo, S.; Hibbert, J.; Wang, G.; Liu, M. TRPM7 Induces Mechanistic Target of Rap1b Through the Downregulation of miR-28-5p in Glioma Proliferation and Invasion. Front. Oncol. 2019, 9, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Inoue, K.; Leng, T.; Guo, S.; Xiong, Z.G. TRPM7 channels regulate glioma stem cell through STAT3 and Notch signaling pathways. Cell. Signal. 2014, 26, 2773–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasper, M.; Schäfer, A.; Piontek, G.; Teufel, J.; Brockhoff, G.; Ringel, F.; Heindl, S.; Zimmer, C.; Schlegel, J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. 2010, 12, 1024–1033. [Google Scholar] [CrossRef]
- Klumpp, D.; Frank, S.C.; Klumpp, L.; Sezgin, E.C.; Eckert, M.; Edalat, L.; Bastmeyer, M.; Zips, D.; Ruth, P.; Huber, S.M. TRPM8 is required for survival and radioresistance of glioblastoma cells. Oncotarget 2017, 8, 95896–95913. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Wu, Y.; Zhuang, S.; Qin, L.; Hua, S.; Mungur, R.; Pan, J.; Zhu, Y.; Zhan, R. Identification of the role of TRPM8 in glioblastoma and its effect on proliferation, apoptosis and invasion of the U251 human glioblastoma cell line. Oncol. Rep. 2019, 42, 1517–1526. [Google Scholar] [CrossRef]
- Ratto, D.; Ferrari, B.; Roda, E.; Brandalise, F.; Siciliani, S.; De Luca, F.; Priori, E.C.; Di Iorio, C.; Cobelli, F.; Veneroni, P.; et al. Squaring the Circle: A New Study of Inward and Outward-Rectifying Potassium Currents in U251 GBM Cells. Cell. Mol. Neurobiol. 2020, 40, 813–828. [Google Scholar] [CrossRef]
- Wondergem, R.; Bartley, J.W. Menthol increases human glioblastoma intracellular Ca2+, BK channel activity and cell migration. J. Biomed. Sci. 2009, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.B.; Amantini, C.; Tomassoni, D.; Nabissi, M.; Arcella, A.; Santoni, G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers 2019, 11, 525. [Google Scholar] [CrossRef] [Green Version]
- Liberati, S.; Morelli, M.B.; Amantini, C.; Santoni, M.; Nabissi, M.; Cardinali, C.; Santoni, G. Advances in transient receptor potential vanilloid-2 channel expression and function in tumor growth and progression. Curr. Protein Pept. Sci. 2014, 15, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef]
- Ou-Yang, Q.; Li, B.; Xu, M.; Liang, H. TRPV4 promotes the migration and invasion of glioma cells via AKT/Rac1 signaling. Biochem. Biophys. Res. Commun. 2018, 503, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Kanugula, A.K.; Adapala, R.K.; Midha, P.; Cappelli, H.C.; Meszaros, J.G.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019, 33, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.B.; Fuller, C.M.; Bubien, J.K.; Benos, D.J. Amiloride-sensitive Na+ channels contribute to regulatory volume increases in human glioma cells. Am. J. Physiol. Cell Physiol. 2007, 293, C1181–C1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Wu, B.; Leng, T.; Zhu, L.; Xiong, Z. Acid-sensing ion channel 1 (ASIC1) mediates weak acid-induced migration of human malignant glioma cells. Am. J. Cancer Res. 2021, 11, 997–1008. [Google Scholar] [PubMed]
- Rooj, A.K.; McNicholas, C.M.; Bartoszewski, R.; Bebok, Z.; Benos, D.J.; Fuller, C.M. Glioma-specific cation conductance regulates migration and cell cycle progression. J. Biol. Chem. 2012, 287, 4053–4065. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, N.; Bartoszewski, R.; Qadri, Y.J.; Bebok, Z.; Bubien, J.K.; Fuller, C.M.; Benos, D.J. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 2009, 284, 24526–24541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, N.; Lee, W.; Clark, E.; Bartoszewski, R.; McNicholas, C.M.; Latham, C.B.; Bebok, Z.; Parpura, V.; Fuller, C.M.; Palmer, C.A.; et al. Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am. J. Physiol. Cell Physiol. 2011, 300, C1246–C1259. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [Green Version]
- Heslop, K.A.; Milesi, V.; Maldonado, E.N. VDAC Modulation of Cancer Metabolism: Advances and Therapeutic Challenges. Front. Physiol. 2021, 12, 742839. [Google Scholar] [CrossRef]
- Zhou, K.; Yao, Y.L.; He, Z.C.; Chen, C.; Zhang, X.N.; Yang, K.D.; Liu, Y.Q.; Liu, Q.; Fu, W.J.; Chen, Y.P.; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.N.; Luo, L.; Ding, D.; Song, S.; Bhuiyan, M.I.H.; Liu, R.; Foley, L.M.; Guan, X.; Kohanbash, G.; Hitchens, T.K.; et al. Blocking NHE1 stimulates glioma tumor immunity by restoring OXPHOS function of myeloid cells. Theranostics 2021, 11, 1295–1309. [Google Scholar] [CrossRef]
- Gomez Zubieta, D.M.; Hamood, M.A.; Beydoun, R.; Pall, A.E.; Kondapalli, K.C. MicroRNA-135a regulates NHE9 to inhibit proliferation and migration of glioblastoma cells. Cell Commun. Signal. 2017, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, K.C.; Llongueras, J.P.; Capilla-González, V.; Prasad, H.; Hack, A.; Smith, C.; Guerrero-Cázares, H.; Quiñones-Hinojosa, A.; Rao, R. A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma. Nat. Commun. 2015, 6, 6289. [Google Scholar] [CrossRef] [Green Version]
- Conti, L.; Palma, E.; Roseti, C.; Lauro, C.; Cipriani, R.; de Groot, M.; Aronica, E.; Limatola, C. Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 2011, 52, 1635–1644. [Google Scholar] [CrossRef]
- Di Angelantonio, S.; Murana, E.; Cocco, S.; Scala, F.; Bertollini, C.; Molinari, M.G.; Lauro, C.; Bregestovski, P.; Limatola, C.; Ragozzino, D. A role for intracellular zinc in glioma alteration of neuronal chloride equilibrium. Cell Death Dis. 2014, 5, e1501. [Google Scholar] [CrossRef] [Green Version]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 2014, 6, 244ra289. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Robel, S.; Cuddapah, V.A.; Robert, S.; Buckingham, S.C.; Kahle, K.T.; Sontheimer, H. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015, 63, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Huberfeld, G.; Vecht, C.J. Seizures and gliomas—Towards a single therapeutic approach. Nat. Rev. Neurol. 2016, 12, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J. Voltage-gated sodium channels and metastatic disease. Channels 2012, 6, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terunuma, M.; Vargas, K.J.; Wilkins, M.E.; Ramírez, O.A.; Jaureguiberry-Bravo, M.; Pangalos, M.N.; Smart, T.G.; Moss, S.J.; Couve, A. Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 13918–13923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Savaskan, N.E.; Heckel, A.; Hahnen, E.; Engelhorn, T.; Doerfler, A.; Ganslandt, O.; Nimsky, C.; Buchfelder, M.; Eyüpoglu, I.Y. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 2008, 14, 629–632. [Google Scholar] [CrossRef]
- Arvind, S.; Arivazhagan, A.; Santosh, V.; Chandramouli, B.A. Differential expression of a novel voltage gated potassium channel—Kv 1.5 in astrocytomas and its impact on prognosis in glioblastoma. Br. J. Neurosurg. 2012, 26, 16–20. [Google Scholar] [CrossRef]
- Preussat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wolfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1.3 and Kv1.5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Grimaldi, A.; Chece, G.; Porzia, A.; Esposito, V.; Santoro, A.; Salvati, M.; Mainiero, F.; Ragozzino, D.; Di Angelantonio, S.; et al. KCa3.1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016, 7, 30781–30796. [Google Scholar] [CrossRef]
- Ge, L.; Hoa, N.T.; Cornforth, A.N.; Bota, D.A.; Mai, A.; Kim, D.I.; Chiou, S.K.; Hickey, M.J.; Kruse, C.A.; Jadus, M.R. Glioma big potassium channel expression in human cancers and possible T cell epitopes for their immunotherapy. J. Immunol. 2012, 189, 2625–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; He, Y.; Dubuc, A.M.; Hashizume, R.; Zhang, W.; Reimand, J.; Yang, H.; Wang, T.A.; Stehbens, S.J.; Younger, S.; et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat. Neurosci. 2015, 18, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Liao, H.; Liu, T.; Zeng, X.; Xiao, F.; Luo, L.; Guo, H.; Guo, L. MiR-296-3p regulates cell growth and multi-drug resistance of human glioblastoma by targeting ether-à-go-go (EAG1). Eur. J. Cancer 2013, 49, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Li, J.Y.; Liu, X.; Yan, X.Y.; Wang, W.; Wu, F.; Liang, T.Y.; Yang, F.; Hu, H.M.; Mao, H.X.; et al. A three ion channel genes-based signature predicts prognosis of primary glioblastoma patients and reveals a chemotherapy sensitive subtype. Oncotarget 2016, 7, 74895–74903. [Google Scholar] [CrossRef]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neurosignals 2017, 25, 26–38. [Google Scholar] [CrossRef]
- Ru, Q.; Tian, X.; Wu, Y.X.; Wu, R.H.; Pi, M.S.; Li, C.Y. Voltage-gated and ATP-sensitive K+ channels are associated with cell proliferation and tumorigenesis of human glioma. Oncol. Rep. 2014, 31, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Ru, Q.; Li, W.L.; Xiong, Q.; Chen, L.; Tian, X.; Li, C.Y. Voltage-gated potassium channel blocker 4-aminopyridine induces glioma cell apoptosis by reducing expression of microRNA-10b-5p. Mol. Biol. Cell 2018, 29, 1125–1136. [Google Scholar] [CrossRef]
- Patt, S.; Preussat, K.; Beetz, C.; Kraft, R.; Schrey, M.; Kalff, R.; Schonherr, K.; Heinemann, S.H. Expression of ether a go-go potassium channels in human gliomas. Neurosci. Lett. 2004, 368, 249–253. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Rudkouskaya, A.; Mongin, A.A.; Kuo, Y.H. Calcium-activated potassium channels BK and IK1 are functionally expressed in human gliomas but do not regulate cell proliferation. PLoS ONE 2010, 5, e12304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoeck, H.-W. Calcium regulation of stem cells. EMBO Rep. 2020, 21, e50028. [Google Scholar] [CrossRef] [PubMed]
- Bordey, A.; Sontheimer, H. Electrophysiological properties of human astrocytic tumor cells In situ: Enigma of spiking glial cells. J. Neurophysiol. 1998, 79, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Grichtchenko, I.I.; Chesler, M. Depolarization-induced alkalinization of astrocytes in gliotic hippocampal slices. Neuroscience 1994, 62, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumour Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Di Martino, S.; Pallini, R.; Larocca, L.M.; Caprodossi, S.; Santoni, M.; et al. The transient receptor potential vanilloid-2 cation channel impairs glioblastoma stem-like cell proliferation and promotes differentiation. Int. J. Cancer 2012, 131, E1067–E1077. [Google Scholar] [CrossRef] [Green Version]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Nazıroğlu, M. Molecular role of catalase on oxidative stress-induced Ca(2+) signaling and TRP cation channel activation in nervous system. J. Recept. Signal Transduct. Res. 2012, 32, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Z.; Liu, X.N.; Fan, R.C.; Jia, Y.P.; Zhang, Q.K.; Gao, X.Q.; Wang, Y.Q.; Yang, M.Q.; Ji, L.Z.; Zhou, Y.Q.; et al. Identification of pimavanserin tartrate as a potent Ca(2+)-calcineurin-NFAT pathway inhibitor for glioblastoma therapy. Acta Pharmacol. Sin. 2021, 42, 1860–1874. [Google Scholar] [CrossRef]
- Xiao, F.; Cheng, Z.; Wang, P.; Gong, B.; Huang, H.; Xing, Y.; Liu, F. MicroRNA-28-5p inhibits the migration and invasion of gastric cancer cells by suppressing AKT phosphorylation. Oncol. Lett. 2018, 15, 9777–9785. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X.Q.; Liu, Y.; Sun, Y.N.; Li, S.; Li, C.M.; Li, J.; Tian, W.; Shang, X.M.; Zhou, Y.T. MicroRNA 28-5p regulates ATP-binding cassette transporter A1 via inhibiting extracellular signal-regulated kinase 2. Mol. Med. Rep. 2016, 13, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Day, J.P.; Wan, S.; Allan, A.K.; Kean, L.; Davies, S.A.; Gray, J.V.; Dow, J.A.T. Identification of two partners from the bacterial Kef exchanger family for the apical plasma membrane V-ATPase of Metazoa. J. Cell Sci. 2008, 121, 2612–2619. [Google Scholar] [CrossRef] [Green Version]
- Saberbaghi, T.; Wong, R.; Rutka, J.T.; Wang, G.L.; Feng, Z.P.; Sun, H.S. Role of Cl(-) channels in primary brain tumour. Cell Calcium 2019, 81, 1–11. [Google Scholar] [CrossRef]
- Poroca, D.R.; Pelis, R.M.; Chappe, V.M. ClC Channels and Transporters: Structure, Physiological Functions, and Implications in Human Chloride Channelopathies. Front. Pharmacol. 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Habela, C.W.; Sontheimer, H. Cytoplasmic volume condensation is an integral part of mitosis. Cell Cycle 2007, 6, 1613–1620. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.; Chen, W.; Zhong, X.; Rutka, J.T.; Feng, Z.P.; Sun, H.S. Swelling-induced chloride current in glioblastoma proliferation, migration, and invasion. J. Cell Physiol. 2018, 233, 363–370. [Google Scholar] [CrossRef]
- Liu, T.; Stauber, T. The Volume-Regulated Anion Channel LRRC8/VRAC Is Dispensable for Cell Proliferation and Migration. Int. J. Mol. Sci. 2019, 20, 2663. [Google Scholar] [CrossRef] [Green Version]
- Crottès, D.; Jan, L.Y. The multifaceted role of TMEM16A in cancer. Cell Calcium 2019, 82, 102050. [Google Scholar] [CrossRef]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Bresenitz, P.; Reska, A.; El Moussaoui, L.; Beier, C.P.; Gründer, S. Glioblastoma cancer stem cell lines express functional acid sensing ion channels ASIC1a and ASIC3. Sci. Rep. 2017, 7, 13674. [Google Scholar] [CrossRef] [PubMed]
- Fuhs, T.; Wetzel, F.; Fritsch, A.W.; Li, X.; Stange, R.; Pawlizak, S.; Kießling, T.R.; Morawetz, E.; Grosser, S.; Sauer, F.; et al. Rigid tumours contain soft cancer cells. Nat. Phys. 2022, 18, 1510–1519. [Google Scholar] [CrossRef]
- Jonietz, E. Mechanics: The forces of cancer. Nature 2012, 491, S56–S57. [Google Scholar] [CrossRef] [PubMed]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Seltmann, K.; Fritsch, A.W.; Käs, J.A.; Magin, T.M. Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc. Natl. Acad. Sci. USA 2013, 110, 18507–18512. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Pfeifer, C.R.; Discher, D.E. Nuclear mechanics during and after constricted migration. Acta Mech. Sin. 2019, 35, 299–308. [Google Scholar] [CrossRef]
- Lan, J.-Y.; Williams, C.; Levin, M.; Black, L.D. Depolarization of Cellular Resting Membrane Potential Promotes Neonatal Cardiomyocyte Proliferation In Vitro. Cell. Mol. Bioeng. 2014, 7, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Piggott, B.J.; Peters, C.J.; He, Y.; Huang, X.; Younger, S.; Jan, L.Y.; Jan, Y.N. Paralytic, the Drosophila voltage-gated sodium channel, regulates proliferation of neural progenitors. Genes Dev. 2019, 33, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef] [PubMed]
- Seker-Polat, F.; Pinarbasi Degirmenci, N.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, P.; Engberg, A.; Eriksson, O.; Fatsis-Kavalopoulos, N.; Stelzl, C.; Sanchez, G.; Idevall-Hagren, O.; Kreuger, J. Piezo1 activation attenuates thrombin-induced blebbing in breast cancer cells. J. Cell Sci. 2022, 135, jcs258809. [Google Scholar] [CrossRef]
- Srivastava, N.; Traynor, D.; Piel, M.; Kabla, A.J.; Kay, R.R. Pressure sensing through Piezo channels controls whether cells migrate with blebs or pseudopods. Proc. Natl. Acad. Sci. USA 2020, 117, 2506–2512. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, P.A.; Puliafito, A.; di Blasio, L.; Chianale, F.; Somale, D.; Seano, G.; Bussolino, F.; Primo, L. Real-time monitoring of cell protrusion dynamics by impedance responses. Sci. Rep. 2015, 5, 10206. [Google Scholar] [CrossRef] [Green Version]
- Alblazi, K.M.; Siar, C.H. Cellular protrusions—Lamellipodia, filopodia, invadopodia and podosomes--and their roles in progression of orofacial tumours: Current understanding. Asian Pac. J. Cancer Prev. 2015, 16, 2187–2191. [Google Scholar] [CrossRef] [Green Version]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepannetier, S.; Zanou, N.; Yerna, X.; Emeriau, N.; Dufour, I.; Masquelier, J.; Muccioli, G.; Tajeddine, N.; Gailly, P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016, 60, 373–383. [Google Scholar] [CrossRef] [PubMed]
- De Felice, D.; Alaimo, A. Mechanosensitive Piezo Channels in Cancer: Focus on altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers 2020, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim-Hashim, A.; Estrella, V. Acidosis and cancer: From mechanism to neutralization. Cancer Metastasis Rev. 2019, 38, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Parent, M.; Akif, A.; Adam, L.C.; Maritim, S.; Mishra, S.K.; Khan, M.H.; Coman, D.; Hyder, F. Imaging Hallmarks of the Tumor Microenvironment in Glioblastoma Progression. Front. Oncol. 2021, 11, 692650. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yi, L.; Hai, L.; Ma, H.; Tao, Z.; Zhang, C.; Abeysekera, I.R.; Zhao, K.; Yang, Y.; Wang, W.; et al. The interactome and spatial redistribution feature of Ca(2+) receptor protein calmodulin reveals a novel role in invadopodia-mediated invasion. Cell Death Dis. 2018, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Keurhorst, D.; Liashkovich, I.; Frontzek, F.; Nitzlaff, S.; Hofschröer, V.; Dreier, R.; Stock, C. MMP3 activity rather than cortical stiffness determines NHE1-dependent invasiveness of melanoma cells. Cancer Cell Int. 2019, 19, 285. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Functional implications for Kir4.1 channels in glial biology: From K+ buffering to cell differentiation. J. Neurochem. 2008, 107, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Morrison, B.M.; Lee, Y.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of axons. Trends Cell Biol. 2013, 23, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afridi, R.; Kim, J.H.; Rahman, M.H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron-Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labak, C.M.; Wang, P.Y.; Arora, R.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. Glucose transport: Meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am. J. Cancer Res. 2016, 6, 1599–1608. [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Ekici, S.; Risk, B.B.; Neill, S.G.; Shu, H.K.; Fleischer, C.C. Characterization of dysregulated glutamine metabolism in human glioma tissue with (1)H NMR. Sci. Rep. 2020, 10, 20435. [Google Scholar] [CrossRef]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef]
- Mair, R.; Wright, A.J.; Ros, S.; Hu, D.E.; Booth, T.; Kreis, F.; Rao, J.; Watts, C.; Brindle, K.M. Metabolic Imaging Detects Low Levels of Glycolytic Activity That Vary with Levels of c-Myc Expression in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2018, 78, 5408–5418. [Google Scholar] [CrossRef] [Green Version]
- Akkulak, A.; Dağdelen, D.N.; Yalçın, A.; Oktay, E.; Diniz, G.; Kahraman, D.S.; Şenoğlu, M.; Yalcin, G.D. The expression of glutamate metabolism modulators in the intracranial tumors and glioblastoma cell line. Mol. Biol. Rep. 2022, 49, 1077–1083. [Google Scholar] [CrossRef]
- Melkonian, E.A.; Schury, M.P. Biochemistry, Anaerobic Glycolysis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Lu, R.O.; Ho, W.S. Mitochondrial Dysfunction, Macrophage, and Microglia in Brain Cancer. Front. Cell Dev. Biol. 2020, 8, 620788. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 2009, 1787, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Gohara, D.W.; Di Cera, E. Molecular Mechanisms of Enzyme Activation by Monovalent Cations. J. Biol. Chem. 2016, 291, 20840–20848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oria-Hernández, J.; Cabrera, N.; Pérez-Montfort, R.; Ramírez-Silva, L. Pyruvate kinase revisited: The activating effect of K+. J. Biol. Chem. 2005, 280, 37924–37929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischof, H.; Burgstaller, S.; Springer, A.; Matt, L.; Rauter, T.; Bachkönig, O.A.; Schmidt, T.; Groschner, K.; Schindl, R.; Madl, T.; et al. Potassium ions promote hexokinase-II dependent glycolysis. iScience 2021, 24, 102346. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, M.; Augustynek, B.; Kulawiak, B.; Koprowski, P.; Bednarczyk, P.; Jarmuszkiewicz, W.; Szewczyk, A. What do we not know about mitochondrial potassium channels? Biochim. Biophys. Acta 2016, 1857, 1247–1257. [Google Scholar] [CrossRef]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.A.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Wrzosek, A.; Augustynek, B.; Żochowska, M.; Szewczyk, A. Mitochondrial Potassium Channels as Druggable Targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef]
- Burgstaller, S.; Bischof, H.; Matt, L.; Lukowski, R. Assessing K(+) ions and K(+) channel functions in cancer cell metabolism using fluorescent biosensors. Free Radic. Biol. Med. 2022, 181, 43–51. [Google Scholar] [CrossRef]
- Teisseyre, A.; Palko-Labuz, A.; Sroda-Pomianek, K.; Michalak, K. Voltage-Gated Potassium Channel Kv1.3 as a Target in Therapy of Cancer. Front. Oncol. 2019, 9, 933. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Ningaraj, N.S. Targeting potassium channels for increasing delivery of imaging agents and therapeutics to brain tumors. Front. Pharmacol. 2013, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Shirmanova, M.V.; Druzhkova, I.N.; Lukina, M.M.; Dudenkova, V.V.; Ignatova, N.I.; Snopova, L.B.; Shcheslavskiy, V.I.; Belousov, V.V.; Zagaynova, E.V. Chemotherapy with cisplatin: Insights into intracellular pH and metabolic landscape of cancer cells in vitro and in vivo. Sci. Rep. 2017, 7, 8911. [Google Scholar] [CrossRef] [PubMed]
- Rocha, P.R.; Medeiros, M.C.; Kintzel, U.; Vogt, J.; Araújo, I.M.; Mestre, A.L.; Mailänder, V.; Schlett, P.; Dröge, M.; Schneider, L.; et al. Extracellular electrical recording of pH-triggered bursts in C6 glioma cell populations. Sci. Adv. 2016, 2, e1600516. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na(+)/H(+) exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective? Semin. Cancer Biol. 2017, 43, 5–16. [Google Scholar] [CrossRef]
- Dong, Y.; Gao, Y.; Ilie, A.; Kim, D.; Boucher, A.; Li, B.; Zhang, X.C.; Orlowski, J.; Zhao, Y. Structure and mechanism of the human NHE1-CHP1 complex. Nat. Commun. 2021, 12, 3474. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofori, A.; Ferrero, S.; Bertolini, I.; Gaudioso, G.; Russo, M.V.; Berno, V.; Vanini, M.; Locatelli, M.; Zavanone, M.; Rampini, P.; et al. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 2015, 6, 17514–17531. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.P.; Forgac, M. Regulation of V-ATPase Assembly in Nutrient Sensing and Function of V-ATPases in Breast Cancer Metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar] [CrossRef] [Green Version]
- Sforna, L.; Cenciarini, M.; Belia, S.; D’Adamo, M.C.; Pessia, M.; Franciolini, F.; Catacuzzeno, L. The role of ion channels in the hypoxia-induced aggressiveness of glioblastoma. Front. Cell. Neurosci. 2014, 8, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Strowitzki, M.J.; Cummins, E.P.; Taylor, C.T. Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 2019, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Tennant, D.A.; Gottlieb, E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J. Mol. Med. 2010, 88, 839–849. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Wei, Y.; Liu, Y.; Ding, X.; Dong, B.; Xu, Y.; Wang, Y. Crucial role of TRPC6 in maintaining the stability of HIF-1α in glioma cells under hypoxia. J. Cell Sci. 2015, 128, 3317–3329. [Google Scholar] [CrossRef] [Green Version]
- Alphandéry, E. Nano-Therapies for Glioblastoma Treatment. Cancers 2020, 12, 242. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J. Dynamics of CNS Barriers: Evolution, Differentiation, and Modulation. Cell. Mol. Neurobiol. 2005, 25, 5–23. [Google Scholar] [CrossRef]
- Wang, K.Y.; Chen, M.M.; Malayil Lincoln, C.M. Adult Primary Brain Neoplasm, including 2016 World Health Organization Classification; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Goddard, L.M.; Iruela-Arispe, M.L. Cellular and molecular regulation of vascular permeability. Thromb. Haemost. 2013, 109, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood–brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, F.; Xiong, N.; Xu, H.; Chai, S.; Wang, H.; Wang, J.; Zhao, H.; Jiang, X.; Fu, P.; et al. Remodelling and Treatment of the Blood-Brain Barrier in Glioma. Cancer Manag. Res. 2021, 13, 4217–4232. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hasan, M.N.; Maniar, S.; Wang, J.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bandyopadhyay, B.C.; Nakamoto, T.; Singh, B.; Liedtke, W.; Melvin, J.E.; Ambudkar, I. A role for AQP5 in activation of TRPV4 by hypotonicity: Concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J. Biol. Chem. 2006, 281, 15485–15495. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, V.; Caprini, M.; Dovizio, M.; Mylonakou, M.N.; Ferroni, S.; Ottersen, O.P.; Amiry-Moghaddam, M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 2563–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badaut, J.; Regli, L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience 2004, 129, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Cavazzin, C.; Ferrari, D.; Facchetti, F.; Russignan, A.; Vescovi, A.L.; La Porta, C.A.; Gritti, A. Unique expression and localization of aquaporin-4 and aquaporin-9 in murine and human neural stem cells and in their glial progeny. Glia 2006, 53, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Fan, Y.; Xie, J.; Ding, J.; Sha, L.; Shi, X.; Sun, X.; Hu, G. AQP4 knockout impairs proliferation, migration and neuronal differentiation of adult neural stem cells. J. Cell Sci. 2008, 121, 4029–4036. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.S.; Gao, K.; Chai, R.C.; Jia, X.H.; Luo, D.P.; Ge, G.; Jiang, Y.W.; Fung, Y.W.; Li, L.; Yu, A.C. Astrocytes in Migration. Neurochem. Res. 2017, 42, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Szu, J.I.; Binder, D.K. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhan, J.; Cai, Q.; Xu, F.; Chai, R.; Lam, K.; Luan, Z.; Zhou, G.; Tsang, S.; Kipp, M.; et al. The Water Transport System in Astrocytes-Aquaporins. Cells 2022, 11, 2564. [Google Scholar] [CrossRef]
- Potokar, M.; Jorgačevski, J.; Zorec, R. Astrocyte Aquaporin Dynamics in Health and Disease. Int. J. Mol. Sci. 2016, 17, 1121. [Google Scholar] [CrossRef] [Green Version]
- van Dijk, B.J.; Vergouwen, M.D.I.; Kelfkens, M.M.; Rinkel, G.J.E.; Hol, E.M. Glial cell response after aneurysmal subarachnoid hemorrhage—Functional consequences and clinical implications. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2016, 1862, 492–505. [Google Scholar] [CrossRef]
- Mou, K.; Chen, M.; Mao, Q.; Wang, P.; Ni, R.; Xia, X.; Liu, Y. AQP-4 in peritumoral edematous tissue is correlated with the degree of glioma and with expression of VEGF and HIF-alpha. J. Neurooncol. 2010, 100, 375–383. [Google Scholar] [CrossRef]
- Aoki, K.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003, 106, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Jain, R.K.; Witwer, B.; Brown, D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc. Res. 1999, 58, 89–98. [Google Scholar] [CrossRef]
- Markert, J.M.; Fuller, C.M.; Gillespie, G.Y.; Bubien, J.K.; McLean, L.A.; Hong, R.L.; Lee, K.; Gullans, S.R.; Mapstone, T.B.; Benos, D.J. Differential gene expression profiling in human brain tumors. Physiol. Genom. 2001, 5, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Krishna, S.; Bell, B.A. Aquaporin-4 expression is increased in oedematous human brain tumours. J. Neurol. Neurosurg. Psychiatry 2002, 72, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.L.; Wang, X.; Lou, J.C.; Ma, X.C.; Zhang, B. The potential roles of aquaporin 4 in malignant gliomas. Oncotarget 2017, 8, 32345–32355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clément, T.; Rodriguez-Grande, B.; Badaut, J. Aquaporins in brain edema. J. Neurosci. Res. 2020, 98, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Huo, Z.; Lomora, M.; Kym, U.; Palivan, C.; Holland-Cunz, S.G.; Gros, S.J. AQP1 Is Up-Regulated by Hypoxia and Leads to Increased Cell Water Permeability, Motility, and Migration in Neuroblastoma. Front. Cell Dev. Biol. 2021, 9, 605272. [Google Scholar] [CrossRef]
- Hayashi, Y.; Edwards, N.A.; Proescholdt, M.A.; Oldfield, E.H.; Merrill, M.J. Regulation and function of aquaporin-1 in glioma cells. Neoplasia 2007, 9, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Noell, S.; Fallier-Becker, P.; Beyer, C.; Kröger, S.; Mack, A.F.; Wolburg, H. Effects of agrin on the expression and distribution of the water channel protein aquaporin-4 and volume regulation in cultured astrocytes. Eur. J. Neurosci. 2007, 26, 2109–2118. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, X.; Zhen, S.; Zhang, S.; Kang, D.; Lin, Z. Aquaporin-4 upregulated expression in glioma tissue is a reaction to glioma-associated edema induced by vascular endothelial growth factor. Oncol. Rep. 2012, 28, 1633–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solar, P.; Hendrych, M.; Barak, M.; Valekova, H.; Hermanova, M.; Jancalek, R. Blood-Brain Barrier Alterations and Edema Formation in Different Brain Mass Lesions. Front. Cell. Neurosci. 2022, 16, 922181. [Google Scholar] [CrossRef]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008, 19, 1–5. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, K.; Liu, Y.; Erokwu, B.O.; Zhao, P.; Flask, C.A.; Ramos-Estebanez, C.; Farr, G.W.; LaManna, J.C.; Boron, W.F.; et al. Increased cerebral vascularization and decreased water exchange across the blood-brain barrier in aquaporin-4 knockout mice. PLoS ONE 2019, 14, e0218415. [Google Scholar] [CrossRef] [Green Version]
- Saadoun, S.; Tait, M.J.; Reza, A.; Davies, D.C.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience 2009, 161, 764–772. [Google Scholar] [CrossRef]
- Warth, A.; Simon, P.; Capper, D.; Goeppert, B.; Tabatabai, G.; Herzog, H.; Dietz, K.; Stubenvoll, F.; Ajaaj, R.; Becker, R.; et al. Expression pattern of the water channel aquaporin-4 in human gliomas is associated with blood-brain barrier disturbance but not with patient survival. J. Neurosci. Res. 2007, 85, 1336–1346. [Google Scholar] [CrossRef]
- Ikota, H.; Kinjo, S.; Yokoo, H.; Nakazato, Y. Systematic immunohistochemical profiling of 378 brain tumors with 37 antibodies using tissue microarray technology. Acta Neuropathol. 2006, 111, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Lan, Y.-L.; Ren, T.; Li, X.; Zhang, L.; Wang, H.; Wang, X. A Bioinformatics Analysis of the Potential Roles of Aquaporin 4 in Human Brain Tumors: An Immune-Related Process. Front. Pharmacol. 2021, 12, 692175. [Google Scholar] [CrossRef]
- da Silva, I.V.; Garra, S.; Calamita, G.; Soveral, G. The Multifaceted Role of Aquaporin-9 in Health and Its Potential as a Clinical Biomarker. Biomolecules 2022, 12, 897. [Google Scholar] [CrossRef]
- Tang, G.; Yang, G.Y. Aquaporin-4: A Potential Therapeutic Target for Cerebral Edema. Int. J. Mol. Sci. 2016, 17, 1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, W.; Ran, J.; Jiang, R.; Guo, P.; Shi, X.; Li, H.; Lv, X.; Li, J.; Chen, D. miRNA-320a inhibits glioma cell invasion and migration by directly targeting aquaporin 4. Oncol. Rep. 2018, 39, 1939–1947. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Li, L.; Chen, J.; Lu, H. Effect of AQP4-RNAi in treating traumatic brain edema: Multi-modal MRI and histopathological changes of early stage edema in a rat model. Exp. Ther. Med. 2020, 19, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E.; Davidson, K.G.; Yasumura, T.; Furman, C.S. Freeze-fracture and immunogold analysis of aquaporin-4 (AQP4) square arrays, with models of AQP4 lattice assembly. Neuroscience 2004, 129, 915–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jullienne, A.; Fukuda, A.M.; Ichkova, A.; Nishiyama, N.; Aussudre, J.; Obenaus, A.; Badaut, J. Modulating the water channel AQP4 alters miRNA expression, astrocyte connectivity and water diffusion in the rodent brain. Sci. Rep. 2018, 8, 4186. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, O.F.; Rodríguez, A.V.; Velasco-España, J.M.; Murillo, L.C.; Sutachan, J.J.; Albarracin, S.L. Role of Connexins 30, 36, and 43 in Brain Tumors, Neurodegenerative Diseases, and Neuroprotection. Cells 2020, 9, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, W.-C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta (BBA)—Biomembr. 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Current Biology 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, M.L. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, G.N.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.G.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef] [Green Version]
- Jung, E.; Alfonso, J.; Osswald, M.; Monyer, H.; Wick, W.; Winkler, F. Emerging intersections between neuroscience and glioma biology. Nat. Neurosci. 2019, 22, 1951–1960. [Google Scholar] [CrossRef]
- Pancholi, S.; Tripathi, A.; Bhan, A.; Acharya, M.M.; Pillai, P. Emerging Concepts on the Role of Extracellular Vesicles and Its Cargo Contents in Glioblastoma-Microglial Crosstalk. Mol. Neurobiol. 2022, 59, 2822–2837. [Google Scholar] [CrossRef]
- Codrici, E.; Popescu, I.D.; Tanase, C.; Enciu, A.M. Friends with Benefits: Chemokines, Glioblastoma-Associated Microglia/Macrophages, and Tumor Microenvironment. Int. J. Mol. Sci. 2022, 23, 2509. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.; Sun, D.; Pedraza, A.M.; Vera, E.; Wang, Z.; Burns, D.K.; Parada, L.F. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 2019, 22, 545–555. [Google Scholar] [CrossRef]
- You, G.; Sha, Z.; Jiang, T. The pathogenesis of tumor-related epilepsy and its implications for clinical treatment. Seizure 2012, 21, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowie, C.J.; Cunningham, M.O. Peritumoral epilepsy: Relating form and function for surgical success. Epilepsy Behav. 2014, 38, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, B. Influences of brain tumor-associated pH changes and hypoxia on epileptogenesis. Acta Neurol. Scand. 2005, 111, 75–83. [Google Scholar] [CrossRef]
- Armstrong, T.S.; Grant, R.; Gilbert, M.R.; Lee, J.W.; Norden, A.D. Epilepsy in glioma patients: Mechanisms, management, and impact of anticonvulsant therapy. Neuro Oncol. 2016, 18, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Buckingham, S.C.; Sontheimer, H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia 2012, 53, 1360–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, S.M.; Sontheimer, H. Glutamate transporters in the biology of malignant gliomas. Cell Mol. Life Sci. 2014, 71, 1839–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, T.I.; Morokoff, A.P.; Bjorksten, A.; D’Abaco, G.; Paradiso, L.; Finch, S.; Wong, D.; Reid, C.A.; Powell, K.L.; Drummond, K.J.; et al. Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology 2012, 79, 883–889. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, G.; O’Toole, K.K.; Moss, S.J.; Maguire, J. Compromised GABAergic inhibition contributes to tumor-associated epilepsy. Epilepsy Res. 2016, 126, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Thews, O.; Riemann, A. Tumor pH and metastasis: A malignant process beyond hypoxia. Cancer Metastasis Rev. 2019, 38, 113–129. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- De Clerck, K.; Elble, R.C. The role of hypoxia and acidosis in promoting metastasis and resistance to chemotherapy. Front. Biosci. FBL 2010, 15, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin. Exp. Metastasis 2009, 26, 19–34. [Google Scholar] [CrossRef]
- Payen, V.L.; Porporato, P.E.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell. Mol. Life Sci. 2016, 73, 1333–1348. [Google Scholar] [CrossRef]
- Jin, C.; Ye, Q.H.; Yuan, F.L.; Gu, Y.L.; Li, J.P.; Shi, Y.H.; Shen, X.M.; Bo, L.; Lin, Z.H. Involvement of acid-sensing ion channel 1α in hepatic carcinoma cell migration and invasion. Tumour Biol. 2015, 36, 4309–4317. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyon, J.; Chapouly, C.; Andrique, L.; Bikfalvi, A.; Daubon, T. The Normal and Brain Tumor Vasculature: Morphological and Functional Characteristics and Therapeutic Targeting. Front. Physiol. 2021, 12, 622615. [Google Scholar] [CrossRef]
- Takano, S.; Yamashita, T.; Ohneda, O. Molecular therapeutic targets for glioma angiogenesis. J. Oncol. 2010, 2010, 351908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacker, S.A.; Caesar, C.; Baldwin, M.E.; Thornton, G.E.; Williams, R.A.; Prevo, R.; Jackson, D.G.; Nishikawa, S.-I.; Kubo, H.; Achen, M.G. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 2001, 7, 186–191. [Google Scholar] [CrossRef]
- Bhargav, A.G.; Domino, J.S.; Chamoun, R.; Thomas, S.M. Mechanical Properties in the Glioma Microenvironment: Emerging Insights and Theranostic Opportunities. Front. Oncol. 2021, 11, 805628. [Google Scholar] [CrossRef]
- Haley, E.M.; Kim, Y. The role of basic fibroblast growth factor in glioblastoma multiforme and glioblastoma stem cells and in their in vitro culture. Cancer Lett. 2014, 346, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, G.; Koushan, K.; Pillo, L.; Shannon, P.; Guha, A. Role of Ang1 and Its Interaction with VEGF-A in Astrocytomas. J. Neuropathol. Exp. Neurol. 2004, 63, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Dunn, I.F.; Heese, O.; Black, P.M. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J. Neurooncol. 2000, 50, 121–137. [Google Scholar] [CrossRef]
- Pullen, N.A.; Anand, M.; Cooper, P.S.; Fillmore, H.L. Matrix metalloproteinase-1 expression enhances tumorigenicity as well as tumor-related angiogenesis and is inversely associated with TIMP-4 expression in a model of glioblastoma. J. Neurooncol. 2012, 106, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-Balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.; Gouverneur, A.; Maumus-Robert, S.; Bérard, X.; Pariente, A.; Bikfalvi, A.; Noize, P. Association Between Antiangiogenic Drugs Used for Cancer Treatment and Artery Dissections or Aneurysms. JAMA Oncol. 2021, 7, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, N.; Suzuki, H.; Itoh, Y.; Muraki, K. TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci. 2013, 92, 317–324. [Google Scholar] [CrossRef]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Rossi, E.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Choublier, N.; Smirnova, M.; Glacial, F.; Perrière, N.; Bourdoulous, S.; et al. Cannabidiol Increases Proliferation, Migration, Tubulogenesis, and Integrity of Human Brain Endothelial Cells through TRPV2 Activation. Mol. Pharm. 2019, 16, 1312–1326. [Google Scholar] [CrossRef]
- Kanugula, A.K.; Adapala, R.K.; Jamaiyar, A.; Lenkey, N.; Guarino, B.D.; Liedtke, W.; Yin, L.; Paruchuri, S.; Thodeti, C.K. Endothelial TRPV4 channels prevent tumor growth and metastasis via modulation of tumor angiogenesis and vascular integrity. Angiogenesis 2021, 24, 647–656. [Google Scholar] [CrossRef]
- Ko, E.A.; Zhou, T. GPCR genes as a predictor of glioma severity and clinical outcome. J. Int. Med. Res. 2022, 50, 3000605221113911. [Google Scholar] [CrossRef] [PubMed]
- Nery de Albuquerque Rego, G.; da Hora Alves, A.; Penteado Nucci, M.; Bustamante Mamani, J.; Anselmo de Oliveira, F.; Gamarra, L.F. Antiangiogenic Targets for Glioblastoma Therapy from a Pre-Clinical Approach, Using Nanoformulations. Int. J. Mol. Sci. 2020, 21, 4490. [Google Scholar] [CrossRef] [PubMed]
- Lucius, A.; Khajavi, N.; Reinach, P.S.; Köhrle, J.; Dhandapani, P.; Huimann, P.; Ljubojevic, N.; Grötzinger, C.; Mergler, S. 3-Iodothyronamine increases transient receptor potential melastatin channel 8 (TRPM8) activity in immortalized human corneal epithelial cells. Cell. Signal. 2016, 28, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Budde, C.; Böhm, A.; Reinach, P.S.; Dhandapani, P.; Ljubojevic, N.; Schweiger, M.W.; von der Waydbrink, H.; Reimers, I.; Köhrle, J.; et al. TRPM8 Activation via 3-Iodothyronamine Blunts VEGF-Induced Transactivation of TRPV1 in Human Uveal Melanoma Cells. Front. Pharmacol. 2018, 9, 1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylli, S.S. Novel Treatment Strategies for Glioblastoma-A Summary. Cancers 2021, 13, 5868. [Google Scholar] [CrossRef]
- Griffin, M.; Khan, R.; Basu, S.; Smith, S. Ion Channels as Therapeutic Targets in High Grade Gliomas. Cancers 2020, 12, 3068. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: Targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Fairhurst, C.; Watt, I.; Martin, F.; Bland, M.; Brackenbury, W.J. Sodium channel-inhibiting drugs and survival of breast, colon and prostate cancer: A population-based study. Sci. Rep. 2015, 5, 16758. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.-C.; Wei, K.-C.; Chen, P.-Y.; Huang, C.-Y.; Chen, K.-T.; Lin, Y.-J.; Cheng, H.-W.; Chen, Y.-R.; Wang, H.-T. Valproic Acid Enhanced Temozolomide-Induced Anticancer Activity in Human Glioma Through the p53–PUMA Apoptosis Pathway. Front. Oncol. 2021, 11, 722754. [Google Scholar] [CrossRef]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Mechanisms of anticarcinogenesis and drug resistance. Mutat. Res. 2005, 591, 247–263. [Google Scholar] [CrossRef]
- Chae, Y.J.; Lee, K.J.; Lee, H.J.; Sung, K.W.; Choi, J.S.; Lee, E.H.; Hahn, S.J. Endoxifen, the active metabolite of tamoxifen, inhibits cloned hERG potassium channels. Eur. J. Pharmacol. 2015, 752, 1–7. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, R.; Yang, S.H.; Yuan, F. Chemotherapeutic effect of tamoxifen on temozolomide-resistant gliomas. Anticancer Drugs 2015, 26, 293–300. [Google Scholar] [CrossRef]
- Waring, W.S. Antidiabetic drugs. Medicine 2016, 44, 138–140. [Google Scholar] [CrossRef]
- Yin, M.; Zhou, J.; Gorak, E.J.; Quddus, F. Metformin is associated with survival benefit in cancer patients with concurrent type 2 diabetes: A systematic review and meta-analysis. Oncologist 2013, 18, 1248–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Zhao, G.; Xie, G.; Zhao, L.; Chen, Y.; Yu, H.; Zhang, Z.; Li, C.; Li, Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget 2015, 6, 32930–32943. [Google Scholar] [CrossRef] [Green Version]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Cohen-Jonathan Moyal, E.; et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Li, S.; Lu, G.; Xue, H.; Kim, D.H.; Zhu, J.J.; Liu, Y. Metformin treatment reduces temozolomide resistance of glioblastoma cells. Oncotarget 2016, 7, 78787–78803. [Google Scholar] [CrossRef] [Green Version]
- Seliger, C.; Meyer, A.L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-β2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef]
- Welch, M.R.; Grommes, C. Retrospective analysis of the effects of steroid therapy and antidiabetic medication on survival in diabetic glioblastoma patients. CNS Oncol. 2013, 2, 237–246. [Google Scholar] [CrossRef]
- Seliger, C.; Genbrugge, E.; Gorlia, T.; Chinot, O.; Stupp, R.; Nabors, B.; Weller, M.; Hau, P. Use of metformin and outcome of patients with newly diagnosed glioblastoma: Pooled analysis. Int. J. Cancer 2020, 146, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Amantini, C.; Maggi, F.; Marinelli, O.; Santoni, M.; Nabissi, M.; Morelli, M.B. The TRPV2 cation channels: From urothelial cancer invasiveness to glioblastoma multiforme interactome signature. Lab. Investig. 2020, 100, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2012, 34, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Pallini, R.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, F. Transient Receptor Potential (TRP) Ion Channels Involved in Malignant Glioma Cell Death and Therapeutic Perspectives. Front. Cell Dev. Biol. 2021, 9, 618961. [Google Scholar] [CrossRef] [PubMed]
- Twelves, C.; Sabel, M.; Checketts, D.; Miller, S.; Tayo, B.; Jove, M.; Brazil, L.; Short, S.C. A phase 1b randomised, placebo-controlled trial of nabiximols cannabinoid oromucosal spray with temozolomide in patients with recurrent glioblastoma. Br J Cancer 2021, 124, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Dall’Stella, P.B.; Docema, M.F.L.; Maldaun, M.V.C.; Feher, O.; Lancellotti, C.L.P. Case Report: Clinical Outcome and Image Response of Two Patients with Secondary High-Grade Glioma Treated with Chemoradiation, PCV, and Cannabidiol. Front. Oncol. 2018, 8, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Likar, R.; Koestenberger, M.; Stultschnig, M.; Nahler, G. Concomitant Treatment of Malignant Brain Tumours with CBD—A Case Series and Review of the Literature. Anticancer Res. 2019, 39, 5797–5801. [Google Scholar] [CrossRef]
- Bostock, M.P.; Prasad, A.R.; Chaouni, R.; Yuen, A.C.; Sousa-Nunes, R.; Amoyel, M.; Fernandes, V.M. An Immobilization Technique for Long-Term Time-Lapse Imaging of Explanted Drosophila Tissues. Front. Cell Dev. Biol. 2020, 8, 590094. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.; Luo, Z.-W.; Ovcjak, A.; Alanazi, R.; Bao, M.-H.; Feng, Z.-P.; Sun, H.-S. Role of TRPM2 in brain tumours and potential as a drug target. Acta Pharmacol. Sin. 2022, 43, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.S. Role of TRPM7 in Cancer: Potential as Molecular Biomarker and Therapeutic Target. Pharmaceuticals 2017, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Chubanov, V.; Ferioli, S.; Gudermann, T. Assessment of TRPM7 functions by drug-like small molecules. Cell Calcium 2017, 67, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Parnas, M.; Peters, M.; Dadon, D.; Lev, S.; Vertkin, I.; Slutsky, I.; Minke, B. Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium 2009, 45, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.L.; Barszczyk, A.; Turlova, E.; Deurloo, M.; Liu, B.; Yang, B.B.; Rutka, J.T.; Feng, Z.P.; Sun, H.S. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget 2015, 6, 16321–16340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanazi, R.; Nakatogawa, H.; Wang, H.; Ji, D.; Luo, Z.; Golbourn, B.; Feng, Z.P.; Rutka, J.T.; Sun, H.S. Inhibition of TRPM7 with carvacrol suppresses glioblastoma functions in vivo. Eur. J. Neurosci. 2022, 55, 1483–1491. [Google Scholar] [CrossRef]
- Fan, M.D.; Zhao, X.Y.; Qi, J.N.; Jiang, Y.; Liu, B.Y.; Dun, Z.P.; Zhang, R.; Wang, C.W.; Pang, Q. TRIM31 enhances chemoresistance in glioblastoma through activation of the PI3K/Akt signaling pathway. Exp. Ther. Med. 2020, 20, 802–809. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhu, J.J. Tumor treating fields: A novel and effective therapy for glioblastoma: Mechanism, efficacy, safety and future perspectives. Chin. Clin. Oncol. 2017, 6, 41. [Google Scholar] [CrossRef]
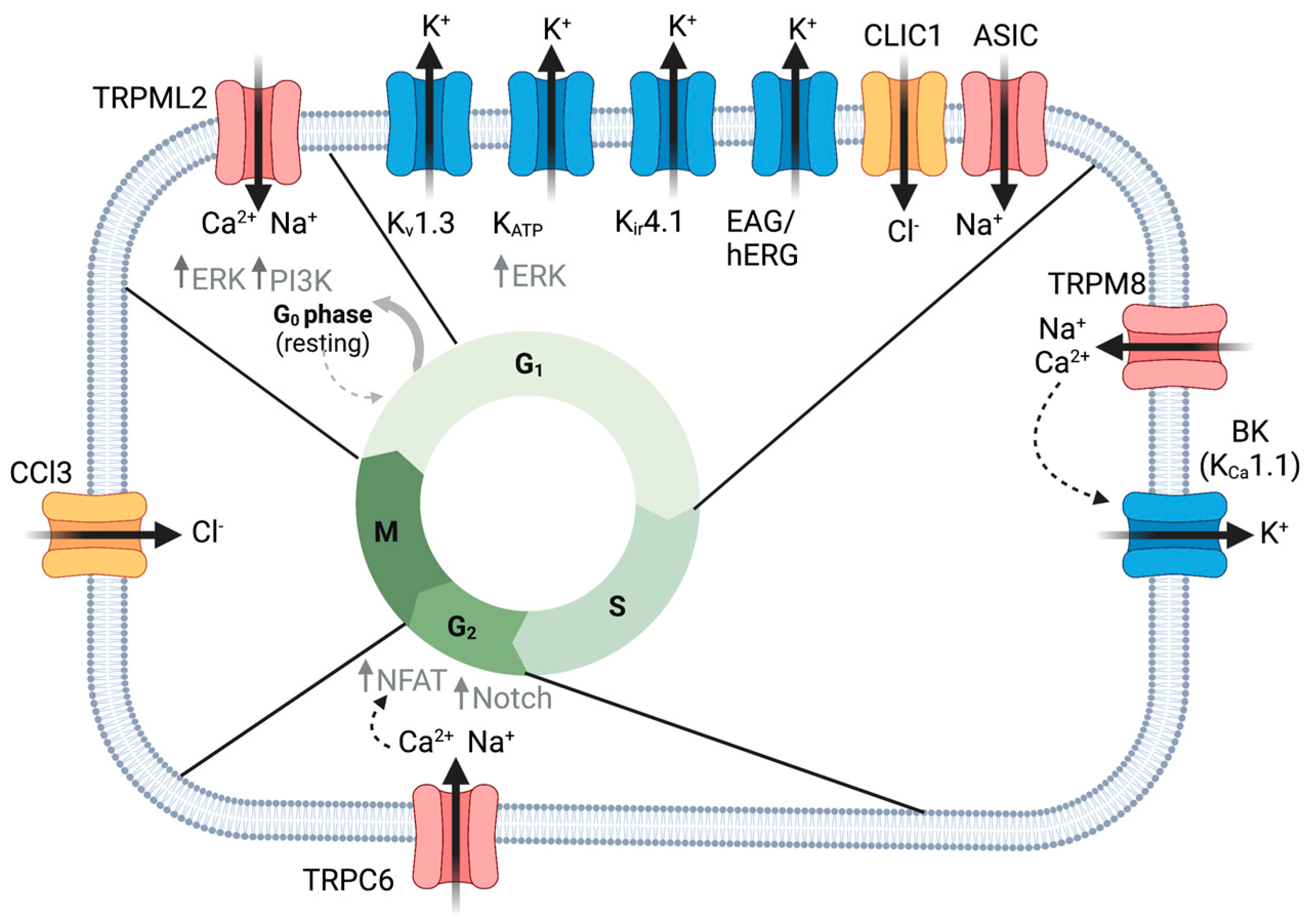

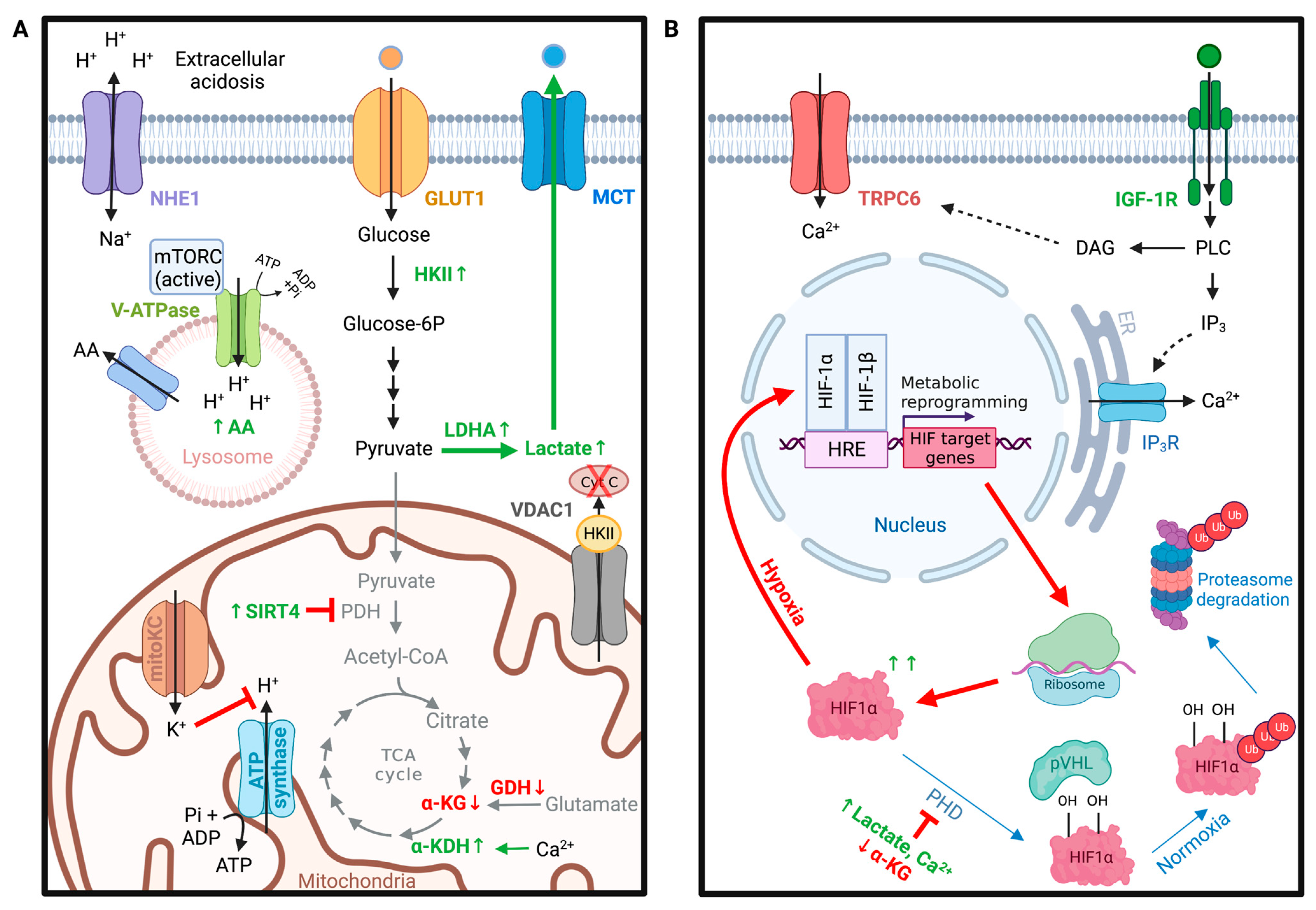
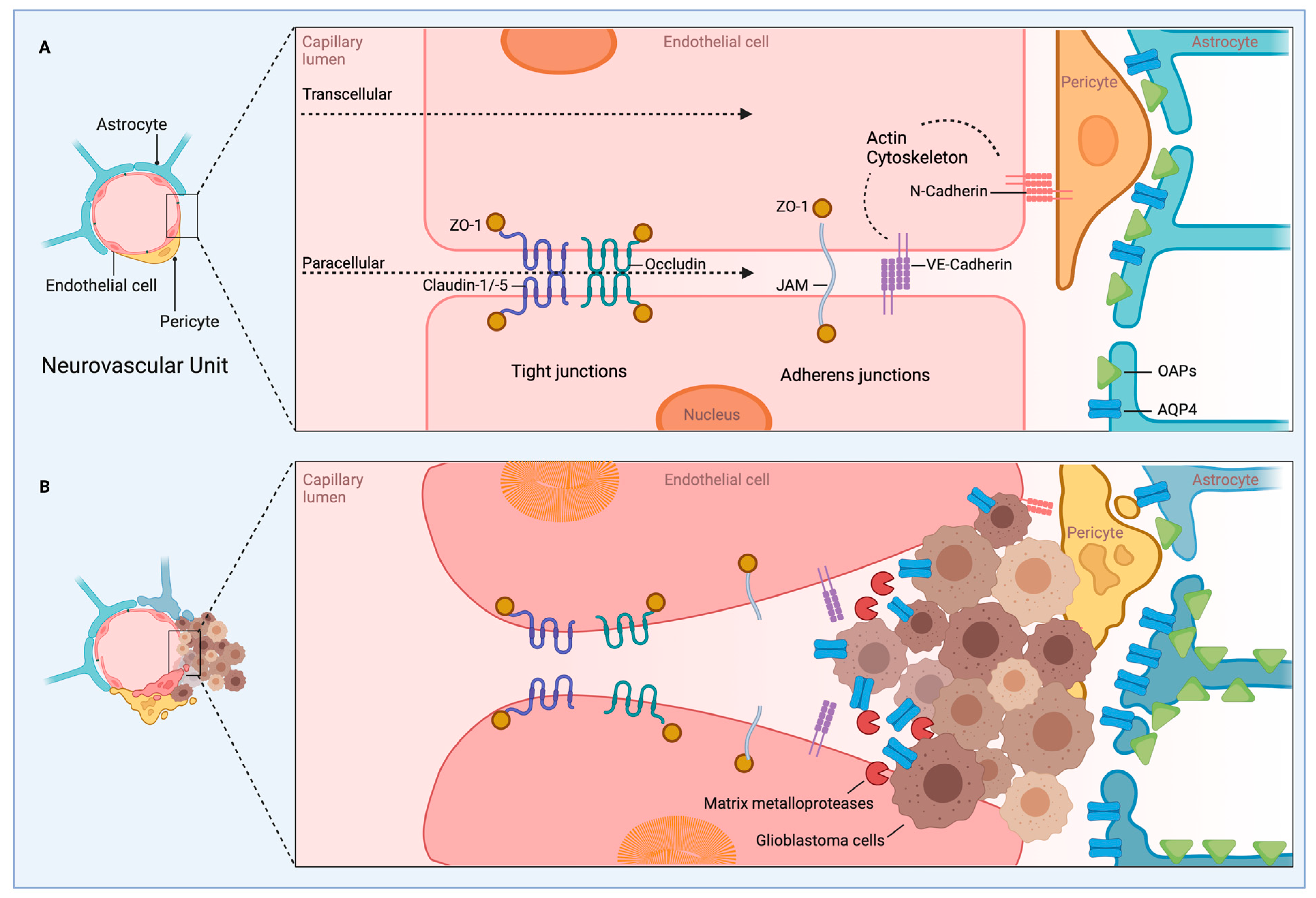

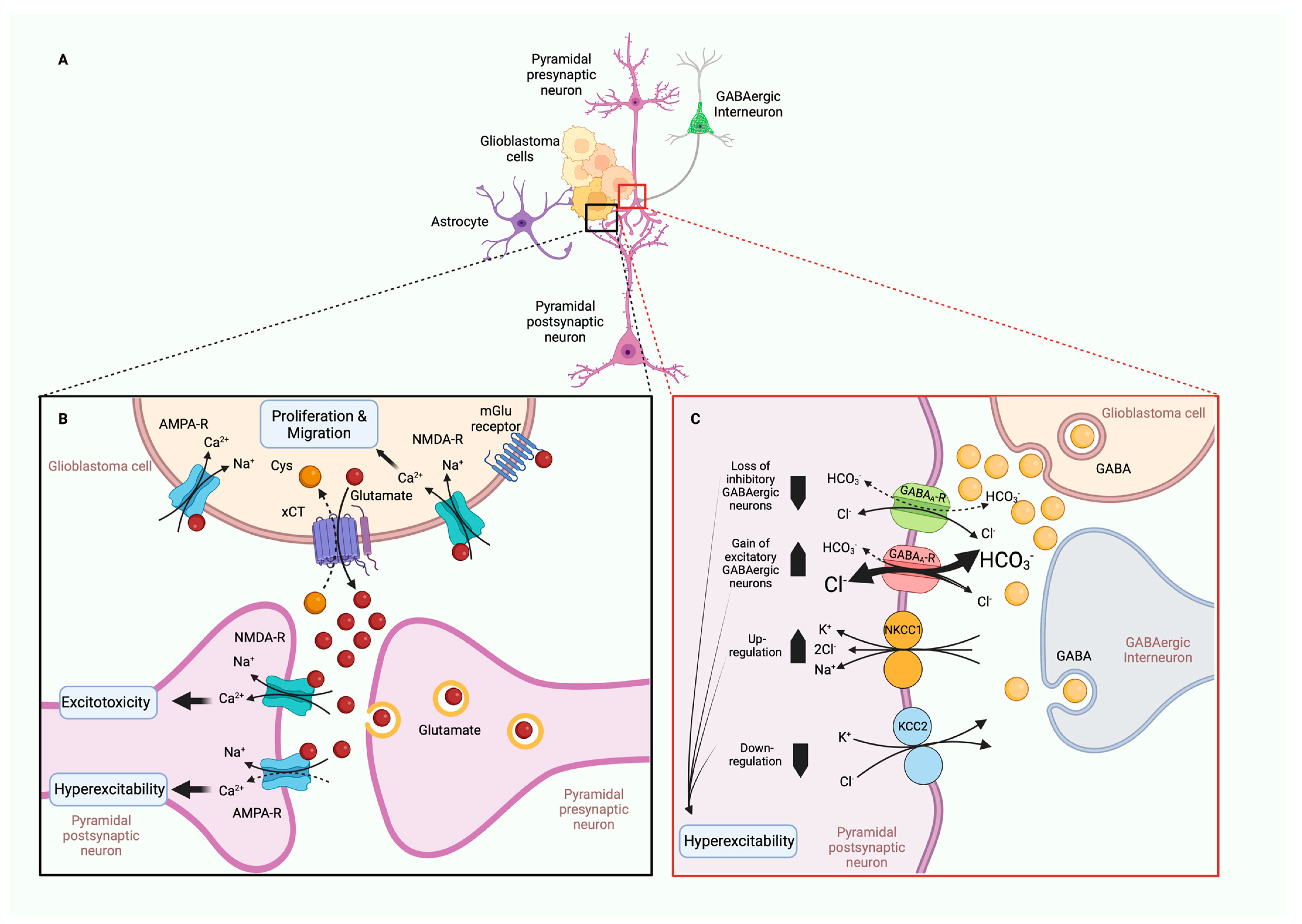

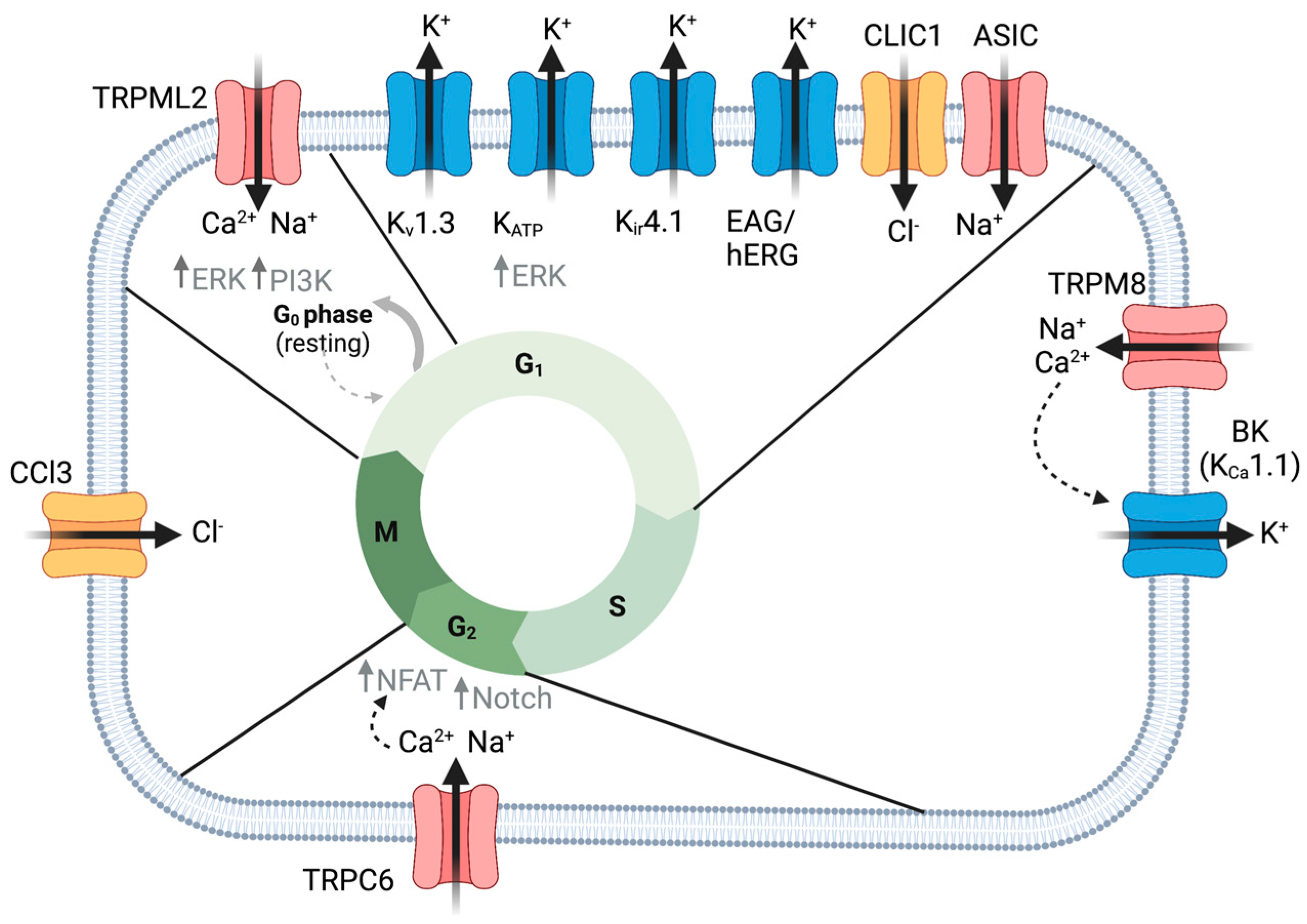{kind=link}
{kind=link}
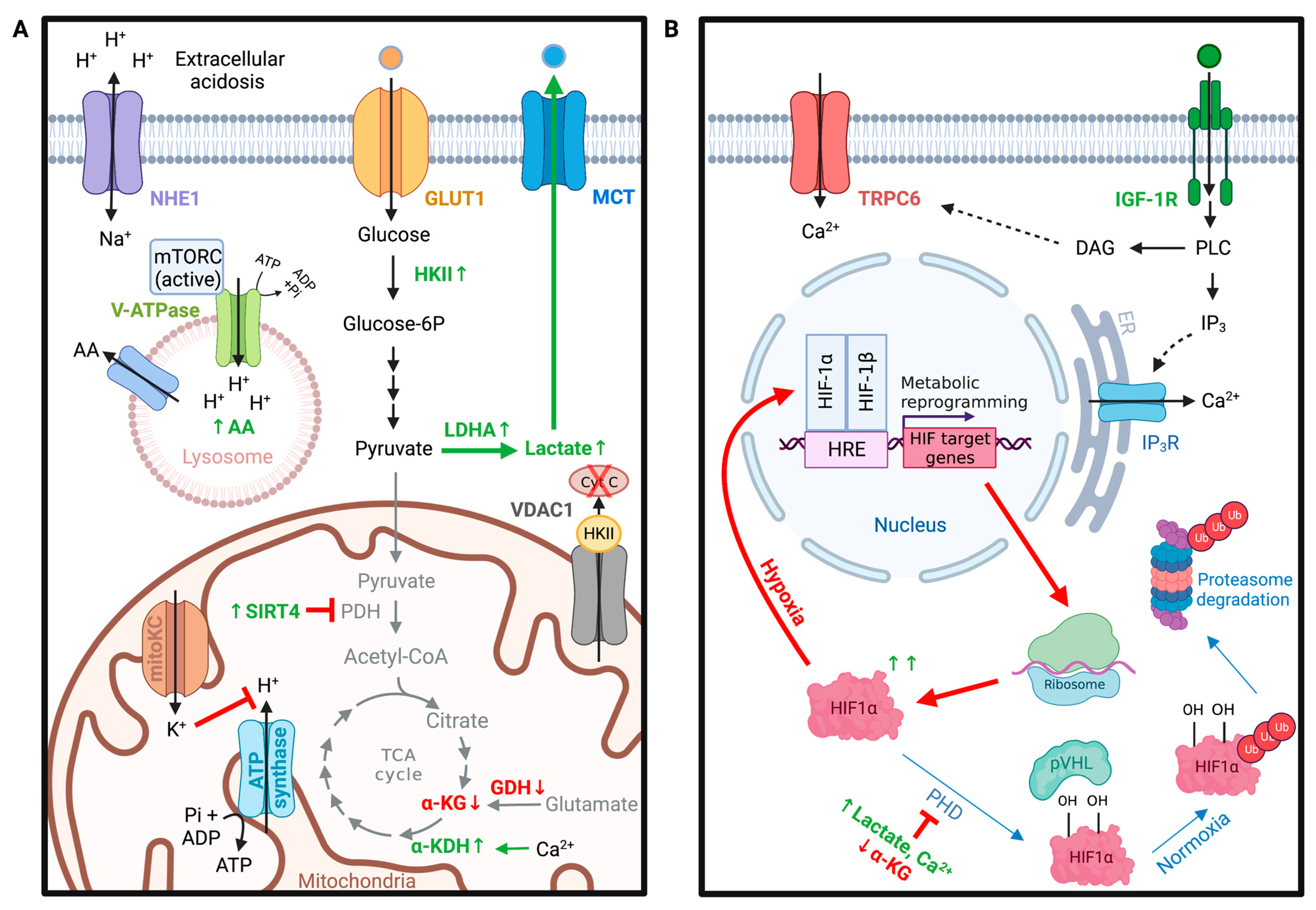{kind=link}
{kind=link}
{kind=link}
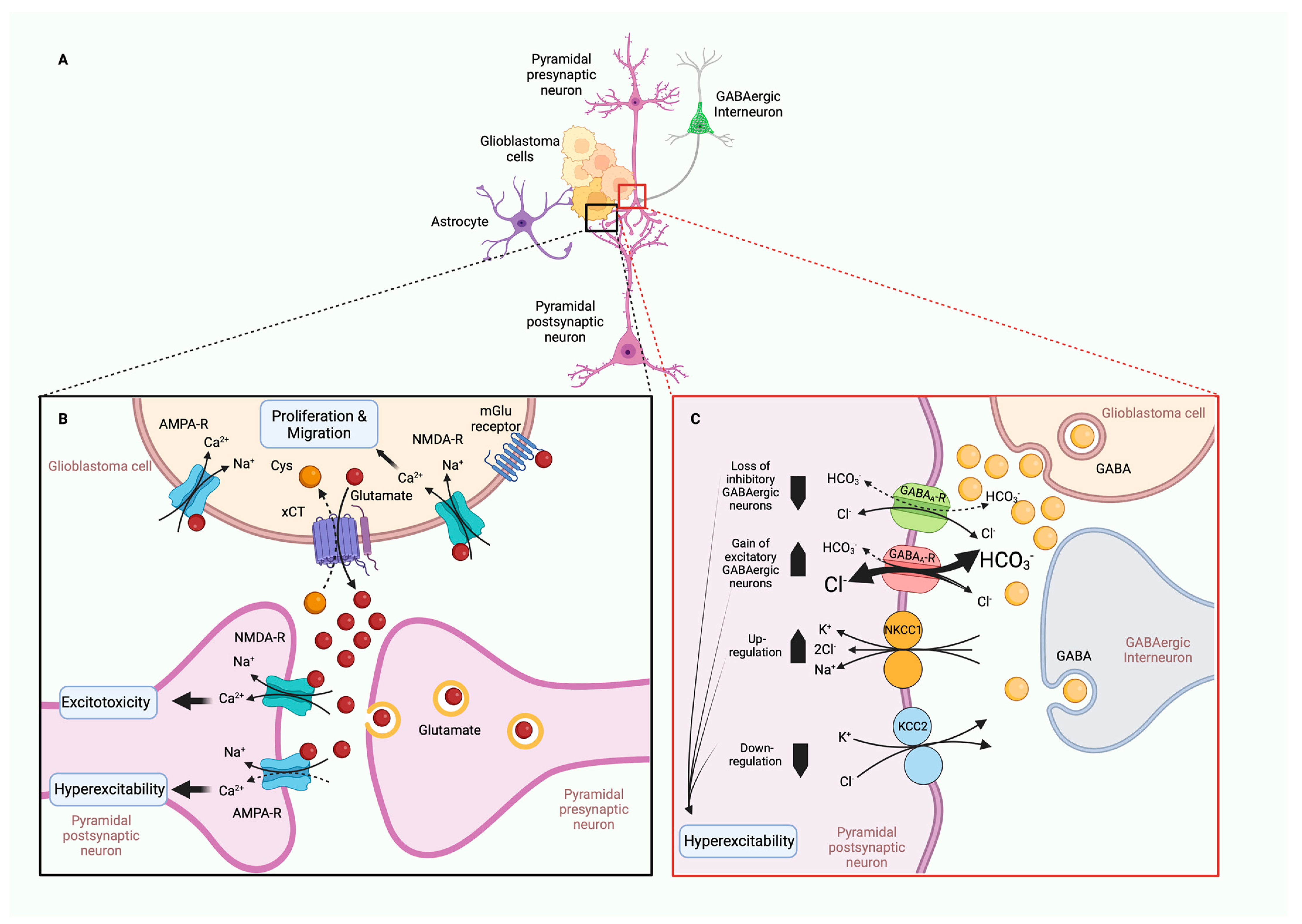{kind=link}
| Ion Channel | Expression | Model | Patient Survival | Reference |
|---|---|---|---|---|
| EAG | Increased | GBM metastases | Expression correlated with worse survival | [9] |
| hERG | Increased | GBM-derived cells | Expression correlated with worse survival | [10] |
| BK | Increased | GBM-derived cells, GBM cell lines U251, U87MG | Expression correlated with malignancy | [11] |
| TRPV1 | Decreased with glioma grade | Normal human astrocytes, U87MG, U737 glioma cell lines | Expression promotes patient survival | [12] |
| TRPV2 | Decreased with glioma grade | U97MG, primary glioma cells | Expression correlated with glioma grade | [13] |
| TRPC6 | Increased | GBM patient samples | N/A | [14] |
| TRPC1 | Increased | Patient glioma samples | Expression positively correlated with patient survival | [12] |
| TRPM2 | Increased | Patient glioma samples | Expression positively correlated with patient survival | [12] |
| TRMPM3 | Increased | Patient glioma samples | Expression positively correlated with patient survival | [12] |
| TRPM7 | Increased | Patient glioma samples | Expression positively correlated with patient survival | [12] |
| TRPM8 | Increased | Patient glioma samples | Expression positively correlated with patient survival | [12] |
| VRAC | Increased | GBM cell lines | N/A | [15,16,17] |
| TMEM16A | Increased | Patient GBM samples, GBM cell lines: U87MG, U118, U251, SHG44 | Increased with grade, less favorable to patient survival | [18,19] |
| CLIC1 | Increased | GBM | Expression correlated with worse prognosis | [20] |
| NHE5 | Increased | C6 glioma cells | N/A | [21] |
| ASIC1 | Decreased | GBM samples | Expression negatively correlated to survival | [22] |
| PIEZO | Increased | Human GBM | Expression inversely correlated to survival | [23,24] |
| NKCC1 | Increased | Primary GBM samples | Expression positively correlated with grade | [25,26] |
| IK | Increased | Patient GBM samples | Expression negatively associated with survival | [27] |
| NHE1 | Increased | Primary human glioma cells | N/A | [28,29,30] |
| Kir4.1 | Decreased or mislocalized | Glioma cell lines | N/A | [31,32] |
| mitoBKCa | Increased | Glioma cell line LN229 | N/A | [33] |
| AQP1 | Increased | Astrocytomas, GBM | N/A | [34,35,36] |
| AQP4 | Mislocalized | Patient GBM samples | N/A | [37,38] |
| TRPM5 | Increased | Patient cells | Expression negatively associated with survival | [39] |
| Ion | GENE | Channel Name(s) | Cell Behavior | Reference |
|---|---|---|---|---|
| K+ | KCNJ10 | Kir4.1 | ↓Proliferation | [48] |
| ↓Fillopodia and invasion | [32] | |||
| Endothelial BBB | [49,50,51,52,53,54,55,56,57] | |||
| K+ | KCNH2 | Kv11.1, hERG | ↑Proliferation, G1→S | [10,58,59] |
| K+ | KCNJ11 | KATP, Kir6.2 | ↑Proliferation, G1→S | [60] |
| K+ | KCNMA1 | KCa1.1, BK, mitoBKCa (splice variant) | ↑Proliferation, S→G2 | [61] |
| ↑Migration, swelling at leading edge | [62,63,64,65,66] | |||
| MitoBK promotes apoptosis | [33] | |||
| K+ | KCNN4 | KCa3.1, IK | Irradiation, ↑Proliferation, G2→M | [27] |
| ↑Migration and invasion, trailing edge | [16,17,27,67,68,69,70,71,72] | |||
| K+ | KCNA1 | KV1.1, Shaker | Endothelial BBB | [49,50,51,52,53,54,55,56,57] |
| K+ | KCNA3 | KV1.3, mitoKV1.3 | MitoKV1.3 promotes apoptosis | [73] |
| K+ | KCNJ8 | (KIR) Kir6.1 | Pericyte BBB expression | [74,75,76,77,78,79] |
| Cl− | CLIC1 | CLIC1 | ↑Proliferation, G1→S | [80] |
| Cl− | CLCN3 | CLC3 | Premitotic condensation, cell cycle speed | [81,82] |
| ↑Migration and invasion, ↑MMP—Extracellular matrix (ECM) remodeling | [69,70,83,84] | |||
| Cl− | LRRC8A | VRAC, IClswell1 | Hypoxia-related proliferation and survival | [85] |
| Cl− | TMEM16A | TMEM16A, ANO1 | ↑Proliferation, ↑Growth factor | [18,19] |
| Cation | TRPC1 | TRPC1 | ↑Proliferation, G2→M | [86] |
| ↑Directional migration, leading edge | [87] | |||
| TRPC6 | TRPC6 | ↑Proliferation, ↑Growth factor | [14,88,89] | |
| ↑Hypoxic migration | [14] | |||
| ↑VEGF-mediated angiogenesis | [90,91] | |||
| Cation | TRPM2 | TRPM2 | ↑Apoptosis | [92,93] |
| Cation | TRPM7 | TRPM7 | ↑Proliferation | [94,95,96] |
| ↑Migration and invasion through kinase domain | [94] | |||
| Cation | TRPM8 | TRPM8 | ↑Proliferation, S→G2 | [97,98,99] |
| ↑Migration mediated by BK activation | [97,100] | |||
| Cation | TRPML1 | MCOLN1 | ↑Apoptosis, ROS sensor? | [101] |
| Cation | TRPML2 | MCOLN2 | ↑Proliferation, G0→G1, ↓Apoptosis | [102,103] |
| Cation | TRPV1 | TRPV1 | ↓Proliferation | [12] |
| Cation | TRPV2 | TRPV2 | ↓Proliferation | [13] |
| Cation | TRPV4 | TRPV4 | ↑Migration and invasion, ↑Cytoskeletal remodeling | [104] |
| ↑Angiogenesis | [105] | |||
| Na+ | ASIC1 | ASIC1 | ↓Proliferation | [22,106] |
| ↑Migration and lamellipodium expansion | [107,108,109,110] | |||
| Na+/Ca2+/K+ | PIEZO1 | PIEZO1 | ↑Proliferation, tissue stiffness | [23] |
| ↑Focal adhesion formation | [23] | |||
| Open: ADP/ATP, Cl−, NADH/NAD+, Closed: Ca2+ | VDAC1 | VDAC1 | ↑Oxidative phosphorylation, translates cellular state to mitochondria function, can drive apoptosis | [111,112,113] |
| VDAC2 | VDAC2 | Negatively regulates glycolysis via PFK direct interaction | [114] | |
| Cl−, HCO3− | SLC4A3 | Cl−/HCO3− exchanger | Pericyte BBB expression | [74,75,76,77,78,79] |
| Cl−, HCO3− | SLC4A4 | Cl−/HCO3− exchanger | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/I− | SLC5A5 | Na+/I− symporter | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/Ca2+ | SLC8A2 | Na+/Ca2+ exchanger | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/Ca2+ | SLC8A3 | Na+/Ca2+ exchanger, | Pericyte BBB endothelial cells | [49,50,51,52,53,54,55,56,57] |
| Na+/H+ | SLC9A1 | NHE1 | ↑Glycolysis | [29,30,115] |
| Na+/H+ | SLC9A3R1 | NHE 3R1 | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/H+ | SLC9A5 | NHE5 | ↑Proliferation | [21] |
| ↑Metastasis, cell adhesion, invasion | [21] | |||
| Na+/H+ | SLC9A9 | NHE9 | ↑Proliferation, ↑Growth factor signaling | [116,117] |
| Na+/K+/Cl− | SLC12A2 | NKCC1 | ↑Migration, invasion, filipodia formation, and focal adhesions | [25,26] |
| Expressed in BBB endothelial cells, role downregulated in GABAergic hyperexcitability | [49,50,51,52,53,54,55,56,57] | |||
| K+, Cl− | SLC12A5 | KCC2 | Downregulated contributes to GABA-mediated hyperexcitability in peritumoral tissue | [118,119,120,121,122] |
| H+ | SLC15A2 | H(+)-coupled peptide transporter | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/K+/Ca2+ | SLC24A3 | Na+/K+/Ca2+ exchanger | Pericyte BBB expression | [74,75,76,77,78,79] |
| Na+/K+ | ATP1A1 | Na+-K+-ATPase | Expressed in BBB endothelial cells, expressed in BBB pericytes | [49,50,51,52,53,54,55,56,57] |
| Ca2+, Na+ | ATP2B2 | Ca2+-ATPases | Pericyte BBB expression | [74,75,76,77,78,79] |
| K+ | ABCC9 | ATP-sensitive K+ channel, ATP-binding cassette | Pericyte BBB expression | [74,75,76,77,78,79] |
| H2O | AQP1 | Aquaporin 1 | ↑Migration | [123] |
| ↑Angiogenesis | [34,35,36] | |||
| H2O | AQP4 | Aquaporin 4 | Contribute to BBB remodeling and brain edema in glioma | [37,38] |
| Na+ | SCN5A | NaV1.5 | ↑Invasion with NHE1 | [124] |
| Ca2+, Na+ | GLUN | NMDAR | Overstimulation of NMDARs leads to excessive Ca2+ excitoxicity, cell death of TME provides room for tumor expansion | [125,126] |
| Na+ | GLUA | AMPAR | ↑Proliferation | [42,127] |
| ↑Invasion | [42,127] | |||
| Hyperexcitable/seizure risk | [128] | |||
| Cl− | GABR | GABA | neurons die and neurotransmitter shifts polarity, causing hyperexcitability | [118,119,120,121,122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elias, A.F.; Lin, B.C.; Piggott, B.J. Ion Channels in Gliomas—From Molecular Basis to Treatment. Int. J. Mol. Sci. 2023, 24, 2530. https://doi.org/10.3390/ijms24032530
Elias AF, Lin BC, Piggott BJ. Ion Channels in Gliomas—From Molecular Basis to Treatment. International Journal of Molecular Sciences. 2023; 24(3):2530. https://doi.org/10.3390/ijms24032530
Chicago/Turabian StyleElias, Abdallah F., Bernice C. Lin, and Beverly J. Piggott. 2023. "Ion Channels in Gliomas—From Molecular Basis to Treatment" International Journal of Molecular Sciences 24, no. 3: 2530. https://doi.org/10.3390/ijms24032530