
Insulin Receptor Substrate 1 Signaling Inhibits Foxp3 Expression and Suppressive Functions in Treg Cells through the mTORC1 Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
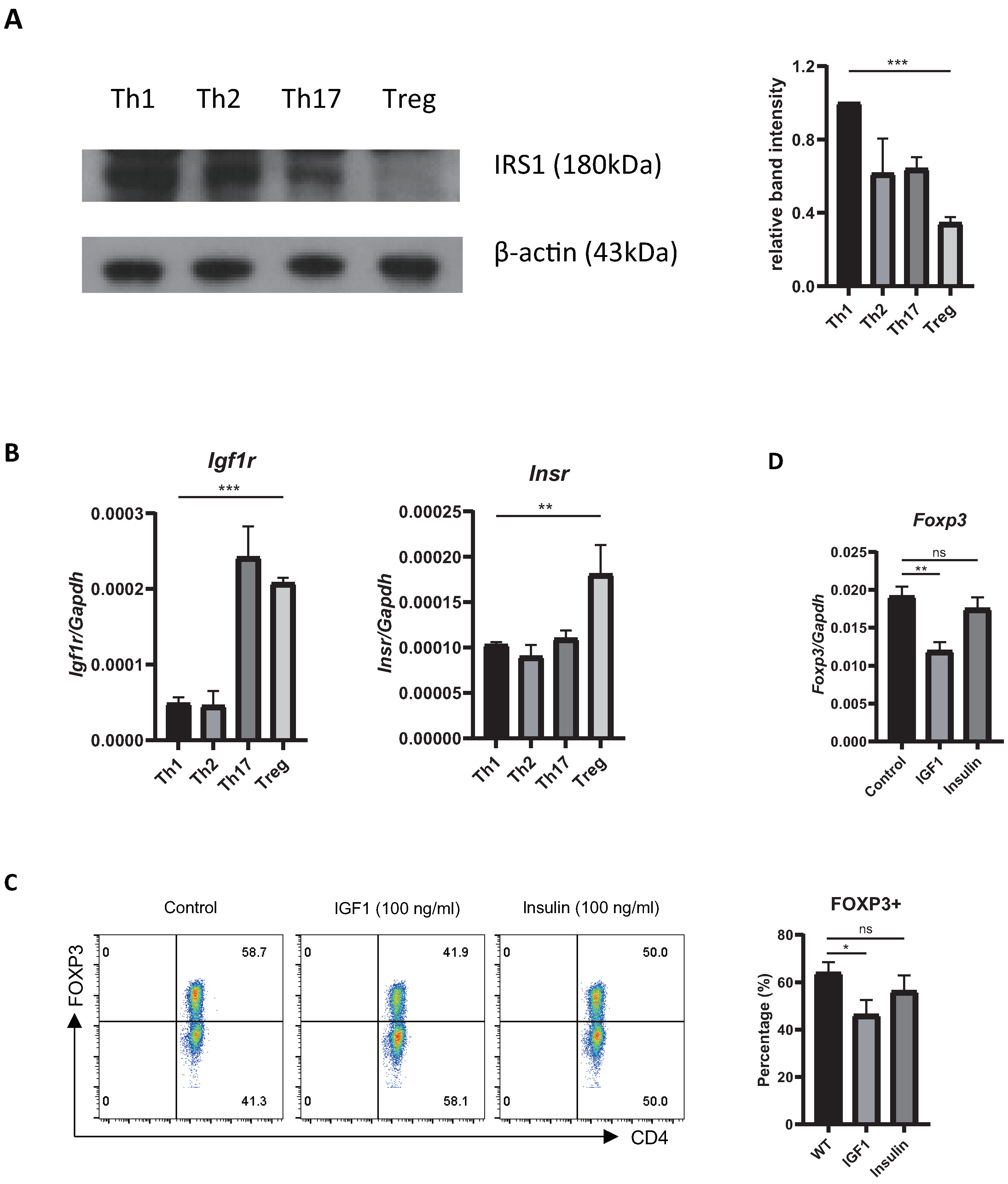
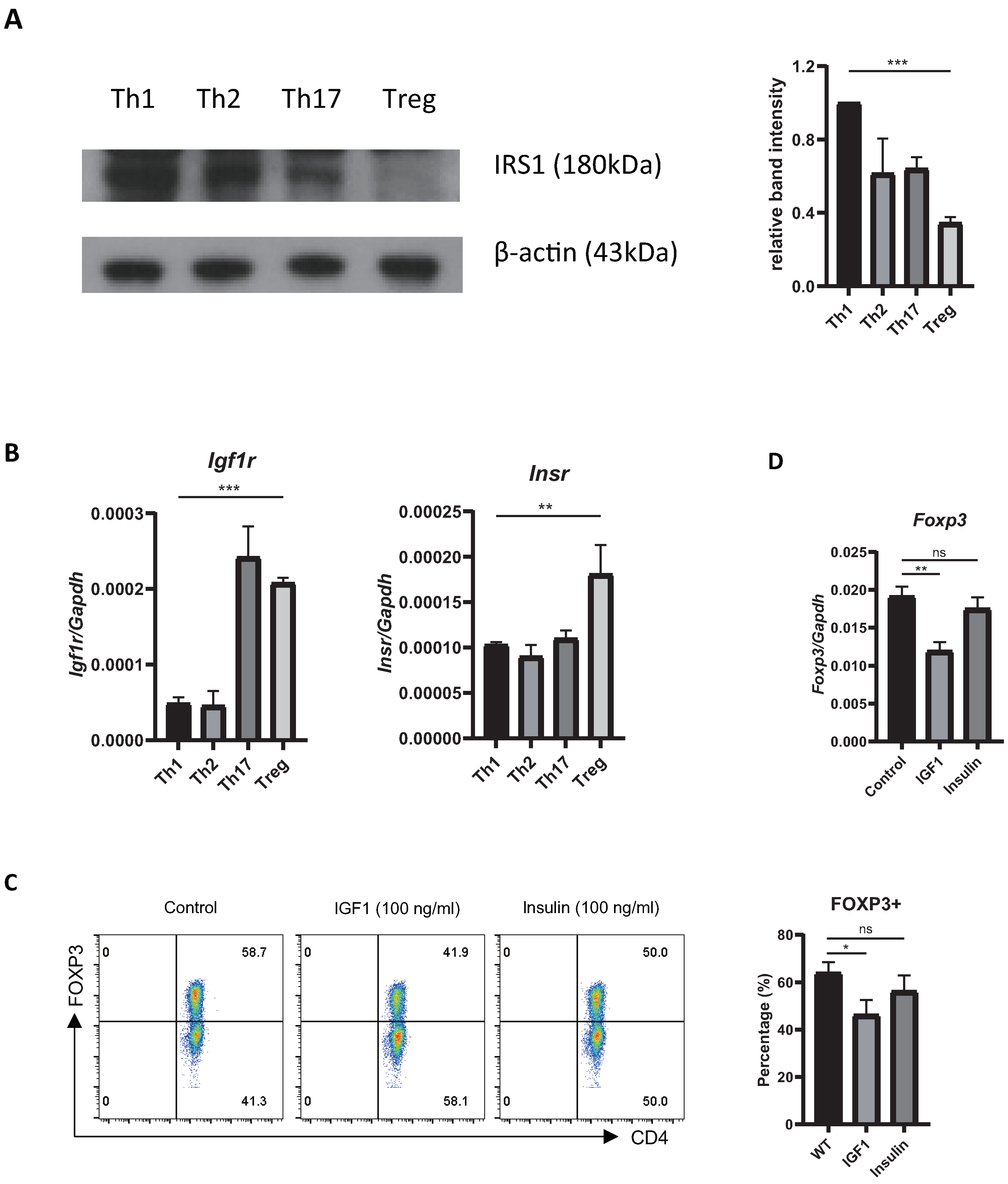
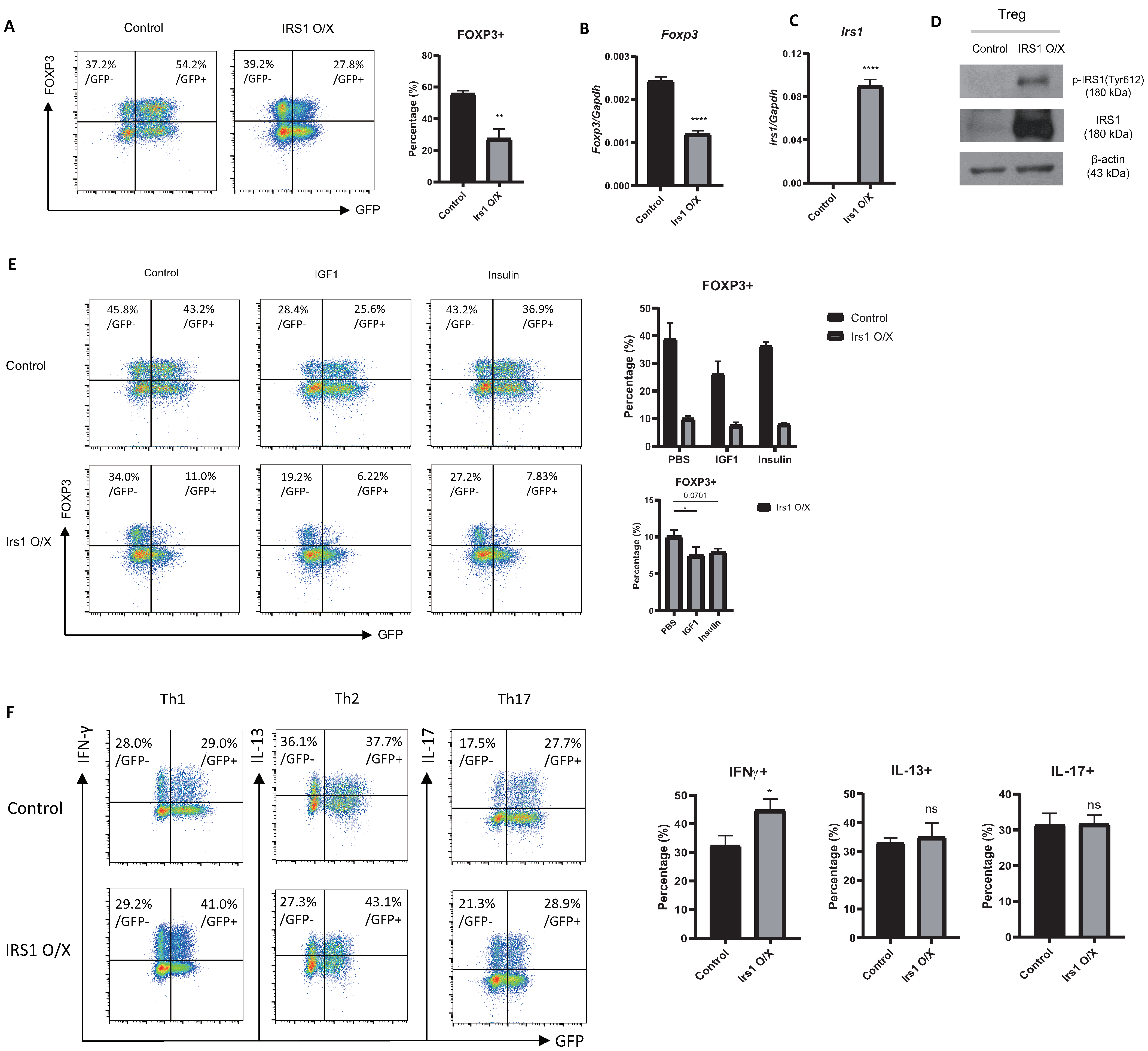
2.1. Treg Cells Show Low Expression of Irs1, and IRS1 Signaling Inhibits Treg Differentiation
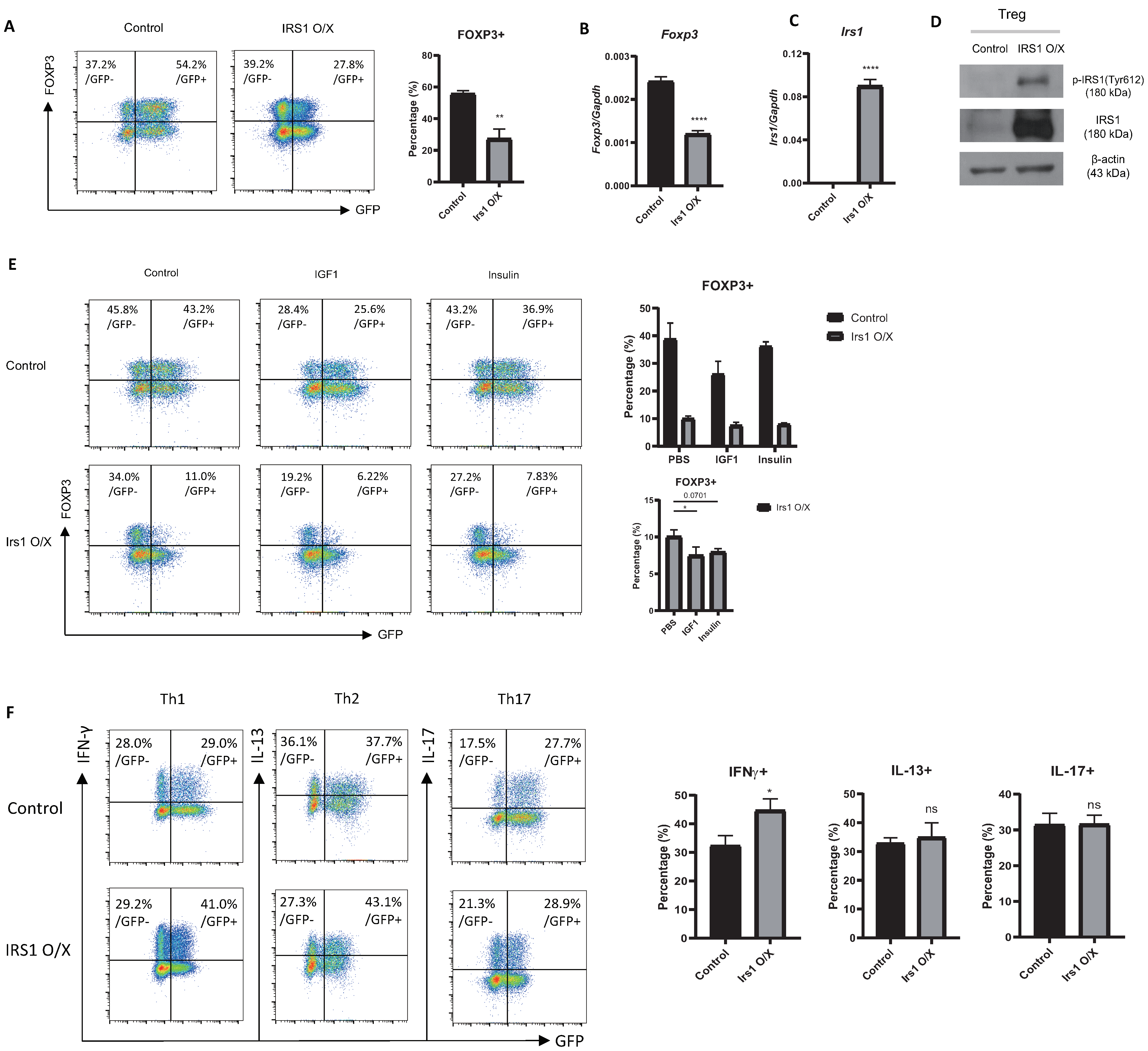
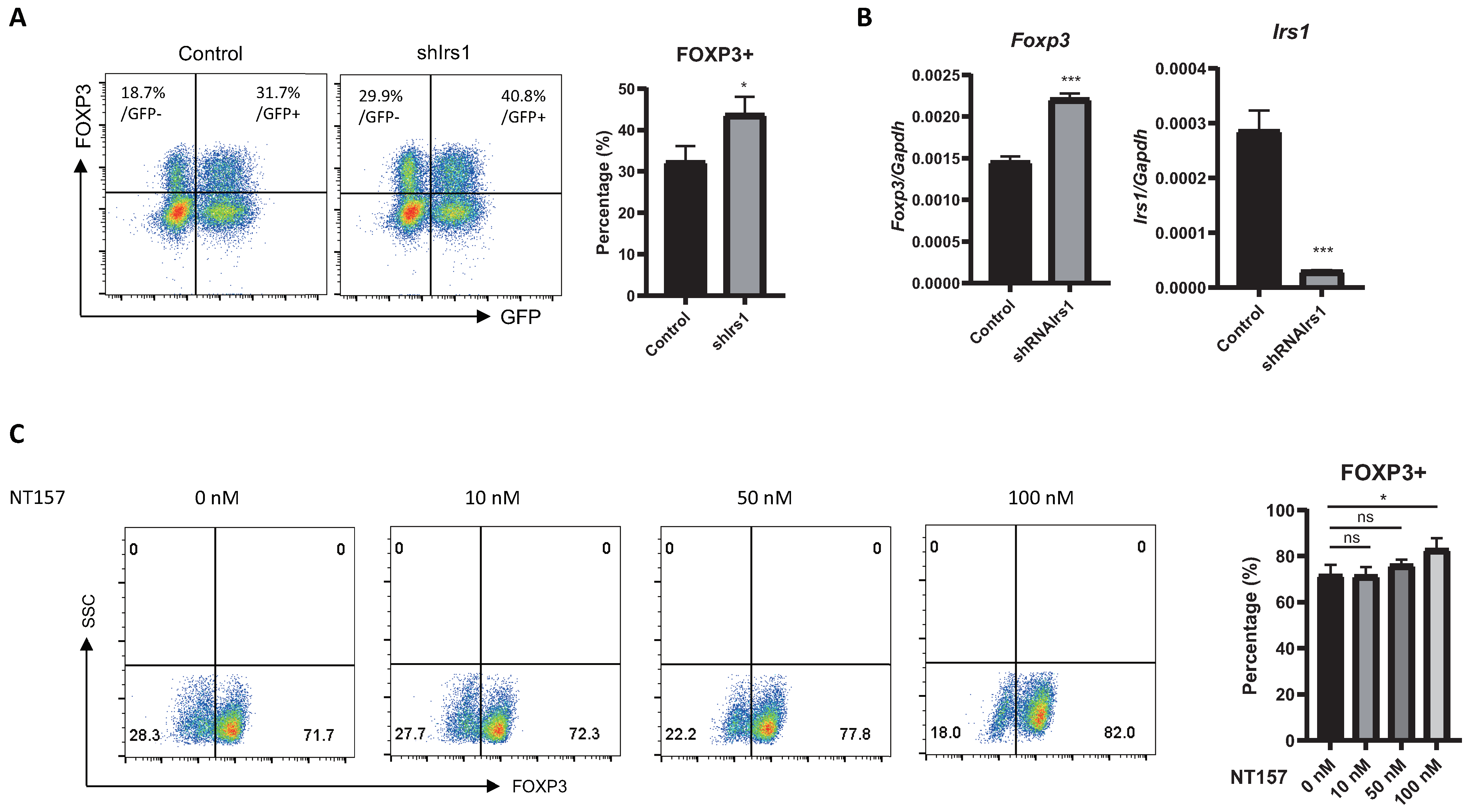
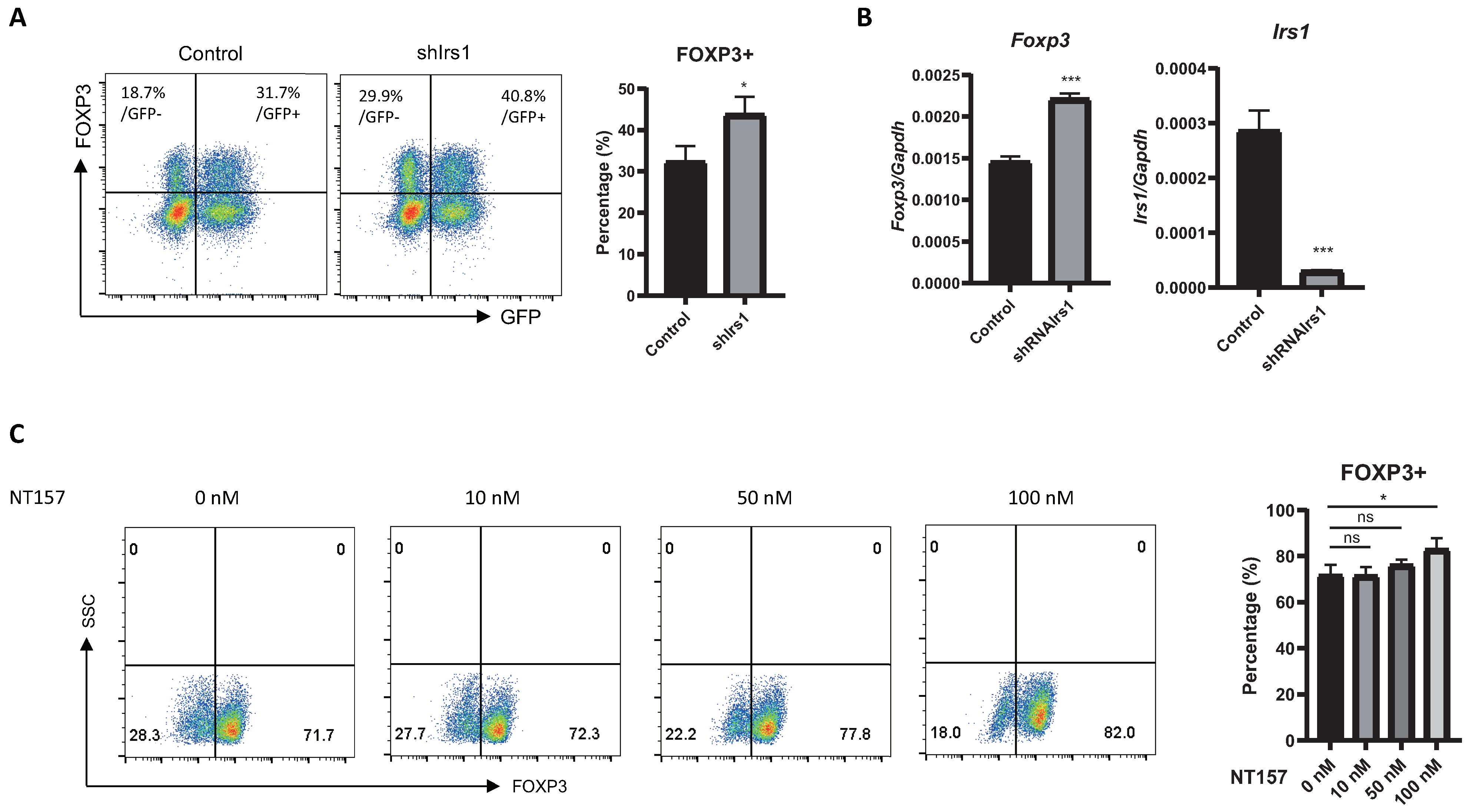
2.2. Overexpression or Knockdown of IRS1 Affects Treg Cell Differentiation
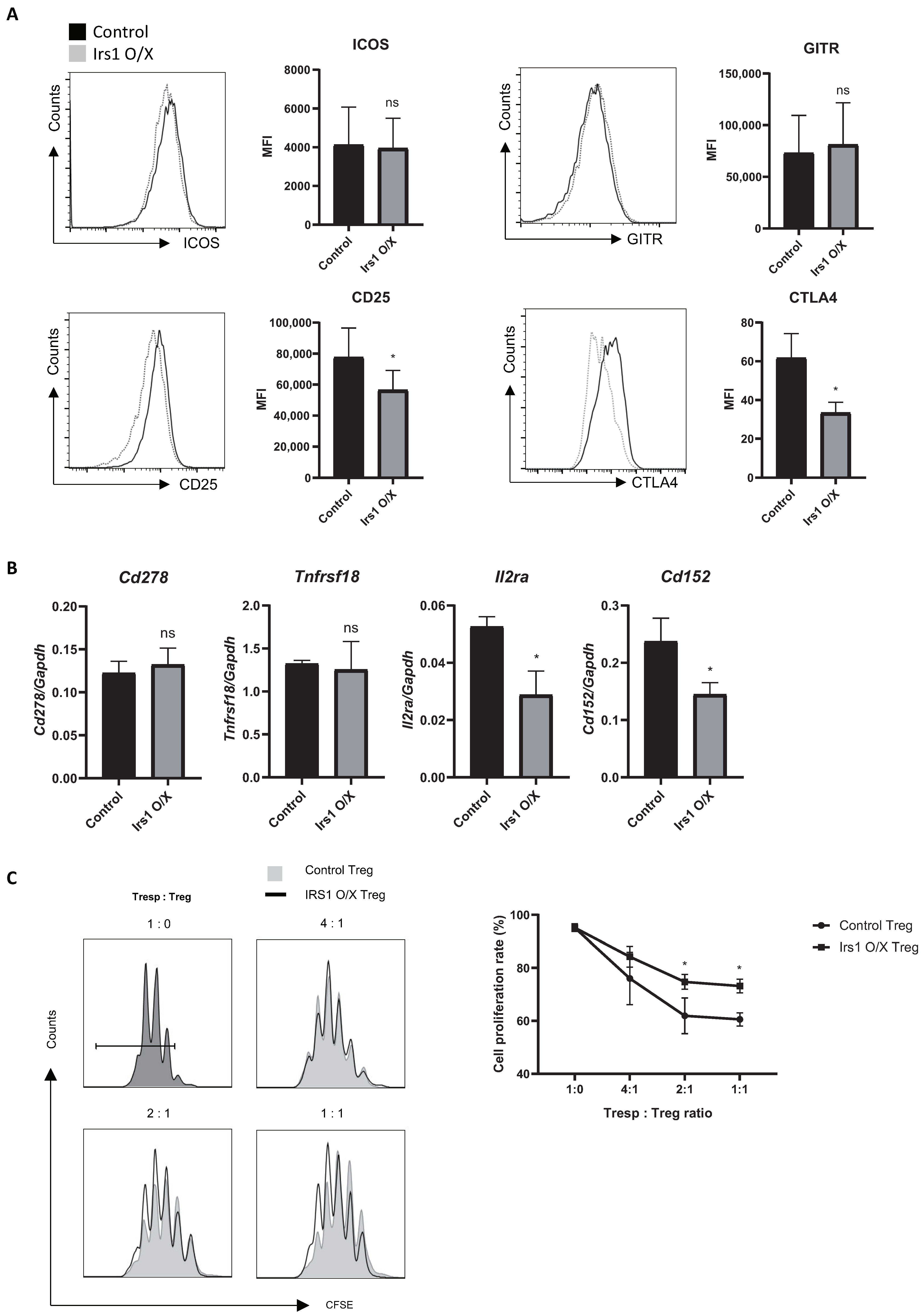
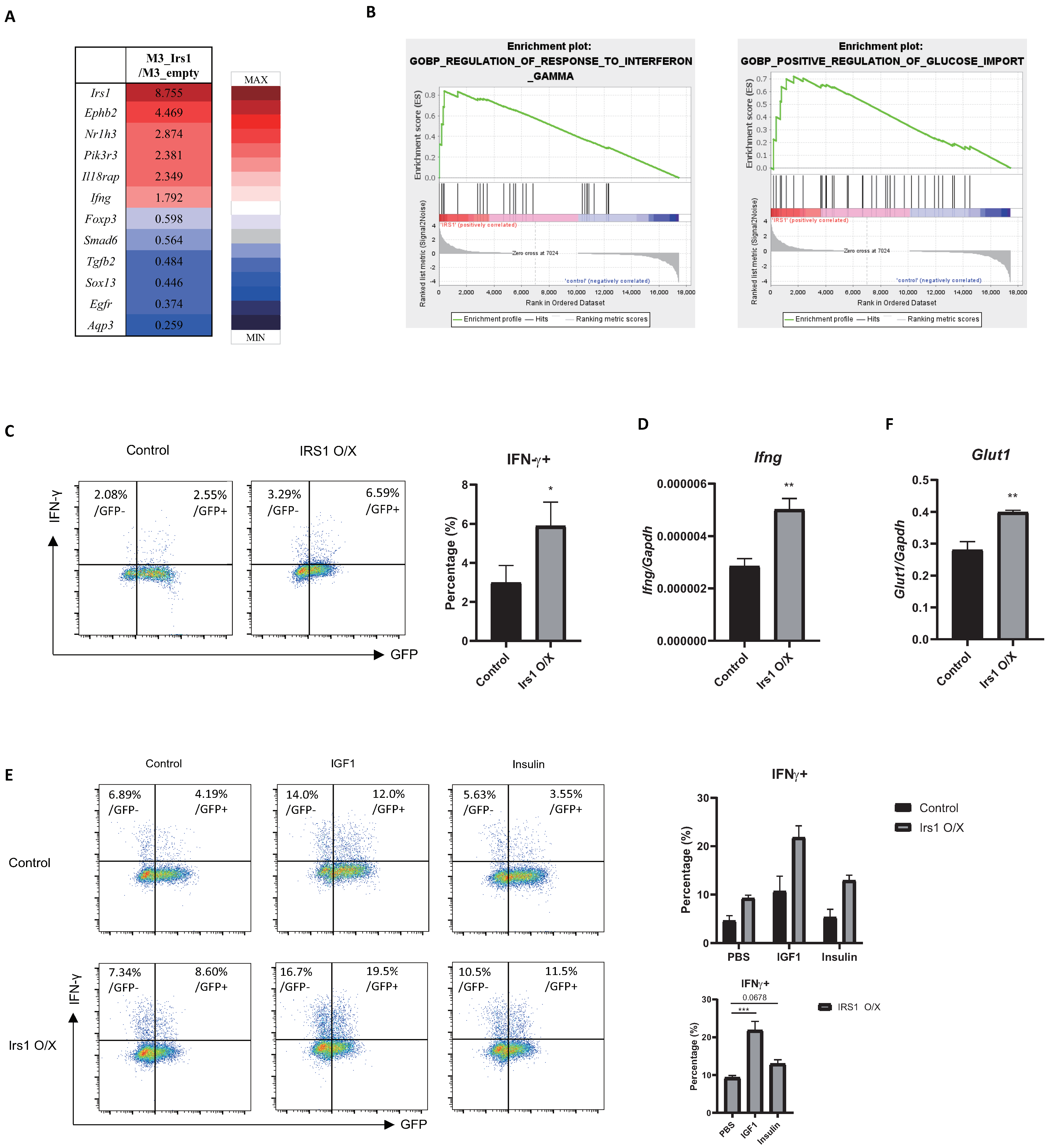
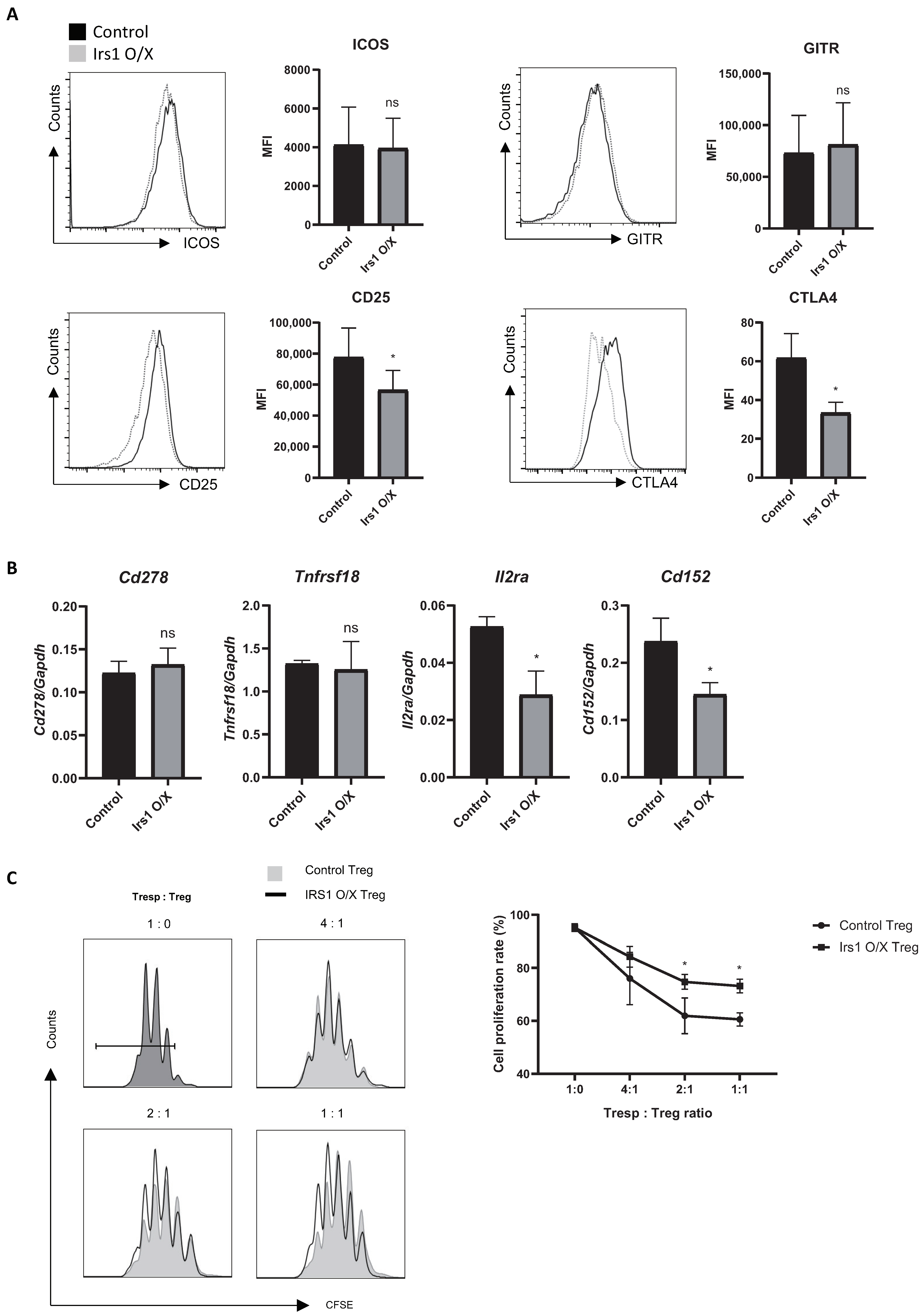
2.3. IRS1 Downregulates Expression of Treg Signature Genes, as Well as Suppressive Activity
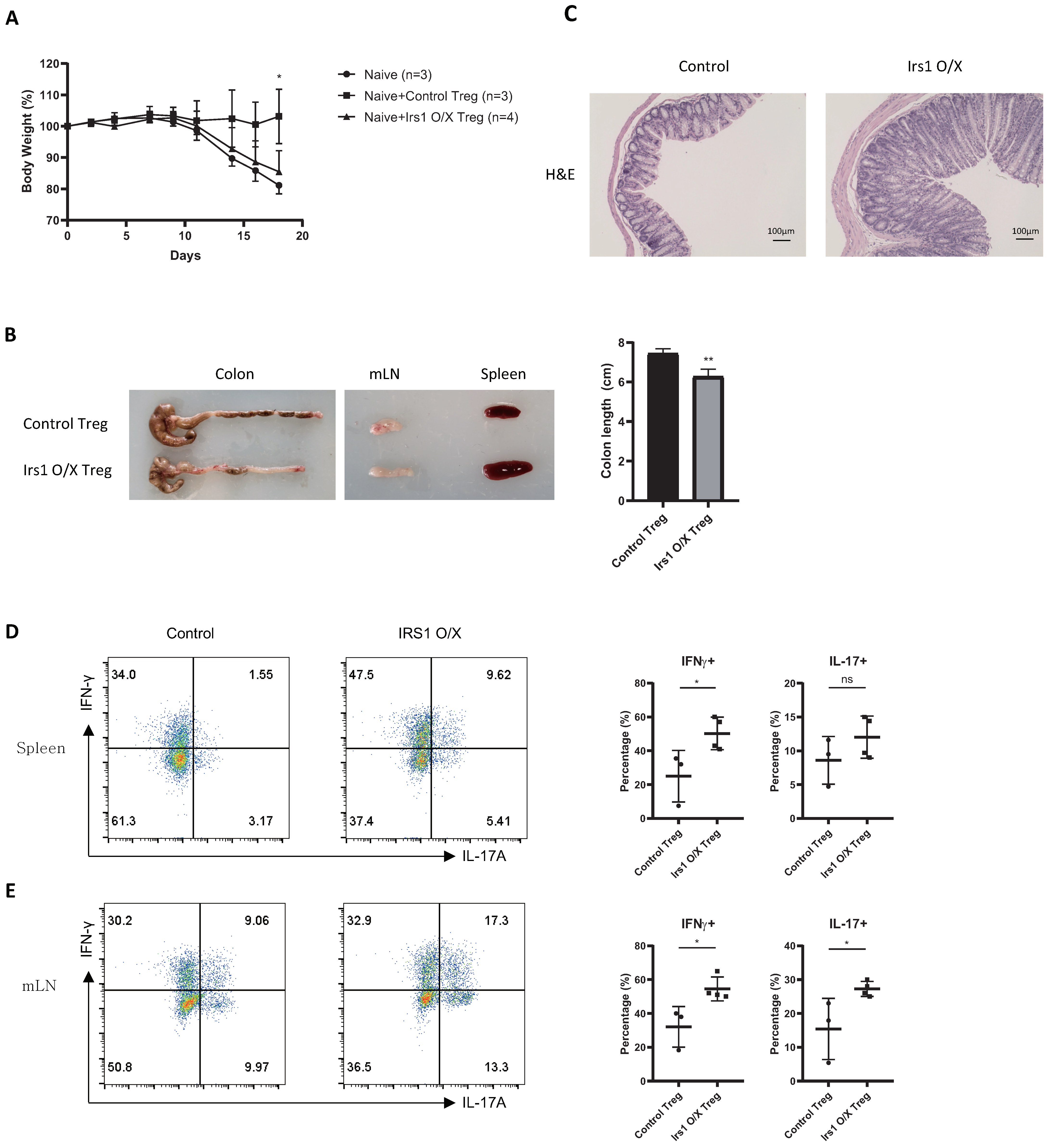
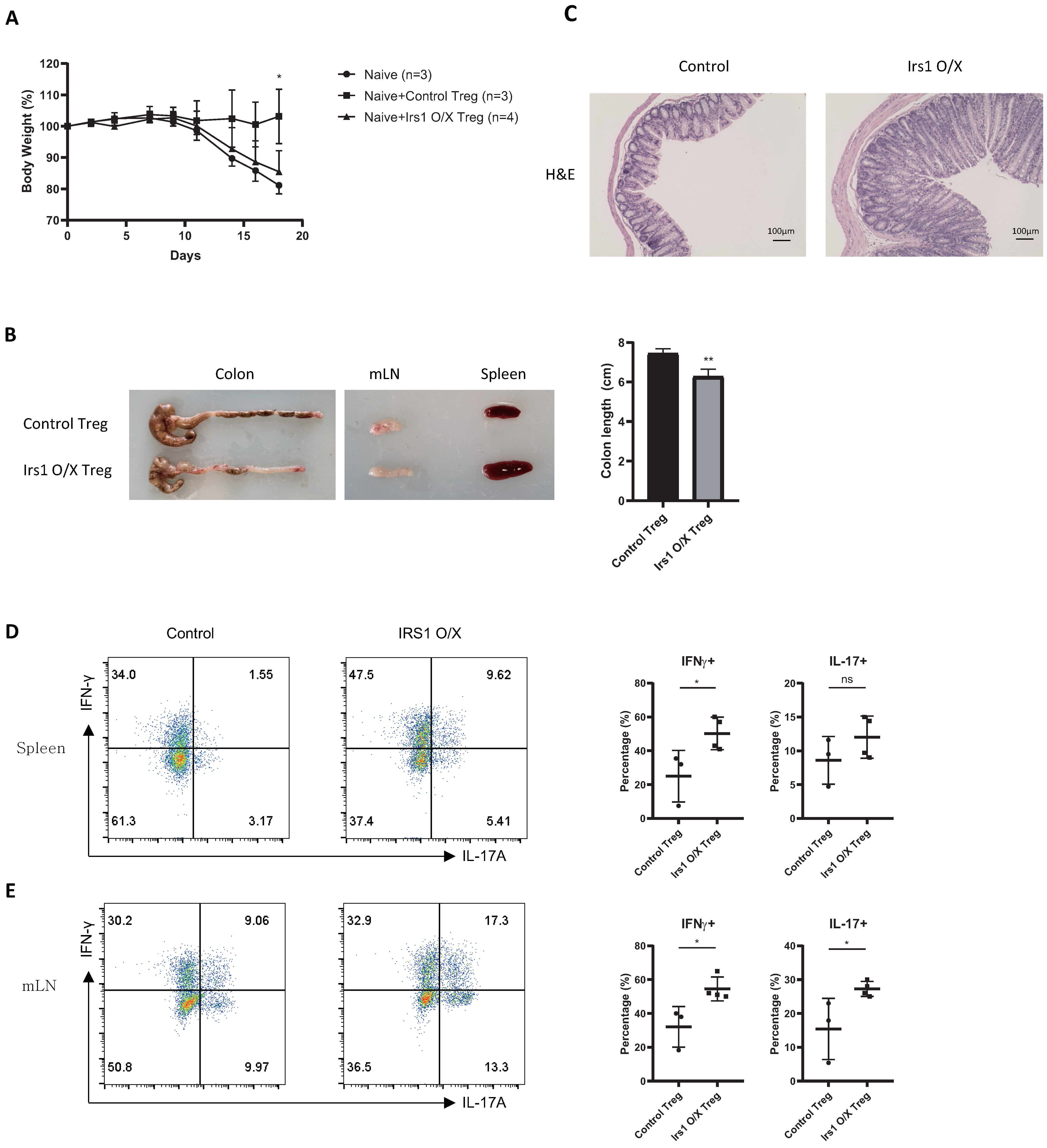
2.4. IRS1-Overexpressing Treg Cells Fail to Control Colitis
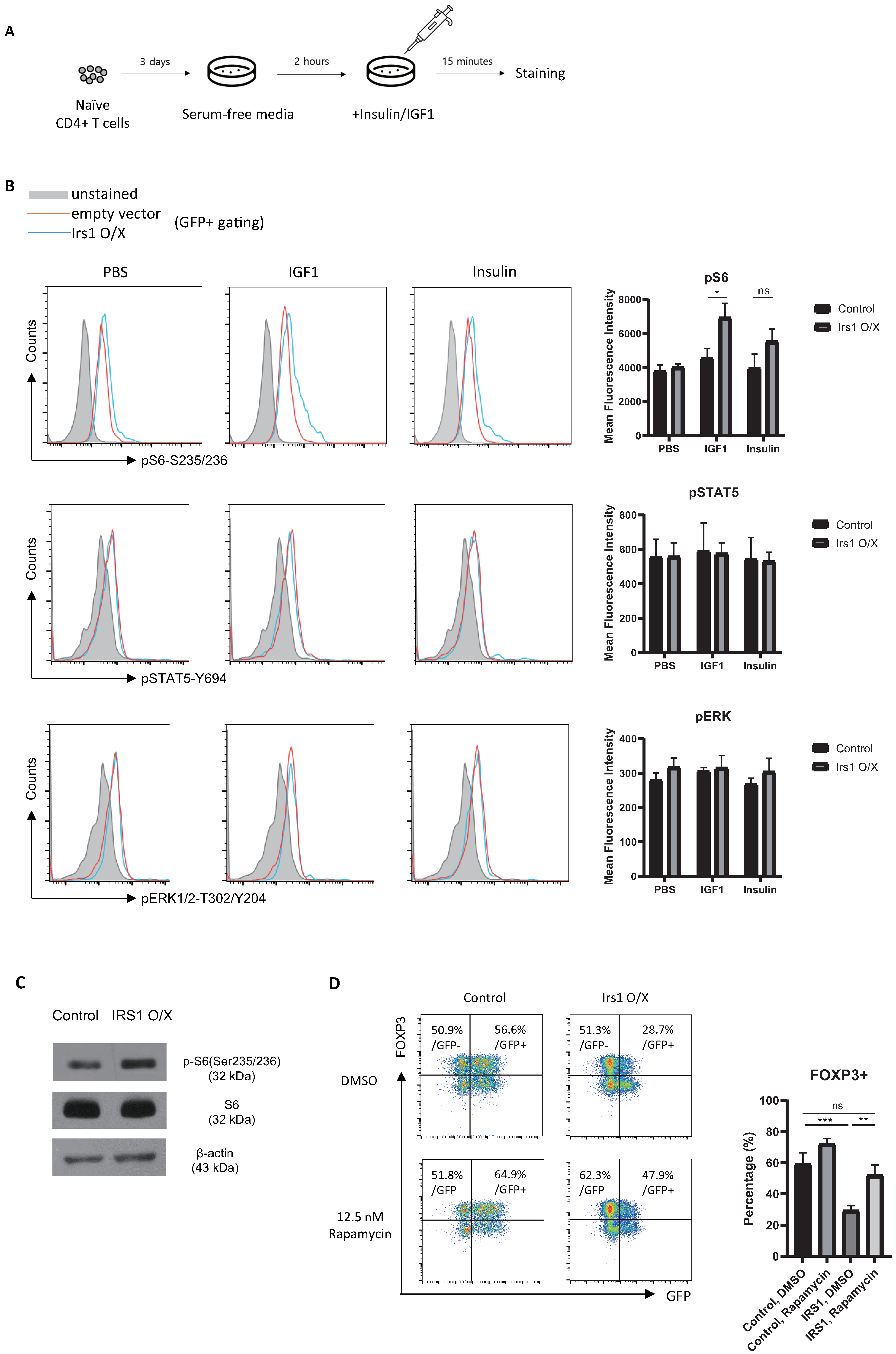
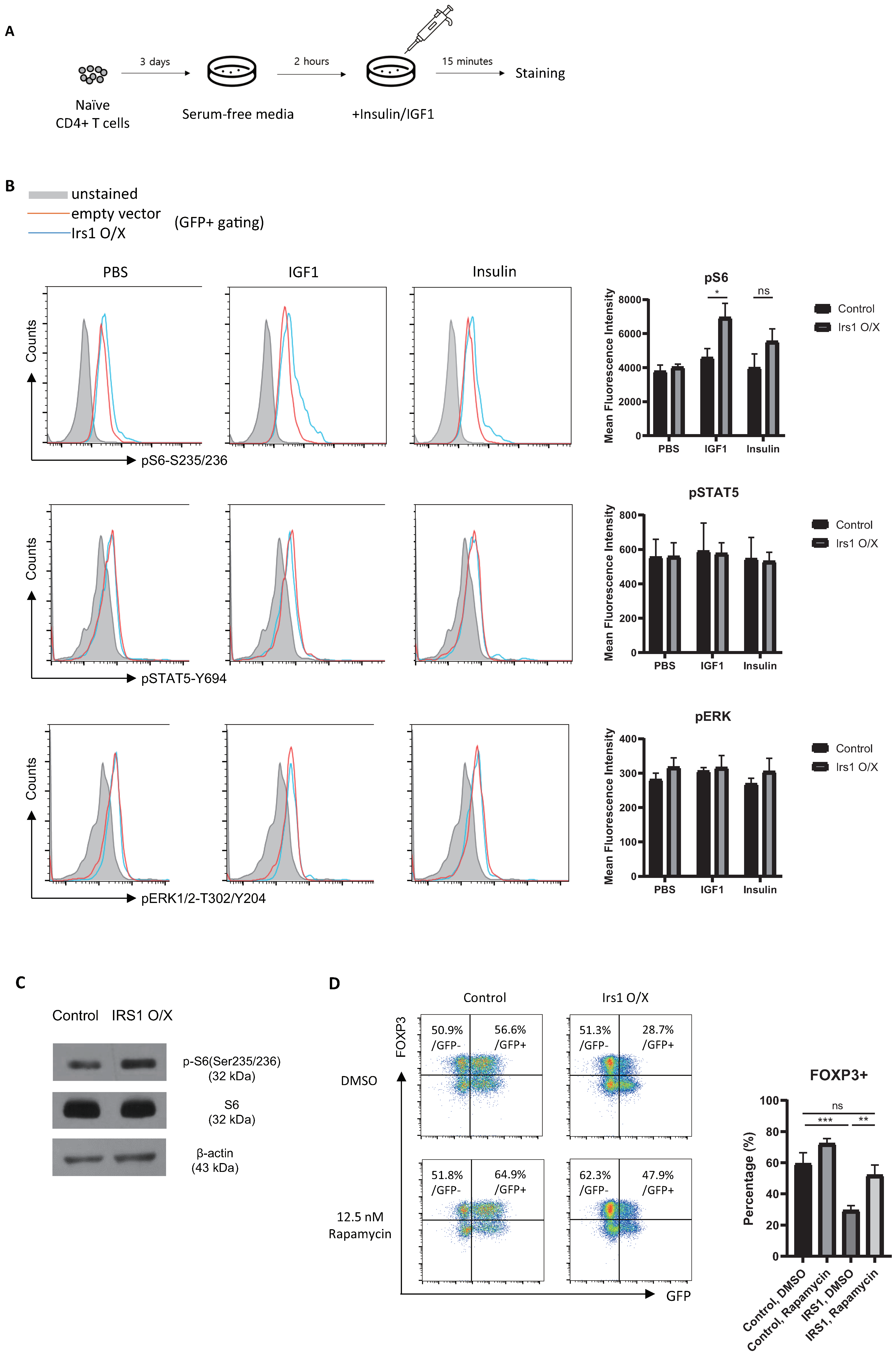
2.5. IRS1 Induces mTORC1 Pathway, but Not the MAPK and JAK2/STAT Pathways, in Treg Cells
2.6. The IGF1/IRS1 Pathway Affects Treg Cell Stability and Upregulates IFN-γ and Glucose Uptake
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Isolation of CD4+ T Cells and In Vitro Differentiation
4.3. Immunoblot Analysis
4.4. Intracellular Staining
4.5. Retroviral (RV) Transduction
4.6. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.7. In Vitro Suppression Assay
4.8. Inflammatory Bowel Disease Model
4.9. Phosphorylation Analysis
4.10. RNA-Seq and Data Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martinez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.E.; Freeman, G.J.; Mathis, D.; Benoist, C. CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J. Exp. Med. 2004, 199, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Josefowicz, S.Z.; Kas, A.; Chu, T.T.; Gavin, M.A.; Rudensky, A.Y. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 2007, 445, 936–940. [Google Scholar] [CrossRef]
- Lee, W.; Lee, G.R. Transcriptional regulation and development of regulatory T cells. Exp. Mol. Med. 2018, 50, e456. [Google Scholar] [CrossRef] [Green Version]
- Sharabi, A.; Tsokos, M.G.; Ding, Y.; Malek, T.R.; Klatzmann, D.; Tsokos, G.C. Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov. 2018, 17, 823–844. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.S.; Lee, H.M.; Lio, C.W. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 2012, 12, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Richards, E.; Srivastava, S.; Thomas, K.R.; Dudda, J.C.; Klonowski, K.D.; Campbell, D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014, 211, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, K.S.; Srivastava, S.; Stolley, J.M.; Campbell, D.J. Regulatory T-cell homeostasis: Steady-state maintenance and modulation during inflammation. Immunol. Rev. 2014, 259, 40–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.D.; Delgoffe, G.M. The mammalian target of rapamycin: Linking T cell differentiation, function, and metabolism. Immunity 2010, 33, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell. Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Shi, H.; Chapman, N.M.; Wen, J.; Guy, C.; Long, L.Y.; Dhungana, Y.; Rankin, S.; Pelletier, S.; Vogel, P.; Wang, H.; et al. Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity 2019, 51, 1012–1027.e7. [Google Scholar] [CrossRef]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef]
- Huang, H.L.; Long, L.Y.; Zhou, P.P.; Chapman, N.M.; Chi, H.B. mTOR signaling at the crossroads of environmental signals and T-cell fate decisions. Immunol. Rev. 2020, 295, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Patterson, S.J.; Speck, M.; Ehses, J.A.; Levings, M.K. Insulin inhibits IL-10-mediated regulatory T cell function: Implications for obesity. J. Immunol. 2014, 192, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Wong, C.K.; Han, J.M.; Orban, P.C.; Huang, Q.; Gillies, J.; Mojibian, M.; Gibson, W.T.; Levings, M.K. T reg-specific insulin receptor deletion prevents diet-induced and age-associated metabolic syndrome. J. Exp. Med. 2020, 217, e20191542. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Y.; Lin, S.H.; Li, N.; Han, Y.; Huang, Q.; Zhao, Y.; Xie, F.; Guo, Y.; Deng, B.; et al. Insulin signaling establishes a developmental trajectory of adipose regulatory T cells. Nat. Immunol. 2021, 22, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- DiToro, D.; Harbour, S.N.; Bando, J.K.; Benavides, G.; Witte, S.; Laufer, V.A.; Moseley, C.; Singer, J.R.; Frey, B.; Turner, H.; et al. Insulin-Like Growth Factors Are Key Regulators of T Helper 17 Regulatory T Cell Balance in Autoimmunity. Immunity 2020, 52, 650–667.e10. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Sun, X.J.; White, M.F. The IRS-1 signaling system. Trends Biochem. Sci. 1994, 19, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Machado-Neto, J.A.; Fenerich, B.A.; Rodrigues Alves, A.P.N.; Fernandes, J.C.; Scopim-Ribeiro, R.; Coelho-Silva, J.L.; Traina, F. Insulin Substrate Receptor (IRS) proteins in normal and malignant hematopoiesis. Clinics 2018, 73, e566s. [Google Scholar] [CrossRef]
- Mardilovich, K.; Pankratz, S.L.; Shaw, L.M. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun. Signal. 2009, 7, 14. [Google Scholar] [CrossRef]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef]
- Li, D.Y.; Xiong, X.Z. ICOS(+) Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, B.; Rui, K.; Wang, S. The Role of GITR/GITRL Interaction in Autoimmune Diseases. Front. Immunol. 2020, 11, 588682. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.S.K. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Passerini, L.; Allan, S.E.; Battaglia, M.; Di Nunzio, S.; Alstad, A.N.; Levings, M.K.; Roncarolo, M.G.; Bacchetta, R. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25- effector T cells. Int. Immunol. 2008, 20, 421–431. [Google Scholar] [CrossRef]
- Davey, H.W.; Xie, T.; McLachlan, M.J.; Wilkins, R.J.; Waxman, D.J.; Grattan, D.R. STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology 2001, 142, 3836–3841. [Google Scholar] [CrossRef]
- Woelfle, J.; Billiard, J.; Rotwein, P. Acute control of insulin-like growth factor-I gene transcription by growth hormone through Stat5b. J. Biol. Chem. 2003, 278, 22696–22702. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yao, S.; Dann, S.M.; Qin, H.; Elson, C.O.; Cong, Y. ERK differentially regulates Th17- and Treg-cell development and contributes to the pathogenesis of colitis. Eur. J. Immunol. 2013, 43, 1716–1726. [Google Scholar] [CrossRef]
- Krook, A.; Zierath, J.R. Specificity of insulin signalling in human skeletal muscle as revealed by small interfering RNA. Diabetologia 2009, 52, 1231–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Multhoff, G.; Vaupel, P. Lactate-avid regulatory T cells: Metabolic plasticity controls immunosuppression in tumour microenvironment. Signal Transduct. Target Ther. 2021, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Handu, S.; Workman, C.J.; Vignali, D.A.A. Interferon gamma production by regulatory T cells is required for response to cancer immunotherapy. J. Immunol. 2020, 204, 244. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-gamma Drives Treg Fragility to Promote Anti-tumor Immunity. Cell 2017, 169, 1130–1141.e11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Luo, Y.; Ding, X.F.; Wang, C.Q.; Yang, X.X.; Liu, M.L. Adipocyte mTORC1 Suppresses Treg Cell Development and Browning of White Adipose Tissue through CRTC2/COX-2/PGs Pathway. Diabetes 2018, 67, 34-OR. [Google Scholar] [CrossRef]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2019, 9, 1–58. [Google Scholar] [CrossRef]
- Lee, W.H.; Jang, S.W.; Kim, H.S.; Kim, S.H.; Heo, J.I.; Kim, G.E.; Lee, G.R. BATF3 is sufficient for the induction of Il9 expression and can compensate for BATF during Th9 cell differentiation. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.H.; Kim, G.E.; Hong, K.J.; Kim, H.S.; Lee, G.R. Insulin Receptor Substrate 1 Signaling Inhibits Foxp3 Expression and Suppressive Functions in Treg Cells through the mTORC1 Pathway. Int. J. Mol. Sci. 2023, 24, 2551. https://doi.org/10.3390/ijms24032551
Lee WH, Kim GE, Hong KJ, Kim HS, Lee GR. Insulin Receptor Substrate 1 Signaling Inhibits Foxp3 Expression and Suppressive Functions in Treg Cells through the mTORC1 Pathway. International Journal of Molecular Sciences. 2023; 24(3):2551. https://doi.org/10.3390/ijms24032551
Chicago/Turabian StyleLee, Woo Ho, Ga Eul Kim, Kyung Jin Hong, Hyeong Su Kim, and Gap Ryol Lee. 2023. "Insulin Receptor Substrate 1 Signaling Inhibits Foxp3 Expression and Suppressive Functions in Treg Cells through the mTORC1 Pathway" International Journal of Molecular Sciences 24, no. 3: 2551. https://doi.org/10.3390/ijms24032551