

Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential

 , ,
, ,  and
and 
Abstract
1. Introduction
2. The overview of Ketone Body Metabolism
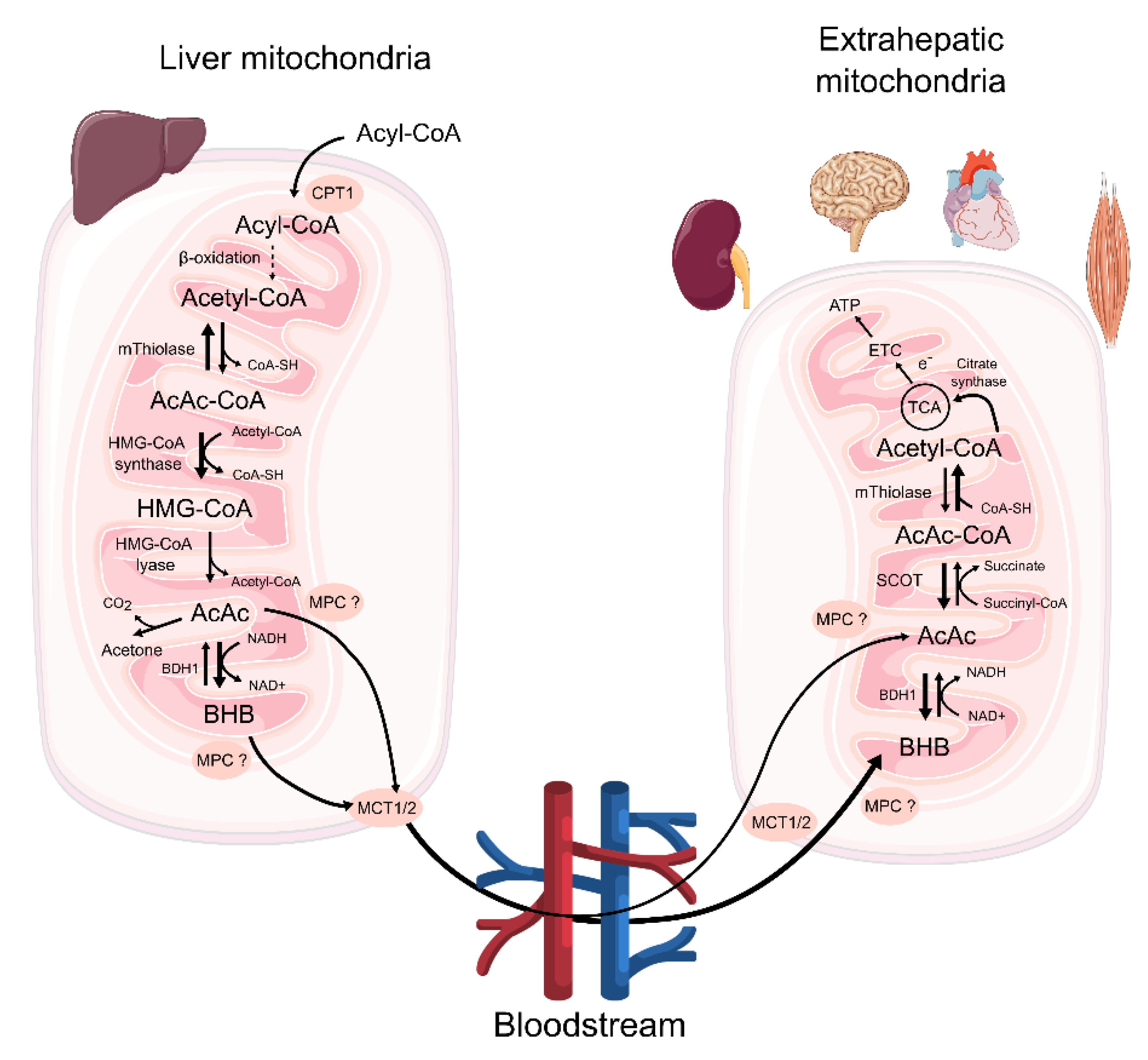
3. The Availability of Ketone Bodies for Extrahepatic Tissues
4. Therapeutic Ketogenic Interventions
4.1. Types of KD
4.2. Association of KD and CR
4.3. KD Mimetics
4.4. Limitations of KD Mimetics Administration
4.4.1. Alterations in Macronutrients Intake
4.4.2. The Influence on Microbiota
4.4.3. The Difference in KD and Mimetic Treatment Duration
4.4.4. Cell Model Limitation
5. Effects of KD and Ketone Bodies on Ischemic Damage
5.1. Cerebral IR Injury
5.2. Renal IR Injury
5.3. Myocardial IR Injury
6. Possible Mechanisms of the Beneficial Effects of KD on IR injury
6.1. Induction of Anti-Inflammatory Response
6.2. Attenuation of Oxidative Stress
6.3. Epigenetic Regulation
6.4. Energy Supply Restoration and Metabolic Adaptations
6.5. Effects on Mitochondria
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- D’Andrea Meira, I.; Romão, T.T.; Pires do Prado, H.J.; Krüger, L.T.; Pires, M.E.P.; da Conceição, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Peterman, M.G. The Ketogenic Diet in Epilepsy. JAMA 1925, 84, 1979–1983. [Google Scholar] [CrossRef]
- Martin, K.; Jackson, C.F.; Levy, R.G.; Cooper, P.N. Ketogenic Diet and Other Dietary Treatments for Epilepsy. Cochrane Database Syst. Rev. 2016, 2, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The Ketogenic Diet for the Treatment of Childhood Epilepsy: A Randomised Controlled Trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Sourbron, J.; Klinkenberg, S.; van Kuijk, S.M.J.; Lagae, L.; Lambrechts, D.; Braakman, H.M.H.; Majoie, M. Ketogenic Diet for the Treatment of Pediatric Epilepsy: Review and Meta-Analysis. Childs. Nerv. Syst. 2020, 36, 1099–1109. [Google Scholar] [CrossRef]
- Guelpa, G. A Lutte Contre L’epilepsie Par La Desintoxication et Par La Reeducation Alimentaire. Rev. Ther. Med. Chir. 1911, 78, 8–13. [Google Scholar]
- Höhn, S.; Dozières-Puyravel, B.; Auvin, S. History of Dietary Treatment: Guelpa & Marie First Report of Intermittent Fasting for Epilepsy in 1911. Epilepsy Behav. 2019, 94, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Rm, W. The Effects of Ketonemia on the Course of Epilepsy. Mayo Clin. Proc. 1921, 2, 307–308. [Google Scholar]
- Wilder, R.M. High Fat Diets in Epilepsy. Mayo Clin. Bull. 1921, 2, 308. [Google Scholar]
- Höhn, S.; Dozières-Puyravel, B.; Auvin, S. History of Dietary Treatment from Wilder’s Hypothesis to the First Open Studies in the 1920s. Epilepsy Behav. 2019, 101, 106588. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic Diet in the Treatment of Cancer—Where Do We Stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic Diet in Cancer Therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef]
- Sremanakova, J.; Sowerbutts, A.M.; Burden, S. A Systematic Review of the Use of Ketogenic Diets in Adult Patients with Cancer. J. Hum. Nutr. Diet. 2018, 31, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Klement, R.J. The Emerging Role of Ketogenic Diets in Cancer Treatment. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 129–134. [Google Scholar] [CrossRef]
- Talib, W.H.; Mahmod, A.I.; Kamal, A.; Rashid, H.M.; Alashqar, A.M.D.; Khater, S.; Jamal, D.; Waly, M. Ketogenic Diet in Cancer Prevention and Therapy: Molecular Targets and Therapeutic Opportunities. Curr. Issues Mol. Biol. 2021, 43, 558–589. [Google Scholar] [CrossRef] [PubMed]
- Castellana, M.; Conte, E.; Cignarelli, A.; Perrini, S.; Giustina, A.; Giovanella, L.; Giorgino, F.; Trimboli, P. Efficacy and Safety of Very Low Calorie Ketogenic Diet (VLCKD) in Patients with Overweight and Obesity: A Systematic Review and Meta-Analysis. Rev. Endocr. Metab. Disord. 2020, 21, 5–16. [Google Scholar] [CrossRef]
- Choi, Y.J.; Jeon, S.-M.; Shin, S. Impact of a Ketogenic Diet on Metabolic Parameters in Patients with Obesity or Overweight and with or without Type 2 Diabetes: A Meta-Analysis of Randomized Controlled Trials. Nutrients 2020, 12, 2005. [Google Scholar] [CrossRef] [PubMed]
- Bruci, A.; Tuccinardi, D.; Tozzi, R.; Balena, A.; Santucci, S.; Frontani, R.; Mariani, S.; Basciani, S.; Spera, G.; Gnessi, L.; et al. Very Low-Calorie Ketogenic Diet: A Safe and Effective Tool for Weight Loss in Patients With Obesity and Mild Kidney Failure. Nutrients 2020, 12, 333. [Google Scholar] [CrossRef]
- Casanueva, F.F.; Castellana, M.; Bellido, D.; Trimboli, P.; Castro, A.I.; Sajoux, I.; Rodriguez-Carnero, G.; Gomez-Arbelaez, D.; Crujeiras, A.B.; Martinez-Olmos, M.A. Ketogenic Diets as Treatment of Obesity and Type 2 Diabetes Mellitus. Rev. Endocr. Metab. Disord. 2020, 21, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Yancy, W.S., Jr.; Olsen, M.K.; Guyton, J.R.; Bakst, R.P.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet versus a Low-Fat Diet to Treat Obesity and Hyperlipidemia: A Randomized, Controlled Trial. Ann. Intern. Med. 2004, 140, 769–777. [Google Scholar] [CrossRef]
- Dashti, H.M.; Mathew, T.C.; Hussein, T.; Asfar, S.K.; Behbahani, A.; Khoursheed, M.A.; Al-Sayer, H.M.; Bo-Abbas, Y.Y.; Al-Zaid, N.S. Long-Term Effects of a Ketogenic Diet in Obese Patients. Exp. Clin. Cardiol. 2004, 9, 200–205. [Google Scholar] [PubMed]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and Efficacy Data from a Ketogenic Diet Intervention in Alzheimer’s Disease. Alzheimers. Dement. 2018, 4, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The Ketogenic Diet as a Potential Treatment and Prevention Strategy for Alzheimer’s Disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-Fat versus Ketogenic Diet in Parkinson’s Disease: A Pilot Randomized Controlled Trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef]
- Shaafi, S.; Najmi, S.; Aliasgharpour, H.; Mahmoudi, J.; Sadigh-Etemad, S.; Farhoudi, M.; Baniasadi, N. The Efficacy of the Ketogenic Diet on Motor Functions in Parkinson’s Disease: A Rat Model. Iran J. Neurol. 2016, 15, 63–69. [Google Scholar]
- Mobbs, C.V.; Mastaitis, J.; Isoda, F.; Poplawski, M. Treatment of Diabetes and Diabetic Complications with a Ketogenic Diet. J. Child Neurol. 2013, 28, 1009–1014. [Google Scholar] [CrossRef]
- Yancy, W.S., Jr.; Foy, M.; Chalecki, A.M.; Vernon, M.C.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet to Treat Type 2 Diabetes. Nutr. Metab. 2005, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Bolla, A.M.; Caretto, A.; Laurenzi, A.; Scavini, M.; Piemonti, L. Low-Carb and Ketogenic Diets in Type 1 and Type 2 Diabetes. Nutrients 2019, 11, 962. [Google Scholar] [CrossRef]
- Rojas-Morales, P.; León-Contreras, J.C.; Sánchez-Tapia, M.; Silva-Palacios, A.; Cano-Martínez, A.; González-Reyes, S.; Jiménez-Osorio, A.S.; Hernández-Pando, R.; Osorio-Alonso, H.; Sánchez-Lozada, L.G.; et al. A Ketogenic Diet Attenuates Acute and Chronic Ischemic Kidney Injury and Reduces Markers of Oxidative Stress and Inflammation. Life Sci. 2022, 289, 120227. [Google Scholar] [CrossRef]
- Thau-Zuchman, O.; Svendsen, L.; Dyall, S.C.; Paredes-Esquivel, U.; Rhodes, M.; Priestley, J.V.; Feichtinger, R.G.; Kofler, B.; Lotstra, S.; Verkuyl, J.M.; et al. A New Ketogenic Formulation Improves Functional Outcome and Reduces Tissue Loss Following Traumatic Brain Injury in Adult Mice. Theranostics 2021, 11, 346–360. [Google Scholar] [CrossRef]
- Guo, M.; Wang, X.; Zhao, Y.; Yang, Q.; Ding, H.; Dong, Q.; Chen, X.; Cui, M. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2018, 11, 86. [Google Scholar] [CrossRef]
- Xu, K.; Ye, L.; Sharma, K.; Jin, Y.; Harrison, M.M.; Caldwell, T.; Berthiaume, J.M.; Luo, Y.; LaManna, J.C.; Puchowicz, M.A. Diet-Induced Ketosis Protects Against Focal Cerebral Ischemia in Mouse. Adv. Exp. Med. Biol. 2017, 977, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guo, M.; Wang, X.; Zhao, Y.; Zhao, Q.; Ding, H.; Dong, Q.; Cui, M. Ischemic Preconditioning with a Ketogenic Diet Improves Brain Ischemic Tolerance through Increased Extracellular Adenosine Levels and Hypoxia-Inducible Factors. Brain Res. 2017, 1667, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and Reperfusion—from Mechanism to Translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Aluja, D.; Barba, I.; Ruiz-Meana, M.; Miró, E.; Poncelas, M.; Vilardosa, Ú.; Castellano, J.; Garcia-Dorado, D. High-Fat Diet Improves Tolerance to Myocardial Ischemia by Delaying Normalization of Intracellular PH at Reperfusion. J. Mol. Cell. Cardiol. 2019, 133, 164–173. [Google Scholar] [CrossRef]
- Rickenbacher, A.; Jang, J.H.; Limani, P.; Ungethüm, U.; Lehmann, K.; Oberkofler, C.E.; Weber, A.; Graf, R.; Humar, B.; Clavien, P.-A. Fasting Protects Liver from Ischemic Injury through Sirt1-Mediated Downregulation of Circulating HMGB1 in Mice. J. Hepatol. 2014, 61, 301–308. [Google Scholar] [CrossRef]
- Guo, Z.; Rousselle, T.; Li, J.I. Caloric Restriction Increases the Resistance of Aged Heart to Myocardial Ischemia/Reperfusion Injury. Diabetes 2018, 67, 153. [Google Scholar] [CrossRef]
- Kamarauskaite, J.; Baniene, R.; Trumbeckas, D.; Strazdauskas, A.; Trumbeckaite, S. Caffeic Acid Phenethyl Ester Protects Kidney Mitochondria against Ischemia/Reperfusion Induced Injury in an In Vivo Rat Model. Antioxidants 2021, 10, 747. [Google Scholar] [CrossRef] [PubMed]
- Skemiene, K.; Liobikas, J.; Borutaite, V. Anthocyanins as Substrates for Mitochondrial Complex I—Protective Effect against Heart Ischemic Injury. FEBS J. 2015, 282, 963–971. [Google Scholar] [CrossRef]
- Boulghobra, D.; Coste, F.; Geny, B.; Reboul, C. Exercise Training Protects the Heart against Ischemia-Reperfusion Injury: A Central Role for Mitochondria? Free Radic. Biol. Med. 2020, 152, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Metabolic and Signaling Roles of Ketone Bodies in Health and Disease. Annu. Rev. Nutr. 2021, 41, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.K.; Ittmann, M.; Cooper, C. The Role of Leucine in Ketogenesis in Starved Rats. Biochem. J. 1982, 204, 399–403. [Google Scholar] [CrossRef]
- Litwack, G. Metabolism of amino acids. In Human Biochemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 359–394. ISBN 9780123838643. [Google Scholar]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial Effects of the Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Comprehensive Review of the Literature. Obes. Rev. 2020, 21, e13024. [Google Scholar] [CrossRef]
- Arnedo, M.; Latorre-Pellicer, A.; Lucia-Campos, C.; Gil-Salvador, M.; Antoñanzas-Peréz, R.; Gómez-Puertas, P.; Bueno-Lozano, G.; Puisac, B.; Pié, J. More Than One HMG-CoA Lyase: The Classical Mitochondrial Enzyme Plus the Peroxisomal and the Cytosolic Ones. Int. J. Mol. Sci. 2019, 20, 6124. [Google Scholar] [CrossRef]
- Dhillon, K.K.; Gupta, S. Biochemistry, ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Liemburg-Apers, D.C.; Imamura, H.; Forkink, M.; Nooteboom, M.; Swarts, H.G.; Brock, R.; Smeitink, J.A.M.; Willems, P.H.G.M.; Koopman, W.J.H. Quantitative Glucose and ATP Sensing in Mammalian Cells. Pharm. Res. 2011, 28, 2745–2757. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying Intracellular Rates of Glycolytic and Oxidative ATP Production and Consumption Using Extracellular Flux Measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The Monocarboxylate Transporter Family--Structure and Functional Characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The Monocarboxylate Transporter Family--Role and Regulation. IUBMB Life 2012, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate Transporters in the Central Nervous System: Distribution, Regulation and Function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, A.; Crone, C. Induction Processes in Blood-Brain Transfer of Ketone Bodies during Starvation. Am. J. Physiol. 1975, 229, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-Induced Ketosis Increases Monocarboxylate Transporter (MCT1) Levels in Rat Brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Jakobsen, J.; Hageman, L.P.; Holm, S.; Paulson, O.B. Brain Metabolism during Short-Term Starvation in Humans. J. Cereb. Blood Flow Metab. 1994, 14, 125–131. [Google Scholar] [CrossRef]
- Pan, J.W.; Rothman, T.L.; Behar, K.L.; Stein, D.T.; Hetherington, H.P. Human Brain Beta-Hydroxybutyrate and Lactate Increase in Fasting-Induced Ketosis. J. Cereb. Blood Flow Metab. 2000, 20, 1502–1507. [Google Scholar] [CrossRef]
- Pan, J.W.; Telang, F.W.; Lee, J.H.; de Graaf, R.A.; Rothman, D.L.; Stein, D.T.; Hetherington, H.P. Measurement of Beta-Hydroxybutyrate in Acute Hyperketonemia in Human Brain. J. Neurochem. 2001, 79, 539–544. [Google Scholar] [CrossRef]
- Barry, D.; Ellul, S.; Watters, L.; Lee, D.; Haluska, R., Jr.; White, R. The Ketogenic Diet in Disease and Development. Int. J. Dev. Neurosci. 2018, 68, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Gentz, J. The Pattern of Blood Lipids, Glycerol and Ketone Bodies during the Neonatal Period, Infancy and Childhood. Acta Paediatr. Scand. 1966, 55, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Crawford, M.A. Survival of the Fattest: Fat Babies Were the Key to Evolution of the Large Human Brain. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003, 136, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C. Ketones, Omega-3 Fatty Acids and the Yin-Yang Balance in the Brain: Insights from Infant Development and Alzheimer’s Disease, and Implications for Human Brain Evolution. OCL 2018, 25, D409. [Google Scholar] [CrossRef]
- Song, F.; Yamaguchi, M. Enzymes of Ketone Body Utilization in Human Tissues: Protein and Messenger RNA Levels of Succinyl-Coenzyme A (CoA): 3-Ketoacid CoA Transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr. Res. 1997, 42, 498–502. [Google Scholar]
- Orii, K.E.; Fukao, T.; Song, X.-Q.; Mitchell, G.A.; Kondo, N. Liver-Specific Silencing of the Human Gene Encoding Succinyl-CoA: 3-Ketoacid CoA Transferase. Tohoku J. Exp. Med. 2008, 215, 227–236. [Google Scholar] [CrossRef]
- Williamson, D.H.; Bates, M.W.; Page, M.A.; Krebs, H.A. Activities of Enzymes Involved in Acetoacetate Utilization in Adult Mammalian Tissues. Biochem. J. 1971, 121, 41–47. [Google Scholar] [CrossRef]
- Zhang, W.W.; Lindahl, R.; Churchill, P. Regulation of Succinyl Coenzyme A:acetoacetyl Coenzyme A Transferase in Rat Hepatoma Cell Lines. Cancer Res. 1990, 50, 5858–5862. [Google Scholar]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for Substrate Utilization in Oxidative Metabolism by Neurons, Astrocytes, and Oligodendrocytes from Developing Brain in Primary Culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Le Foll, C.; Dunn-Meynell, A.A.; Miziorko, H.M.; Levin, B.E. Regulation of Hypothalamic Neuronal Sensing and Food Intake by Ketone Bodies and Fatty Acids. Diabetes 2014, 63, 1259–1269. [Google Scholar] [CrossRef]
- Blázquez, C.; Woods, A.; de Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-Activated Protein Kinase Is Involved in the Regulation of Ketone Body Production by Astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-Chain Fatty Acids Inhibit Mitochondrial Metabolism in Astrocytes Promoting Astrocyte-Neuron Lactate and Ketone Body Shuttle Systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Fan, T.W.-M.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis Contributes to Intestinal Cell Differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, H.; Kong, X.; Wang, K.; Mao, X.; Yan, X.; Wang, Y.; Liu, S.; Zhang, X.; Li, J.; et al. Proteomics Analysis Reveals Diabetic Kidney as a Ketogenic Organ in Type 2 Diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E287–E295. [Google Scholar] [CrossRef]
- Takagi, A.; Kume, S.; Kondo, M.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.-I.; Koya, D.; Haneda, M.; Chano, T.; et al. Mammalian Autophagy Is Essential for Hepatic and Renal Ketogenesis during Starvation. Sci. Rep. 2016, 6, 18944. [Google Scholar] [CrossRef]
- Barzegar, M.; Afghan, M.; Tarmahi, V.; Behtari, M.; Rahimi Khamaneh, S.; Raeisi, S. Ketogenic Diet: Overview, Types, and Possible Anti-Seizure Mechanisms. Nutr. Neurosci. 2021, 24, 307–316. [Google Scholar] [CrossRef]
- Caprio, M.; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; Migliaccio, S.; et al. Very-Low-Calorie Ketogenic Diet (VLCKD) in the Management of Metabolic Diseases: Systematic Review and Consensus Statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Invest. 2019, 42, 1365–1386. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Helen Cross, J.; Dahlin, M.G.; et al. Optimal Clinical Management of Children Receiving the Ketogenic Diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.M.; Vining, E.P.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The Efficacy of the Ketogenic Diet-1998: A Prospective Evaluation of Intervention in 150 Children. Pediatrics 1998, 102, 1358–1363. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Helen Cross, J.; Dahlin, M.G.; et al. Optimal Clinical Management of Children Receiving Dietary Therapies for Epilepsy: Updated Recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Y.; Wang, Y.; Tang, H.; Zhang, F.; Zhang, Y.; Zhao, Y. Ketogenic Diet for Treatment of Intractable Epilepsy in Adults: A Meta-Analysis of Observational Studies. Epilepsia Open 2018, 3, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Schon, H.; Lippack, I.; Gelpke, W. Mischglycerid Der Fettsauren Mittlerer Kettenlange. II. Untersuchungen Iiber Die Veranderungen Des Ketonkorpergehmaltes von Blut Und Urin Nach Zufuhr des Mischglycerides. Gastroenterologia 1959, 91, 199. [Google Scholar] [PubMed]
- Bergen, S.S., Jr.; Hashim, S.A.; Van Itallie, T.B. Hyperketonemia Induced in Man by Medium-Chain Triglyceride. Diabetes 1966, 15, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-Chain Triglycerides as a Therapy for Intractable Childhood Epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Iapadre, G.; Di Francesco, L.; Zagaroli, L.; Farello, G. Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients 2020, 12, 2645. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Krauss, G.L.; McGrogan, J.R.; Freeman, J.M. Efficacy of the Atkins Diet as Therapy for Intractable Epilepsy. Neurology 2003, 61, 1789–1791. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Dorward, J.L. The Modified Atkins Diet. Epilepsia 2008, 49 (Suppl. 8), 37–41. [Google Scholar] [CrossRef]
- Pfeifer, H.H.; Thiele, E.A. Low-Glycemic-Index Treatment: A Liberalized Ketogenic Diet for Treatment of Intractable Epilepsy. Neurology 2005, 65, 1810–1812. [Google Scholar] [CrossRef]
- Barrea, L.; Caprio, M.; Camajani, E.; Verde, L.; Elce, A.; Frias-Toral, E.; Ceriani, F.; Cucalón, G.; Garcia-Velasquez, E.; El Ghoch, M.; et al. Clinical and Nutritional Management of Very-Low-Calorie Ketogenic Diet (VLCKD) in Patients with Psoriasis and Obesity: A Practical Guide for the Nutritionist. Crit. Rev. Food Sci. Nutr. 2022, 62, 398–414. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Barrea, L.; Laudisio, D.; Pugliese, G.; Salzano, C.; Savastano, S.; Colao, A. The Management of Very Low-Calorie Ketogenic Diet in Obesity Outpatient Clinic: A Practical Guide. J. Transl. Med. 2019, 17, 356. [Google Scholar] [CrossRef] [PubMed]
- Goday, A.; Bellido, D.; Sajoux, I.; Crujeiras, A.B.; Burguera, B.; García-Luna, P.P.; Oleaga, A.; Moreno, B.; Casanueva, F.F. Short-Term Safety, Tolerability and Efficacy of a Very Low-Calorie-Ketogenic Diet Interventional Weight Loss Program versus Hypocaloric Diet in Patients with Type 2 Diabetes Mellitus. Nutr. Diabetes 2016, 6, e230. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, J.; Mohan, V. Ketogenic Diets: Boon or Bane? Indian J. Med. Res. 2018, 148, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Rakhra, V.; Galappaththy, S.L.; Bulchandani, S.; Cabandugama, P.K. Obesity and the Western Diet: How We Got Here. MO Med. 2020, 117, 536–538. [Google Scholar] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and Evolution of the Western Diet: Health Implications for the 21st Century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric Restriction Reduces Age-Related and All-Cause Mortality in Rhesus Monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of Caloric Restriction on Health and Survival in Rhesus Monkeys from the NIA Study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef]
- Singh, G.; Krishan, P. Dietary Restriction Regimens for Fighting Kidney Disease: Insights from Rodent Studies. Exp. Gerontol. 2019, 128, 110738. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Verweij, M.; Brand, K.; van de Ven, M.; Goemaere, N.; van den Engel, S.; Chu, T.; Forrer, F.; Müller, C.; de Jong, M.; et al. Short-Term Dietary Restriction and Fasting Precondition against Ischemia Reperfusion Injury in Mice. Aging Cell 2010, 9, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Andrianova, N.V.; Zorova, L.D.; Pevzner, I.B.; Popkov, V.A.; Chernikov, V.P.; Silachev, D.N.; Plotnikov, E.Y.; Zorov, D.B. Resemblance and Differences in Dietary Restriction Nephroprotective Mechanisms in Young and Old Rats. Aging 2020, 12, 18693–18715. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, B.; Nelson, J.F.; Colston, J.T.; Freeman, G.L. Calorie Restriction Attenuates Inflammatory Responses to Myocardial Ischemia-Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2094–H2102. [Google Scholar] [CrossRef]
- Ciobanu, O.; Elena Sandu, R.; Tudor Balseanu, A.; Zavaleanu, A.; Gresita, A.; Petcu, E.B.; Uzoni, A.; Popa-Wagner, A. Caloric Restriction Stabilizes Body Weight and Accelerates Behavioral Recovery in Aged Rats after Focal Ischemia. Aging Cell 2017, 16, 1394–1403. [Google Scholar] [CrossRef]
- Manzanero, S.; Gelderblom, M.; Magnus, T.; Arumugam, T.V. Calorie Restriction and Stroke. Exp. Transl. Stroke Med. 2011, 3, 8. [Google Scholar] [CrossRef]
- Menezes-Filho, S.L.; Amigo, I.; Prado, F.M.; Ferreira, N.C.; Koike, M.K.; Pinto, I.F.D.; Miyamoto, S.; Montero, E.F.S.; Medeiros, M.H.G.; Kowaltowski, A.J. Caloric Restriction Protects Livers from Ischemia/reperfusion Damage by Preventing Ca2+-Induced Mitochondrial Permeability Transition. Free Radic. Biol. Med. 2017, 110, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-T.; Mao, Y.-Q.; Han, B.; Zhang, Z.-Y.; Chen, H.-L.; Li, Z.-M.; Kong, C.-Y.; Xu, J.-Q.; Cai, P.-R.; Zeng, Y.-P.; et al. Calorie Restriction Conferred Improvement Effect on Long-Term Rehabilitation of Ischemic Stroke via Gut Microbiota. Pharmacol. Res. 2021, 170, 105726. [Google Scholar] [CrossRef] [PubMed]
- Balasse, E.O. Kinetics of Ketone Body Metabolism in Fasting Humans. Metabolism 1979, 28, 41–50. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef]
- Bueno, N.B.; de Melo, I.S.V.; de Oliveira, S.L.; da Rocha Ataide, T. Very-Low-Carbohydrate Ketogenic Diet v. Low-Fat Diet for Long-Term Weight Loss: A Meta-Analysis of Randomised Controlled Trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef]
- Paoli, A. Ketogenic Diet for Obesity: Friend or Foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.A.; Seimon, R.V.; Lee, C.M.Y.; Ayre, J.; Franklin, J.; Markovic, T.P.; Caterson, I.D.; Sainsbury, A. Do Ketogenic Diets Really Suppress Appetite? A Systematic Review and Meta-Analysis. Obes. Rev. 2015, 16, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Roekenes, J.; Martins, C. Ketogenic Diets and Appetite Regulation. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 359–363. [Google Scholar] [CrossRef]
- Deemer, S.E.; Plaisance, E.P.; Martins, C. Impact of Ketosis on Appetite Regulation-a Review. Nutr. Res. 2020, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.B.; Hill, C.M.; Bitto, A.; Kaeberlein, M. Antiaging Diets: Separating Fact from Fiction. Science 2021, 374, eabe7365. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; Zou, L.; Qu, J.; Litovsky, S.; Umeda, P.; Zhou, L.; Chatham, J.; Marsh, S.A.; Dell’Italia, L.J.; et al. High-Fat, Low-Carbohydrate Diet Promotes Arrhythmic Death and Increases Myocardial Ischemia-Reperfusion Injury in Rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H598–H608. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lloyd, S.G. High-Fat, Low-Carbohydrate Diet Alters Myocardial Oxidative Stress and Impairs Recovery of Cardiac Function after Ischemia and Reperfusion in Obese Rats. Nutr. Res. 2013, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Izuta, Y.; Imada, T.; Hisamura, R.; Oonishi, E.; Nakamura, S.; Inagaki, E.; Ito, M.; Soga, T.; Tsubota, K. Ketone Body 3-Hydroxybutyrate Mimics Calorie Restriction via the Nrf2 Activator, Fumarate, in the Retina. Aging Cell 2018, 17, e12699. [Google Scholar] [CrossRef]
- Robertson, C.; Goodman, J.C.; Grossman, R.G.; Claypool, M.; White, A. Dietary Nonprotein Calories and Cerebral Infarction Size in Rats. Stroke 1992, 23, 564–568. [Google Scholar] [CrossRef]
- Koch, K.; Berressem, D.; Konietzka, J.; Thinnes, A.; Eckert, G.P.; Klein, J. Hepatic Ketogenesis Induced by Middle Cerebral Artery Occlusion in Mice. J. Am. Heart Assoc. 2017, 6, e005556. [Google Scholar] [CrossRef]
- Xu, K.; Sun, X.; Eroku, B.O.; Tsipis, C.P.; Puchowicz, M.A.; LaManna, J.C. Diet-induced ketosis improves cognitive performance in aged rats. In Oxygen Transport to Tissue XXXI; Springer: Boston, MA, USA, 2010; pp. 71–75. [Google Scholar]
- Al-Zaid, N.S.; Dashti, H.M.; Mathew, T.C.; Juggi, J.S. Low Carbohydrate Ketogenic Diet Enhances Cardiac Tolerance to Global Ischaemia. Acta Cardiol. 2007, 62, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Rennison, J.H.; McElfresh, T.A.; Okere, I.C.; Patel, H.V.; Foster, A.B.; Patel, K.K.; Stoll, M.S.; Minkler, P.E.; Fujioka, H.; Hoit, B.D.; et al. Enhanced Acyl-CoA Dehydrogenase Activity Is Associated with Improved Mitochondrial and Contractile Function in Heart Failure. Cardiovasc. Res. 2008, 79, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Rennison, J.H.; McElfresh, T.A.; Okere, I.C.; Vazquez, E.J.; Patel, H.V.; Foster, A.B.; Patel, K.K.; Chen, Q.; Hoit, B.D.; Tserng, K.-Y.; et al. High-Fat Diet Postinfarction Enhances Mitochondrial Function and Does Not Exacerbate Left Ventricular Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1498–H1506. [Google Scholar] [CrossRef] [PubMed]
- Shalamu, A.; Dong, Z.; Liu, B.; Pan, L.; Cai, Y.; Liu, L.; Ma, X.; Hu, K.; Sun, A.; Ge, J. Effects of the Ketogenic Diet in Mice with Hind Limb Ischemia. Nutr. Metab. 2022, 19, 59. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dong, Z.; Liu, J.; Ma, L.; Sun, X.; Gao, R.; Pan, L.; Zhang, J.; Dilan, A.; An, J.; et al. β-Hydroxybutyrate Exacerbates Hypoxic Injury by Inhibiting HIF-1α-Dependent Glycolysis in Cardiomyocytes-Adding Fuel to the Fire? Cardiovasc. Drugs Ther. 2022, 36, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, A.; Rao, P.; Pranay, A.; Xu, K.; LaManna, J.C.; Puchowicz, M.A. Chronic ketosis modulates HIF1α-mediated inflammatory response in rat brain. In Oxygen Transport to Tissue XLII; Nemoto, E.M., Harrison, E.M., Pias, S.C., Bragin, D.E., Harrison, D.K., LaManna, J.C., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 3–7. ISBN 9783030482381. [Google Scholar]
- Shaafi, S.; Sharifi-Bonab, M.; Ghaemian, N.; Mokhtarkhani, M.; Akbari, H. Early Motor-Behavioral Outcome of Ischemic Stroke with Ketogenic Diet Preconditioning: Interventional Animal Study. J. Stroke Cerebrovasc. Dis. 2019, 28, 1032–1039. [Google Scholar] [CrossRef]
- Watanabe, T.; Fukatsu, K.; Murakoshi, S.; Mochizuki, S.; Higashizono, K.; Watkins, A.; Noguchi, M.; Yasuhara, H.; Yamamoto, J.; Ueno, H. Modulation of Nitric Oxide Production by a Low-Carbohydrate High -Fat Diet in a Murine Gut Ischemia Reperfusion Model: Mechanism Possibly Underlying Improved Survival. Clin. Nutr. 2018, 37, S43–S44. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Douglas, S.L.; Tate, J.M.; Sham, S.; Lloyd, S.G. Impact of High-Fat, Low-Carbohydrate Diet on Myocardial Substrate Oxidation, Insulin Sensitivity, and Cardiac Function after Ischemia-Reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1–H10. [Google Scholar] [CrossRef]
- Watanabe, T.; Fukatsu, K.; Murakoshi, S.; Yamamoto, J.; Hase, K.; Yasuhara, H. SUN-PP224: The Influence of Short-Term Low Carbohydratehigh Fat Diet on Survival After Gut Ischemia Reperfusion in Mice. Clin. Nutr. 2015, 34, S106. [Google Scholar] [CrossRef]
- Tai, K.-K.; Nguyen, N.; Pham, L.; Truong, D.D. Ketogenic Diet Prevents Cardiac Arrest-Induced Cerebral Ischemic Neurodegeneration. J. Neural Transm. 2008, 115, 1011–1017. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Zechel, J.L.; Valerio, J.; Emancipator, D.S.; Xu, K.; Pundik, S.; LaManna, J.C.; Lust, W.D. Neuroprotection in Diet-Induced Ketotic Rat Brain after Focal Ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Lehto, A.; Koch, K.; Barnstorf-Brandes, J.; Viel, C.; Fuchs, M.; Klein, J. ß-Hydroxybutyrate Improves Mitochondrial Function After Transient Ischemia in the Mouse. Neurochem. Res. 2022, 47, 3241–3249. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Ma, A.; Kang, Y. Rational Application of β-Hydroxybutyrate Attenuates Ischemic Stroke by Suppressing Oxidative Stress and Mitochondrial-Dependent Apoptosis via Activation of the Erk/CREB/eNOS Pathway. ACS Chem. Neurosci. 2021, 12, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, T.; Uchida, Y.; Kadono, K.; Hirao, H.; Kawasoe, J.; Watanabe, T.; Ueda, S.; Okajima, H.; Terajima, H.; Uemoto, S. Up-Regulation of FOXO1 and Reduced Inflammation by β-Hydroxybutyric Acid Are Essential Diet Restriction Benefits against Liver Injury. Proc. Natl. Acad. Sci. USA 2019, 116, 13533–13542. [Google Scholar] [CrossRef]
- Tajima, T.; Yoshifuji, A.; Matsui, A.; Itoh, T.; Uchiyama, K.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. β-Hydroxybutyrate Attenuates Renal Ischemia-Reperfusion Injury through Its Anti-Pyroptotic Effects. Kidney Int. 2019, 95, 1120–1137. [Google Scholar] [CrossRef]
- Lee, B.S.; Woo, D.-C.; Woo, C.-W.; Kim, K.-S. Exogenous β-Hydroxybutyrate Treatment and Neuroprotection in a Suckling Rat Model of Hypoxic-Ischemic Encephalopathy. Dev. Neurosci. 2018, 40, 73–83. [Google Scholar] [CrossRef]
- Bazzigaluppi, P.; Lake, E.M.; Beckett, T.L.; Koletar, M.M.; Weisspapir, I.; Heinen, S.; Mester, J.; Lai, A.; Janik, R.; Dorr, A.; et al. Imaging the Effects of β-Hydroxybutyrate on Peri-Infarct Neurovascular Function and Metabolism. Stroke 2018, 49, 2173–2181. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, Y.; Zhang, Y.; Zhang, Z.; An, W.; Zhao, X. Treatment with D-β-Hydroxybutyrate Protects Heart from Ischemia/reperfusion Injury in Mice. Eur. J. Pharmacol. 2018, 829, 121–128. [Google Scholar] [CrossRef]
- Qian, J.; Zhu, W.; Lu, M.; Ni, B.; Yang, J. D-β-Hydroxybutyrate Promotes Functional Recovery and Relieves Pain Hypersensitivity in Mice with Spinal Cord Injury. Br. J. Pharmacol. 2017, 174, 1961–1971. [Google Scholar] [CrossRef]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 Mediates Neuroprotection of Ketones against Ischemic Stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef]
- Massieu, L.; Haces, M.L.; Montiel, T.; Hernández-Fonseca, K. Acetoacetate Protects Hippocampal Neurons against Glutamate-Mediated Neuronal Damage during Glycolysis Inhibition. Neuroscience 2003, 120, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Sasaguri, S.; Rajesh, K.G.; Suzuki, R. Dl-3-Hydroxybutyrate Administration Prevents Myocardial Damage after Coronary Occlusion in Rat Hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1968–H1974. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Suzuki, M.; Kitamura, Y.; Mori, S.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Beta-Hydroxybutyrate, a Cerebral Function Improving Agent, Protects Rat Brain against Ischemic Damage Caused by Permanent and Transient Focal Cerebral Ischemia. Jpn. J. Pharmacol. 2002, 89, 36–43. [Google Scholar] [CrossRef]
- Suzuki, M.; Suzuki, M.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Effect of Beta-Hydroxybutyrate, a Cerebral Function Improving Agent, on Cerebral Hypoxia, Anoxia and Ischemia in Mice and Rats. Jpn. J. Pharmacol. 2001, 87, 143–150. [Google Scholar] [CrossRef]
- Gueldry, S.; Marie, C.; Rochette, L.; Bralet, J. Beneficial Effect of 1,3-Butanediol on Cerebral Energy Metabolism and Edema Following Brain Embolization in Rats. Stroke 1990, 21, 1458–1463. [Google Scholar] [CrossRef]
- Lammerant, J.; Huynh-Thu, T.; Kolanowski, J. Stabilization of Left Ventricular Function with D-(-)-3-Hydroxybutyrate after Coronary Occlusion in the Intact Dog. J. Mol. Cell Cardiol. 1988, 20, 579–583. [Google Scholar] [CrossRef]
- Eiger, S.M.; Kirsch, J.R.; D’Alecy, L.G. Hypoxic Tolerance Enhanced by Beta-Hydroxybutyrate-Glucagon in the Mouse. Stroke 1980, 11, 513–517. [Google Scholar] [CrossRef]
- Bergqvist, A.G.C.; Schall, J.I.; Gallagher, P.R.; Cnaan, A.; Stallings, V.A. Fasting versus Gradual Initiation of the Ketogenic Diet: A Prospective, Randomized Clinical Trial of Efficacy. Epilepsia 2005, 46, 1810–1819. [Google Scholar] [CrossRef]
- Kossoff, E.H.; McGrogan, J.R.; Bluml, R.M.; Pillas, D.J.; Rubenstein, J.E.; Vining, E.P. A Modified Atkins Diet Is Effective for the Treatment of Intractable Pediatric Epilepsy. Epilepsia 2006, 47, 421–424. [Google Scholar] [CrossRef]
- Carrette, E.; Vonck, K.; de Herdt, V.; Dewaele, I.; Raedt, R.; Goossens, L.; Van Zandijcke, M.; Wadman, W.; Thadani, V.; Boon, P. A Pilot Trial with Modified Atkins’ Diet in Adult Patients with Refractory Epilepsy. Clin. Neurol. Neurosurg. 2008, 110, 797–803. [Google Scholar] [CrossRef]
- Rezaei, S.; Harsini, S.; Kavoosi, M.; Badv, R.S.; Mahmoudi, M. Efficacy of Low Glycemic Index Treatment in Epileptic Patients: A Systematic Review. Acta Neurol. Belg. 2018, 118, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Desrochers, S.; David, F.; Garneau, M.; Jetté, M.; Brunengraber, H. Metabolism of R- and S-1,3-Butanediol in Perfused Livers from Meal-Fed and Starved Rats. Biochem. J. 1992, 285 Pt 2, 647–653. [Google Scholar] [CrossRef]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T. A New Way to Produce Hyperketonemia: Use of Ketone Ester in a Case of Alzheimer’s Disease. Alzheimers. Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Brunengraber, H. Potential of Ketone Body Esters for Parenteral and Oral Nutrition. Nutrition 1997, 13, 233–235. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Norwitz, N.G.; Clarke, K. Why a D-β-Hydroxybutyrate Monoester? Biochem. Soc. Trans. 2020, 48, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; VanItallie, T.B.; et al. Kinetics, Safety and Tolerability of (R)-3-Hydroxybutyl (R)-3-Hydroxybutyrate in Healthy Adult Subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, S.W.; et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef]
- Kawamura, M., Jr.; Ruskin, D.N.; Masino, S.A. Metabolic Autocrine Regulation of Neurons Involves Cooperation among Pannexin Hemichannels, Adenosine Receptors, and KATP Channels. J. Neurosci. 2010, 30, 3886–3895. [Google Scholar] [CrossRef]
- Spindler, S.R. Caloric Restriction: From Soup to Nuts. Ageing Res. Rev. 2010, 9, 324–353. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Lazarow, A.; Luhovyy, B.; Wehrli, S.; Nissim, I. Response of Brain Amino Acid Metabolism to Ketosis. Neurochem. Int. 2005, 47, 119–128. [Google Scholar] [CrossRef]
- Dahlin, M.; Elfving, A.; Ungerstedt, U.; Amark, P. The Ketogenic Diet Influences the Levels of Excitatory and Inhibitory Amino Acids in the CSF in Children with Refractory Epilepsy. Epilepsy Res. 2005, 64, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.J.; Linder, B.A.; Hester, S.; Fontes, M.; Pernomian, L.; Wenceslau, C.F.; Robinson, A.T.; McCarthy, C.G. The Janus Face of Ketone Bodies in Hypertension. J. Hypertens. 2022, 40, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Poff, A.M.; Koutnik, A.P.; Egan, B. Nutritional Ketosis with Ketogenic Diets or Exogenous Ketones: Features, Convergence, and Divergence. Curr. Sports Med. Rep. 2020, 19, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhou, Q.; Qiu, C.-Z.; Dai, W.-K.; Wang, H.-P.; Li, Y.-H.; Liao, J.-X.; Lu, X.-G.; Lin, S.-F.; Ye, J.-H.; et al. Ketogenic Diet Poses a Significant Effect on Imbalanced Gut Microbiota in Infants with Refractory Epilepsy. World J. Gastroenterol. 2017, 23, 6164–6171. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 174, 497. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Iñiguez, A.J.; Yang, G.E.; Fang, P.; Pronovost, G.N.; Jameson, K.G.; Rendon, T.K.; Paramo, J.; Barlow, J.T.; Ismagilov, R.F.; et al. Alterations in the Gut Microbiota Contribute to Cognitive Impairment Induced by the Ketogenic Diet and Hypoxia. Cell Host Microbe 2021, 29, 1378–1392.e6. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Kiebish, M.A.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Metabolic Management of Brain Cancer. Biochim. Biophys. Acta 2011, 1807, 577–594. [Google Scholar] [CrossRef]
- Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tisdale, M.J.; Brennan, R.A. Loss of Acetoacetate Coenzyme A Transferase Activity in Tumours of Peripheral Tissues. Br. J. Cancer 1983, 47, 293–297. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Mukherjee, P. Targeting Energy Metabolism in Brain Cancer: Review and Hypothesis. Nutr. Metab. 2005, 2, 30. [Google Scholar] [CrossRef]
- Zhou, W.; Mukherjee, P.; Kiebish, M.A.; Markis, W.T.; Mantis, J.G.; Seyfried, T.N. The Calorically Restricted Ketogenic Diet, an Effective Alternative Therapy for Malignant Brain Cancer. Nutr. Metab. 2007, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Brucker, D.P.; Bähr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential Utilization of Ketone Bodies by Neurons and Glioma Cell Lines: A Rationale for Ketogenic Diet as Experimental Glioma Therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef]
- Xu, S.; Tao, H.; Cao, W.; Cao, L.; Lin, Y.; Zhao, S.-M.; Xu, W.; Cao, J.; Zhao, J.-Y. Ketogenic Diets Inhibit Mitochondrial Biogenesis and Induce Cardiac Fibrosis. Signal. Transduct. Target Ther. 2021, 6, 54. [Google Scholar] [CrossRef]
- Zhao, Z.Q.; Nakamura, M.; Wang, N.P.; Wilcox, J.N.; Shearer, S.; Ronson, R.S.; Guyton, R.A.; Vinten-Johansen, J. Reperfusion Induces Myocardial Apoptotic Cell Death. Cardiovasc. Res. 2000, 45, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Q.; Velez, D.A.; Wang, N.P.; Hewan-Lowe, K.O.; Nakamura, M.; Guyton, R.A.; Vinten-Johansen, J. Progressively Developed Myocardial Apoptotic Cell Death during Late Phase of Reperfusion. Apoptosis 2001, 6, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Algoet, M.; Janssens, S.; Himmelreich, U.; Gsell, W.; Pusovnik, M.; Van den Eynde, J.; Oosterlinck, W. Myocardial Ischemia-Reperfusion Injury and the Influence of Inflammation. Trends Cardiovasc. Med. 2022. [Google Scholar] [CrossRef]
- Lau, A.; Rahn, J.J.; Chappellaz, M.; Chung, H.; Benediktsson, H.; Bihan, D.; von Mässenhausen, A.; Linkermann, A.; Jenne, C.N.; Robbins, S.M.; et al. Dipeptidase-1 Governs Renal Inflammation during Ischemia Reperfusion Injury. Sci. Adv. 2022, 8, eabm0142. [Google Scholar] [CrossRef] [PubMed]
- Franke, M.; Bieber, M.; Kraft, P.; Weber, A.N.R.; Stoll, G.; Schuhmann, M.K. The NLRP3 Inflammasome Drives Inflammation in Ischemia/reperfusion Injury after Transient Middle Cerebral Artery Occlusion in Mice. Brain Behav. Immun. 2021, 92, 223–233. [Google Scholar] [CrossRef]
- Guo, J.; Guan, Q.; Liu, X.; Wang, H.; Gleave, M.E.; Nguan, C.Y.C.; Du, C. Relationship of Clusterin with Renal Inflammation and Fibrosis after the Recovery Phase of Ischemia-Reperfusion Injury. BMC Nephrol. 2016, 17, 133. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate Immune Signaling in Cardiac Ischemia. Nat. Rev. Cardiol. 2011, 8, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release Mechanisms of Major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Mitroulis, I.; von Renesse, J.; Hajishengallis, G.; Chavakis, T. From Leukocyte Recruitment to Resolution of Inflammation: The Cardinal Role of Integrins. J. Leukoc. Biol. 2017, 102, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte Integrins: Role in Leukocyte Recruitment and as Therapeutic Targets in Inflammatory Disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil Chemoattractant Receptors in Health and Disease: Double-Edged Swords. Cell Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Xin, Q.; Ji, B.; Cheng, B.; Wang, C.; Liu, H.; Chen, X.; Chen, J.; Bai, B. Endoplasmic Reticulum Stress in Cerebral Ischemia. Neurochem. Int. 2014, 68, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, F. Endoplasmic Reticulum Stress in Brain Ischemia. Int. J. Neurosci. 2016, 126, 681–691. [Google Scholar] [CrossRef]
- Nakka, V.P.; Gusain, A.; Raghubir, R. Endoplasmic Reticulum Stress Plays Critical Role in Brain Damage after Cerebral Ischemia/reperfusion in Rats. Neurotox. Res. 2010, 17, 189–202. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER Stress Activates the NLRP3 Inflammasome via an UPR-Independent Pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Seifert, H.A.; Pennypacker, K.R. Molecular and Cellular Immune Responses to Ischemic Brain Injury. Transl. Stroke Res. 2014, 5, 543–553. [Google Scholar] [CrossRef]
- Tong, Y.; Ding, Z.-H.; Zhan, F.-X.; Cai, L.; Yin, X.; Ling, J.-L.; Ye, J.-J.; Hou, S.-Y.; Lu, Z.; Wang, Z.-H.; et al. The NLRP3 Inflammasome and Stroke. Int. J. Clin. Exp. Med. 2015, 8, 4787–4794. [Google Scholar]
- Quadri, S.A.; Farooqui, M.; Ikram, A.; Zafar, A.; Khan, M.A.; Suriya, S.S.; Claus, C.F.; Fiani, B.; Rahman, M.; Ramachandran, A.; et al. Recent Update on Basic Mechanisms of Spinal Cord Injury. Neurosurg. Rev. 2020, 43, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome–mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, T.; Iwata, M.; Kamiya, N.; Tsunetomi, K.; Kajitani, N.; Wada, N.; Iitsuka, T.; Yamauchi, T.; Miura, A.; Pu, S.; et al. Beta-Hydroxybutyrate, an Endogenic NLRP3 Inflammasome Inhibitor, Attenuates Stress-Induced Behavioral and Inflammatory Responses. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzúa, S.; Rojas, I.; Líbano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr. Pharm. Des. 2020, 26, 4246–4260. [Google Scholar] [CrossRef] [PubMed]
- Kurian, G.A.; Rajagopal, R.; Vedantham, S.; Rajesh, M. The Role of Oxidative Stress in Myocardial Ischemia and Reperfusion Injury and Remodeling: Revisited. Oxid. Med. Cell. Longev. 2016, 2016, 1656450. [Google Scholar] [CrossRef]
- Tabriziani, H.; Lipkowitz, M.S.; Vuong, N. Chronic Kidney Disease, Kidney Transplantation and Oxidative Stress: A New Look to Successful Kidney Transplantation. Clin. Kidney J. 2018, 11, 130–135. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Oxidative Stress and Acute Hepatic Injury. Curr. Opin. Toxicol. 2018, 7, 17–21. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.-P.; Patel, M. Acute Oxidative Stress and Systemic Nrf2 Activation by the Ketogenic Diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef] [PubMed]
- G Bardallo, R.; Panisello-Roselló, A.; Sanchez-Nuno, S.; Alva, N.; Roselló-Catafau, J.; Carbonell, T. Nrf2 and Oxidative Stress in Liver Ischemia/reperfusion Injury. FEBS J. 2021. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in Myocardial Ischemia and Reperfusion Injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, M.; Wang, Y.; Xie, F.; Zhang, G.; Qin, X. Nrf2-a Promising Therapeutic Target for Defensing Against Oxidative Stress in Stroke. Mol. Neurobiol. 2017, 54, 6006–6017. [Google Scholar] [CrossRef]
- Chakravarty, S.; Jhelum, P.; Bhat, U.A.; Rajan, W.D.; Maitra, S.; Pathak, S.S.; Patel, A.B.; Kumar, A. Insights into the Epigenetic Mechanisms Involving Histone Lysine Methylation and Demethylation in Ischemia Induced Damage and Repair Has Therapeutic Implication. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 152–164. [Google Scholar] [CrossRef]
- Su, Y.; Zhang, L.; Zhou, Y.; Ding, L.; Li, L.; Wang, Z. The Progress of Research on Histone Methylation in Ischemic Stroke Pathogenesis. J. Physiol. Biochem. 2022, 78, 1–8. [Google Scholar] [CrossRef]
- Mar, D.; Gharib, S.A.; Zager, R.A.; Johnson, A.; Denisenko, O.; Bomsztyk, K. Heterogeneity of Epigenetic Changes at Ischemia/reperfusion- and Endotoxin-Induced Acute Kidney Injury Genes. Kidney Int. 2015, 88, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhong, B.; Tan, J.; Chen, C.; Lei, Q.; Zeng, L. The Emerging Role of Epigenetics in Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 1887–1905. [Google Scholar] [CrossRef] [PubMed]
- Kao, M.-H.; Lin, T.-N. Histone Deacetylases in Stroke. Chin. J. Physiol. 2019, 62, 95–107. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Q.; Chen, J.; Ma, Y.; Liu, X. Updating a Strategy for Histone Deacetylases and Its Inhibitors in the Potential Treatment of Cerebral Ischemic Stroke. Dis. Markers 2020, 2020, 8820803. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lin, Y.-H.; Ni, H.-Y.; Dong, J.; Yuan, H.-J.; Zhang, Y.; Liang, H.-Y.; Yao, M.-C.; Zhou, Q.-G.; Wu, H.-Y.; et al. Inhibiting Histone Deacetylase 2 (HDAC2) Promotes Functional Recovery From Stroke. J. Am. Heart Assoc. 2017, 6, e007236. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Ramadoss, M.; Mahadevan, V. Histone Deacetylase (HDAC) Inhibitors—Emerging Roles in Neuronal Memory, Learning, Synaptic Plasticity and Neural Regeneration. Curr. Neuropharmacol. 2016, 14, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, J.; Ismail, M.; Tweeten, S.; Zeng, F.; Gao, L.; Ballinger, S.; Young, M.; Prabhu, S.D.; Rowe, G.C.; et al. HDAC Inhibition Induces Autophagy and Mitochondrial Biogenesis to Maintain Mitochondrial Homeostasis during Cardiac Ischemia/reperfusion Injury. J. Mol. Cell Cardiol. 2019, 130, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tang, Y.; Hill, J.A. HDAC Inhibition as a Therapeutic Strategy in Myocardial Ischemia/reperfusion Injury. J. Mol. Cell Cardiol. 2019, 129, 188–192. [Google Scholar] [CrossRef]
- Costalonga, E.C.; Silva, F.M.O.; Noronha, I.L. Valproic Acid Prevents Renal Dysfunction and Inflammation in the Ischemia-Reperfusion Injury Model. Biomed. Res. Int. 2016, 2016, 5985903. [Google Scholar] [CrossRef]
- Pickell, Z.; Williams, A.M.; Alam, H.B.; Hsu, C.H. Histone Deacetylase Inhibitors: A Novel Strategy for Neuroprotection and Cardioprotection Following Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2020, 9, e016349. [Google Scholar] [CrossRef]
- Levine, M.H.; Wang, Z.; Bhatti, T.R.; Wang, Y.; Aufhauser, D.D.; McNeal, S.; Liu, Y.; Cheraghlou, S.; Han, R.; Wang, L.; et al. Class-Specific Histone/protein Deacetylase Inhibition Protects against Renal Ischemia Reperfusion Injury and Fibrosis Formation. Am. J. Transplant. 2015, 15, 965–973. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and Toxicity of Histone Deacetylase Inhibitors in Relapsed/refractory Multiple Myeloma: Systematic Review and Meta-Analysis of Clinical Trials. Exp. Ther. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.-D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.-H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.-M. Histone Deacetylase Inhibitors Induce Autophagy through FOXO1-Dependent Pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, Y.-S. Longevity Factor FOXO3: A Key Regulator in Aging-Related Vascular Diseases. Front Cardiovasc Med 2021, 8, 778674. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.E.; McCoy, D.M. Platelet-Derived Growth Factor-A Regulates Lung Fibroblast S-Phase Entry through p27(kip1) and FoxO3a. Respir. Res. 2013, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-F.; Pandey, S.; Day, C.H.; Chen, Y.-F.; Jiang, A.-Z.; Ho, T.-J.; Chen, R.-J.; Padma, V.V.; Kuo, W.-W.; Huang, C.-Y. Synergistic Effect of HIF-1α and FoxO3a Trigger Cardiomyocyte Apoptosis under Hyperglycemic Ischemia Condition. J. Cell Physiol. 2018, 233, 3660–3671. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Mangal, R.; Dandu, C.; Geng, X.; Ding, Y. Role of Forkhead Box Protein O1 (FoxO1) in Stroke: A Literature Review. Aging Dis. 2022, 13, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of Inflammation by the Antioxidant Haem Oxygenase. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Du, S.; Zheng, H. Role of FoxO Transcription Factors in Aging and Age-Related Metabolic and Neurodegenerative Diseases. Cell Biosci. 2021, 11, 188. [Google Scholar] [CrossRef]
- Li, L.; Kang, H.; Zhang, Q.; D’Agati, V.D.; Al-Awqati, Q.; Lin, F. FoxO3 Activation in Hypoxic Tubules Prevents Chronic Kidney Disease. J. Clin. Invest. 2019, 129, 2374–2389. [Google Scholar] [CrossRef]
- Gu, X.; Raman, A.; Susztak, K. Going from Acute to Chronic Kidney Injury with FoxO. J. Clin. Invest. 2019, 129, 2192–2194. [Google Scholar] [CrossRef] [PubMed]
- Lin, F. Molecular Regulation and Function of FoxO3 in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhong, X.; Li, J.; Liu, H.; Ma, X.; He, R.; Zhao, Y. MicroRNA-30c-5p Inhibits NLRP3 Inflammasome-Mediated Endothelial Cell Pyroptosis through FOXO3 down-Regulation in Atherosclerosis. Biochem. Biophys. Res. Commun. 2018, 503, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 Deacetylates Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase 2 and Regulates Ketone Body Production. Cell Metab. 2010, 12, 654–661. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 Regulates Mitochondrial Fatty-Acid Oxidation by Reversible Enzyme Deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.-H. Function of the SIRT3 Mitochondrial Deacetylase in Cellular Physiology, Cancer, and Neurodegenerative Disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 Blocks the Cardiac Hypertrophic Response by Augmenting Foxo3a-Dependent Antioxidant Defense Mechanisms in Mice. J. Clin. Invest. 2009, 119, 2758–2771. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Morris, G.; Puri, B.K.; Carvalho, A.; Maes, M.; Berk, M.; Ruusunen, A.; Olive, L. Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought? Int. J. Neuropsychopharmacol. 2020, 23, 366–384. [Google Scholar] [CrossRef]
- Yu, W.; Qin, J.; Chen, C.; Fu, Y.; Wang, W. Moderate Calorie Restriction Attenuates Age-associated Alterations and Improves Cardiac Function by Increasing SIRT1 and SIRT3 Expression. Mol. Med. Rep. 2018, 18, 4087–4094. [Google Scholar] [CrossRef]
- Gonzalez, S.V.; Nguyen, N.H.T.; Rise, F.; Hassel, B. Brain Metabolism of Exogenous Pyruvate. J. Neurochem. 2005, 95, 284–293. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Lactate Metabolism: The Discoveries and the Controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.; Fekete, A.D.; Kashem, M.A.; Nasrallah, F.A.; Bröer, S. Metabolism, Compartmentation, Transport and Production of Acetate in the Cortical Brain Tissue Slice. Neurochem. Res. 2012, 37, 2541–2553. [Google Scholar] [CrossRef] [PubMed]
- Künnecke, B.; Cerdan, S.; Seelig, J. Cerebral Metabolism of [1,2-13C2]glucose and [U-13C4]3-Hydroxybutyrate in Rat Brain as Detected by 13C NMR Spectroscopy. NMR Biomed. 1993, 6, 264–277. [Google Scholar] [CrossRef]
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-Glucose Treatment Induces Ketogenesis, Sustains Mitochondrial Function, and Reduces Pathology in Female Mouse Model of Alzheimer’s Disease. PLoS ONE 2011, 6, e21788. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-L.; Liu, Z.; Wang, C.; Li, Y.; Cai, Z. Insulin Resistance in Ischemic Stroke. Metab. Brain Dis. 2017, 32, 1323–1334. [Google Scholar] [CrossRef]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-Activated Protein Kinase Mediates Ischemic Glucose Uptake and Prevents Postischemic Cardiac Dysfunction, Apoptosis, and Injury. J. Clin. Invest. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Seo-Mayer, P.W.; Thulin, G.; Zhang, L.; Alves, D.S.; Ardito, T.; Kashgarian, M.; Caplan, M.J. Preactivation of AMPK by Metformin May Ameliorate the Epithelial Cell Damage Caused by Renal Ischemia. Am. J. Physiol. Renal Physiol. 2011, 301, F1346–F1357. [Google Scholar] [CrossRef]
- Cai, C.-C.; Zhu, J.-H.; Ye, L.-X.; Dai, Y.-Y.; Fang, M.-C.; Hu, Y.-Y.; Pan, S.-L.; Chen, S.; Li, P.-J.; Fu, X.-Q.; et al. Glycine Protects against Hypoxic-Ischemic Brain Injury by Regulating Mitochondria-Mediated Autophagy via the AMPK Pathway. Oxid. Med. Cell Longev. 2019, 2019, 4248529. [Google Scholar] [CrossRef]
- Hu, H.; Li, X.; Ren, D.; Tan, Y.; Chen, J.; Yang, L.; Chen, R.; Li, J.; Zhu, P. The Cardioprotective Effects of Carvedilol on Ischemia and Reperfusion Injury by AMPK Signaling Pathway. Biomed. Pharmacother. 2019, 117, 109106. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Physiological and Pathological Responses to Hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef]
- Sethi, K.; Rao, K.; Bolton, D.; Patel, O.; Ischia, J. Targeting HIF-1α to Prevent Renal Ischemia-Reperfusion Injury: Does It Work? Int. J. Cell Biol. 2018, 2018, 9852791. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Satriano, J.; Thomas, J.L.; Miyamoto, S.; Sharma, K.; Pastor-Soler, N.M.; Hallows, K.R.; Singh, P. Interactions between HIF-1α and AMPK in the Regulation of Cellular Hypoxia Adaptation in Chronic Kidney Disease. Am. J. Physiol. Renal Physiol. 2015, 309, F414–F428. [Google Scholar] [CrossRef]
- Dengler, F. Activation of AMPK under Hypoxia: Many Roads Leading to Rome. Int. J. Mol. Sci. 2020, 21, 2428. [Google Scholar] [CrossRef]
- Sun, X.; Wang, D.; Zhang, T.; Lu, X.; Duan, F.; Ju, L.; Zhuang, X.; Jiang, X. Eugenol Attenuates Cerebral Ischemia-Reperfusion Injury by Enhancing Autophagy via AMPK-mTOR-P70S6K Pathway. Front. Pharmacol. 2020, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.-H.; Chen, T.; Li, X.; Yue, K.-Y.; Luo, P.; Yang, L.-K.; Zhu, J.; Wang, Y.-H.; Fei, Z.; Jiang, X.-F. Sirt3 Confers Protection against Neuronal Ischemia by Inducing Autophagy: Involvement of the AMPK-mTOR Pathway. Free Radic. Biol. Med. 2017, 108, 345–353. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The Role of the Autophagy in Myocardial Ischemia/reperfusion Injury. Biochim. Biophys. Acta 2015, 1852, 271–276. [Google Scholar] [CrossRef]
- Hindawi Autophagy and Liver Ischemia-Reperfusion Injury. Available online: https://www.hindawi.com/journals/bmri/2015/417590/ (accessed on 20 September 2022).
- Decuypere, J.-P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the Kidney: Implications for Ischemia-Reperfusion Injury and Therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef]
- Steiner, C.A.; Cartwright, I.M.; Taylor, C.T.; Colgan, S.P. Hypoxia-Inducible Factor as a Bridge between Healthy Barrier Function, Wound Healing, and Fibrosis. Am. J. Physiol. Cell Physiol. 2022, 323, C866–C878. [Google Scholar] [CrossRef]
- Haase, V.H. Pathophysiological Consequences of HIF Activation: HIF as a Modulator of Fibrosis. Ann. NY Acad. Sci. 2009, 1177, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.A.; Hewitson, T.D. Hypoxia in Tissue Repair and Fibrosis. Cell Tissue Res. 2016, 365, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Scorrano, L.; Colonna, R.; Petronilli, V.; Di Lisa, F. Mitochondria and Cell Death. Mechanistic Aspects and Methodological Issues. Eur. J. Biochem. 1999, 264, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Mattson, M.P.; Cheng, A. Permeability Transition Pore-Mediated Mitochondrial Superoxide Flashes Regulate Cortical Neural Progenitor Differentiation. PLoS ONE 2013, 8, e76721. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Scorrano, L. Mitochondria: From Cell Death Executioners to Regulators of Cell Differentiation. Trends Cell Biol. 2014, 24, 761–770. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef]
- Reynolds, I.J. Mitochondrial Membrane Potential and the Permeability Transition in Excitotoxicity. Ann. NY Acad. Sci. 1999, 893, 33–41. [Google Scholar] [CrossRef]
- Crompton, M. The Mitochondrial Permeability Transition Pore and Its Role in Cell Death. Biochem. J. 1999, 341 Pt 2, 233–249. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of Cyclophilin D Reveals a Critical Role for Mitochondrial Permeability Transition in Cell Death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Büki, A.; Okonkwo, D.O.; Wang, K.K.; Povlishock, J.T. Cytochrome c Release and Caspase Activation in Traumatic Axonal Injury. J. Neurosci. 2000, 20, 2825–2834. [Google Scholar] [CrossRef]
- Zoratti, M.; Szabò, I. The Mitochondrial Permeability Transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Duchen, M.R.; McGuinness, O.; Brown, L.A.; Crompton, M. On the Involvement of a Cyclosporin A Sensitive Mitochondrial Pore in Myocardial Reperfusion Injury. Cardiovasc. Res. 1993, 27, 1790–1794. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Halestrap, A.P. Protection by Cyclosporin A of Ischemia/reperfusion-Induced Damage in Isolated Rat Hearts. J. Mol. Cell Cardiol. 1993, 25, 1461–1469. [Google Scholar] [CrossRef]
- Halestrap, A.P. A Pore Way to Die: The Role of Mitochondria in Reperfusion Injury and Cardioprotection. Biochem. Soc. Trans. 2010, 38, 841–860. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Panel, M.; Ruiz, I.; Brillet, R.; Lafdil, F.; Teixeira-Clerc, F.; Nguyen, C.T.; Calderaro, J.; Gelin, M.; Allemand, F.; Guichou, J.-F.; et al. Small-Molecule Inhibitors of Cyclophilins Block Opening of the Mitochondrial Permeability Transition Pore and Protect Mice From Hepatic Ischemia/Reperfusion Injury. Gastroenterology 2019, 157, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Hånell, A.; Greer, J.E.; McGinn, M.J.; Povlishock, J.T. Traumatic Brain Injury-Induced Axonal Phenotypes React Differently to Treatment. Acta Neuropathol. 2015, 129, 317–332. [Google Scholar] [CrossRef]
- Fournier, N.; Ducet, G.; Crevat, A. Action of Cyclosporine on Mitochondrial Calcium Fluxes. J. Bioenerg. Biomembr. 1987, 19, 297–303. [Google Scholar] [CrossRef]
- Kim, D.Y.; Davis, L.M.; Sullivan, P.G.; Maalouf, M.; Simeone, T.A.; van Brederode, J.; Rho, J.M. Ketone Bodies Are Protective against Oxidative Stress in Neocortical Neurons. J. Neurochem. 2007, 101, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone Bodies Mediate Antiseizure Effects through Mitochondrial Permeability Transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Barsoum, M.J.; Godzik, A.; Schwarzenbacher, R.; Lipton, S.A. Mitochondrial Fission in Apoptosis, Neurodegeneration and Aging. Curr. Opin. Cell Biol. 2003, 15, 706–716. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal Mitochondrial Dynamics and Neurodegenerative Diseases. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2010, 1802, 135–142. [Google Scholar] [CrossRef]
- Pradeep, H.; Sharma, B.; Rajanikant, G.K. Drp1 in Ischemic Neuronal Death: An Unusual Suspect. Curr. Med. Chem. 2014, 21, 2183–2189. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Diet | Macronutrients, P:C:F *, % | Calorie Intake Normalization | Duration before/after Injury | Animal, Organ | Model | Positive Effect | Results | References |
|---|---|---|---|---|---|---|---|---|
| KD | 9.5:4.7:85.8 | No | 14 d/28 d | Mice Hind-limb | Unilateral femoral artery ligation | − − | Long-term KD decreased blood perfusion and aggravated inflammation of ischemic tissue, delayed muscle recovery and wound healing at the surgical site, induced muscle atrophy of non-ischemic tissue. | [127] |
| KD | 4.7:1.8:93.5 | N/D ** | 28 d/28 d | Mice Heart | Left anterior descending artery ligation | − − | Long-term KD exacerbated cardiac dysfunction. | [128] |
| KD | 10.4:0.1:89.5 | N/D | 28 d/- | Rats Brain | MCAO | + | KD-induced stabilization of HIF-1α in rat brain under normoxic conditions was associated with inflammatory response activation and neuroprotection during IR. | [129] |
| KD | 11.3:1.5:87.2 | Yes | 3 d/- | Rats Kidney | Left renal artery occlusion | ++ | Acute KD feeding caused protein acetylation, liver AMPK activation, and increased resistance to IR-induced kidney injury. KD attenuated oxidative damage, increased antioxidant defenses, and reduced inflammation after kidney IR. KD prevented interstitial fibrosis development at two weeks, upregulation of HSP70, and chronic Klotho deficiency. | [31] |
| “New-KD” | 12:5:84 | Yes | -/70 d | Rats Brain | Controlled cortical impact | + | The new KD attenuated sensorimotor deficits, corrected spatial memory deficit, reduced the lesion size, perilesional inflammation, and oxidative damage, enhanced the mTOR pathway, and increased histone acetylation and methylation. | [32] |
| KD | N/D:N/D:80 | No | 3 d/7 d | Rats Brain | endothelin-1 injection | + | KD led to a reduction in motor behavior impairment in the model of ischemic stroke. | [130] |
| KD | 10.4:0.1:89.5 | Yes | 21 d/- | Mice Brain | MCAO | + | KD provided tolerance to MCAO/R, inhibited endoplasmic reticulum stress, and suppressed TXNIP/NLRP3 inflammasome activation in the brain. | [33] |
| LCHFD | N/D:N/D:60 | N/D | 21 d/- | Mice Gut | N/D | +/− | LCHFD feeding increased gut NO levels before gut IR, but decreased them after gut IR. | [131] |
| KD | 10.4:0.1:89.5 | No | 28 d/- | Mice Brain | MCAO | +++ | KD was neuroprotective against focal cerebral ischemia in a concentration-dependent manner, and upregulated cytoprotective pathways associated with HIF-1α, pAKT, and AMPK. | [34] |
| KD | 10.4:0.1:89.5 | No | 21 d/- | Mice Brain | MCAO | +++ | KD led to a reduction in infarct volume and an increase in regional cerebral blood flow and extracellular adenosine levels in both the ischemic and the reperfusion phases. KD increased Akt and ERK1/2 phosphorylation via A1R activation, and upregulated HIF-1α/HIF-2α, VEGF, and EPO. | [35] |
| Fat-rich diet | 17:48:35 | Yes | 21 d/- | Mice Brain, liver | MCAO | +/− | Fat-rich diets increased BHB levels in liver, blood, brain microdialysate, and brain homogenate 90 min after MCAO. Glucose levels were changed in the opposite manner. Reperfusion decreased BHB and increased glucose within 60 min. Citrate and succinate were moderately increased by the fat-rich diet and unchanged after stroke. | [122] |
| HFLCD | 30:10:60 | No | 14 d/- | Rats Heart | IR in the isolated heart | − − | HFLCD led to an increase in free fatty acid (FFA) oxidation and a decrease in carbohydrate or ketone oxidation, both in control and IR. HFLCD led to decreased recovery of left ventricular function and reduced insulin sensitivity. | [132] |
| HFLCD | 15:20:65 | N/D | 5 d/- | Mice Gut | Superior mesenteric artery occlusion | +/− | Short-term HFLCD did not affect survival after gut IR. | [133] |
| HFLCD | 30:10:60 | Yes | 14 d/- | Rats Heart | Left anterior descending coronary artery ligation | − − | HFLCD did not affect nonischemic left ventricular function but led to greater myocardial injury during IR, with increased risk of death by pump failure and ventricular arrhythmias. | [118] |
| HFLCD | 30:10:60 | Yes | 14 d/- | Rats Heart | IR in the isolated heart | − − | HFLCD led to increased ischemic myocardial injury, impaired recovery of function after reperfusion, and enhanced oxidative stress. | [119] |
| KD | 10.4:0.1:89.5 | N/D | 21 d/- | Rats Brain | Hypoxia in hypobaric chambers | + | In the aged rats, KD improved cognitive performance under normoxic and hypoxic conditions, while motor performance remained unchanged. Capillary density and HIF-1α levels were elevated in the aged KD group independent of hypoxic challenge. | [123] |
| KD | 10:2:78 | No | 25 d/- | Rats Brain | Cardiac arrest-induced cerebral ischemia | + | KD prevented cardiac-arrest-induced cerebral ischemic neurodegeneration in several brain regions. | [134] |
| KD | 10.4:0.1:89.5 | No | 21 d/- | Rats Brain | MCAO | + | KD reduced infarct volumes following IR. | [135] |
| HFD | 20:20:60 | No | -/56 d | Rats Heart | Coronary artery ligation | + | HFD *** increased state 3 respiration and acyl-CoA dehydrogenase activity, but did not normalize levels of acyl-CoA dehydrogenases in IR-induced heart failure. | [125] |
| HFD | 20:20:60 | No | 14 d/- or -/56 d | Rats Heart | Coronary artery ligation | +/− − | HFD following cardiac IR did not exacerbate left ventricular dysfunction and remodeling, but increased surgical mortality. HFD increased mitochondrial oxidative phosphorylation and ETC complex activity. HFD before surgery resulted in an increased surgical mortality rate | [126] |
| LCKD | 60:10:30 | No | 133 d/- | Rats Heart | IR in the isolated heart | + | LCKD increased the number of mitochondria and provided a tolerance to ischemia and a faster recovery of cardiac function following reperfusion. | [124] |
| five experimental diets | 17:21:62 **** | Yes | 12 h/- | Rats Brain | MCAO | + | Infarct volumes were significantly smaller after the 1,3-butanediol diet and after the triacetin–tributyrin diet. Infarct volume correlated with the plasma glucose, but not lactate, ketone body, or acetate concentration before ischemia. | [121] |
| Mimetic | Dose | Duration before/ after Injury | Animal/ Cells, Organ | Model | Positive Effect | Results | References |
|---|---|---|---|---|---|---|---|
| BHB | one intraperitoneal injection of 30 mg/kg | -/90 min | Mice | MCAO | ++ | Single acute BHB injection improved the neurological score determined and mitochondrial respiratory complex I and II activity after 24 h but not at later time points. | [136] |
| BHB sodium salt | 1–100 mM | 12 h/- | Mouse cardiomyocytes | Incubation under hypoxic conditions | − − | Treatment with BHB enhanced cardiomyocyte death and decreased glucose absorption and glycolysis under hypoxic conditions. | [128] |
| BHB | in vivo: 4 μL of BHB (250–1000 μg/kg) injected once into lateral ventricle | -/1 h | Rats Brain | MCAO | ++ | BHB enhanced mitochondrial respiratory chain complex I activity, reduced oxidative stress, inhibited apoptosis, improved neurological scores, and reduced infarct volume after ischemia. BHB acted through upregulation of BHB transporter SMCT1 and activation of the Erk/CREB/eNOS pathway. | [137] |
| in vitro: 2–100 mM | 24 h/- | Rat Neuronal cells | OGD | ++ | |||
| DL-BHB | one intraperitoneal injection of 10 mmoL/kg | 30 min/- | Mice Liver | Partial warm hepatic IR | +++ | BHB reduced hepatocellular necrosis after IR treatment. Exogenous BHB induced acetylation of histone-3, which resulted in higher expressions of FOXO1 and HO-1 upregulation. The expression of NLRP3 in the liver and serum levels of IL-1β was suppressed by BHB. | [138] |
| BHB | in vivo: 8 μL/h, 1 g/mL; osmotic pumps intraperitoneally | 1 d/1 d | Mice Kidney | Left renal artery and vein occlusion | + | Renal IR injury was attenuated by BHB treatment. BHB reduced the number of TUNEL-positive cells in kidney, increased expression of FOXO3, and decreased the expression of caspase-1 and proinflammatory cytokines. In an HK-2 cell line exposed to hypoxia and reoxygenation, BHB reduced cell death in a FOXO3-dependent fashion. Histone acetylation was decreased in kidneys exposed to IR and in HK-2 cells exposed to hypoxia and reoxygenation, though this effect was ameliorated by BHB through the inhibition of histone deacetylases. | [139] |
| in vitro: 1–40 mM | 0/12 h | HK-2 cells | OGD | + | |||
| D-BHB sodium salt | 4 doses of intraperitoneal injection of 5.0 mmol/kg | -/0–6 h | Rats Brain | Hypoxic ischemic encephalopathy | + | The BHB group demonstrated significantly lower brain pathological scores after hypoxic ischemic injury. The intact residual hemispheric and hippocampal volumes were also greater in this group. Neurological functions were unaffected. | [140] |
| DL-BHB | one intraperitoneal injection of 500 mg/kg | 0/1 h | Rats Brain | endothelin-1 injection | ++ | BHB treatment reduced oxidative stress, diminished astrogliosis and neuronal death, preserved neuronal functioning, normalized perilesional perfusion, and ameliorated cerebrovascular tolerance to hypercapnia. | [141] |
| D-BHB | 1.6 mmol/kg; osmotic pumps subcutaneously | 0/1 d | Mice Heart | Left anterior descending coronary artery occlusion | +++ | Treatment with BHB reduced infarct size and levels of cardiac troponin I, creatine kinase, and lactate dehydrogenase in serum, attenuated apoptosis in myocardium, and preserved cardiac function in IR mice, reduced mitochondrial formation of ROS and swelling, enhanced ATP production, partly restored mitochondrial membrane potential, and attenuated ER stress in myocardium. | [142] |
| BHB | 10 mM | N/D * | SH-SY-5Y cells | OGD | +++ | 10 mM BHB prevented the mitochondrial translocation of Drp1 to inhibit mitochondrial fission, decreased ROS generation, and suppressed ER stress-induced NLRP3 inflammasome activation in OGD-injured cells. | [33] |
| D-BHB | 0.4–1.6 mmol/kg/day; osmotic pumps subcutaneously | 0/84 d | Mice Brain | Spinal cord injury | + | BHB promoted functional recovery and relieved pain hypersensitivity in mice with spinal cord injury, possibly through inhibition of histone deacetylases, suppression of NLRP3 inflammasome, and protection of mitochondrial function. | [143] |
| D-BHB sodium salt | 7 days of twice-daily or single-dose subcutaneous injection of 1000 mg/kg | 0–7 d/0 | Rats Eye | Optic nerve and central retinal blood vessel transection | +++ | BHB provides retinal antioxidant defense by the antioxidant Nrf2 pathway activation via modification of a fumarate metabolism. | [120] |
| BHB | 10 mM | 12 h/0 | N2a cells | OGD | ++ | Pretreatment with BHB reduced OGD-induced cell death. Transfection with small interfering RNA (siRNA) targeting the A1 adenosine receptor before BHB pretreatment reversed the protective effect, indicating adenosine signaling pathways involved in neuronal cell tolerance to ischemic injury. | [35] |
| BHB+AcAc | BHB (0.4 mmol/kg), AcAc (0.45 mmol/kg), 7 doses of subcutaneous injection every hour | 0/7 h | Mice Brain | MCAO | ++ | Ketone body treatment enhanced mitochondria function, reduced oxidative stress, and infarct volume, led to improved neurologic function after ischemia, including the neurologic scores in Rotarod and open field tests. Ketone bodies’ effects were achieved by upregulating of SIRT3 and downstream effectors, FoxO3a and SOD2, in the penumbra region. | [144] |
| N/D | - | Murine neuronal cells | Rotenone treatment | ++ | |||
| D-BHB sodium salt | intraventricular infusion; 10 mM 1 ul/hr | 4 d/- | Rats Brain | MCAO | + | A 55–70% reduction in infarct volume was observed with BHB infusion or diet-induced ketosis. HIF-1α and Bcl-2 protein levels increased after BHB infusions. Succinate content increased 4-fold with BHB infusion | [135] |
| Lithium salt of AcAc | 4 groups: (I) 600 mM AcAc 0.5 μL/h/14 d by osmotic pump subcutaneously; (II) daily intraperitoneal injections 250 mg/kg AcAc for 7 days; (III) intravenously 500 μL of a 200 mM AcAc; (IV) three intraperitoneal injections of 250 mg/kg AcAc | (I) 10 d/4 d; (II) 4 d/3 d; (III) 0/15 min; (IV) 0/0.5–1.5 h | Rats Brain | Iodoacetate+L-trans-pyrrolydine-2,4-dicarboxylate injection | + | AcAc efficiently protects against glutamate neurotoxicity both in vivo and in vitro. | [145] |
| DL-BHB | 25 μmol/kg/min; intravenously using a syringe pump | 60 min/- | Rats Heart | Left coronary artery occlusion | ++ | DL-BHB infusion after prolonged fasting reduced infarct volume and apoptosis after heart IR, possibly by increasing myocardial ATP levels. | [146] |
| D-BHB sodium salt | 30 mg/kg/h; intravenous catheter | 0/1–3 d | Rats Brain | MCAO | ++ | BHB reduced infarct area at 24 h, but not at 72 h after permanent MCAO. In rats with 2 h transient MCAO followed by 22 h reperfusion, BHB significantly reduced cerebral infarct area, edema formation, lipid peroxidation, and neurological deficits. | [147] |
| D-BHB sodium salt | 3–100 mg/kg/h; intravenous catheter | 30 min/0 or 0/3 h | Rats and mice Brain | N2-induced hypoxia in mice KCN-induced anoxia and global cerebral ischemia induced by bilateral carotid artery ligation in rats | ++ | BHB administered immediately after a bilateral carotid artery ligation significantly suppressed the elevation of cerebral water and sodium contents as well as maintaining high ATP and low lactate levels. BHB demonstrated protective effects on cerebral hypoxia, anoxia, and ischemia-induced metabolic change. | [148] |
| 1,3-Butanediol | from 1 to 4 intraperitoneal injections injections; 25 mmol/kg | 30 min/0–9 h | Rats Brain | Embolization by microspheres | ++ | 1,3-Butanediol attenuated ischemia-induced metabolic changes by increasing the concentrations of phosphocreatine, ATP, and glycogen and by reducing the concentrations of pyruvate and lactate. | [149] |
| D-BHB L-arginine salt | 20 μmol/kg/min; intravenous catheter | -/90 min after 40 min occlusion | Dogs Heart | Left anterior descending coronary artery occlusion | + | The BHB treatment stabilized the left ventricular function. | [150] |
| BHB | one 12 mg/mouse intravenous or 30–120 mg/mouse intraperitoneal injection | 30 min/0 | Mice Global hypoxia | N2-induced hypoxia | ++ | BHB with glucagon combination increased survival time in global hypoxia. | [151] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makievskaya, C.I.; Popkov, V.A.; Andrianova, N.V.; Liao, X.; Zorov, D.B.; Plotnikov, E.Y. Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 2576. https://doi.org/10.3390/ijms24032576
Makievskaya CI, Popkov VA, Andrianova NV, Liao X, Zorov DB, Plotnikov EY. Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential. International Journal of Molecular Sciences. 2023; 24(3):2576. https://doi.org/10.3390/ijms24032576
Chicago/Turabian StyleMakievskaya, Ciara I., Vasily A. Popkov, Nadezda V. Andrianova, Xinyu Liao, Dmitry B. Zorov, and Egor Y. Plotnikov. 2023. "Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential" International Journal of Molecular Sciences 24, no. 3: 2576. https://doi.org/10.3390/ijms24032576
APA StyleMakievskaya, C. I., Popkov, V. A., Andrianova, N. V., Liao, X., Zorov, D. B., & Plotnikov, E. Y. (2023). Ketogenic Diet and Ketone Bodies against Ischemic Injury: Targets, Mechanisms, and Therapeutic Potential. International Journal of Molecular Sciences, 24(3), 2576. https://doi.org/10.3390/ijms24032576