_Putnam.png)
Recent Advances in the Applications of Small Molecules in the Treatment of Multiple Myeloma


Abstract
:1. Introduction
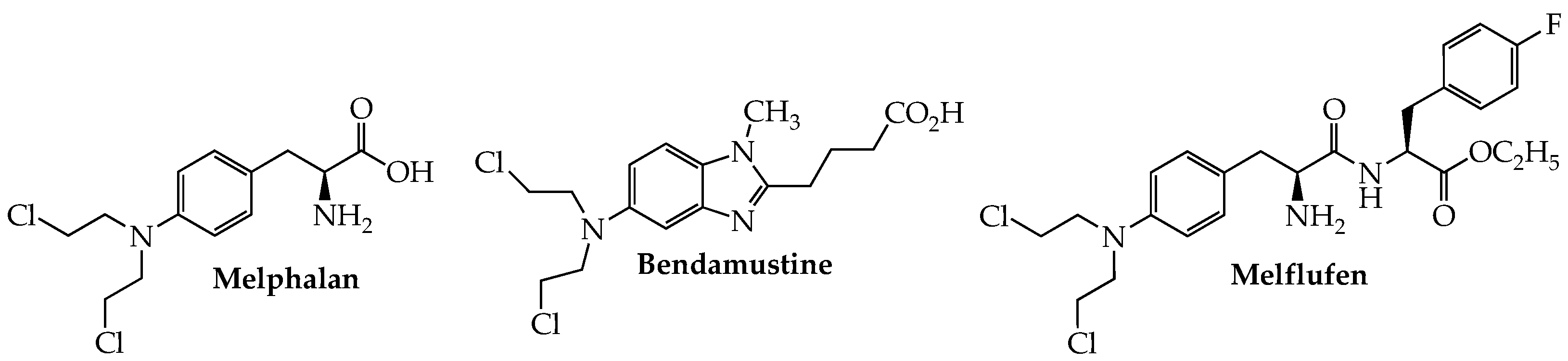
2. Alkylating Agents
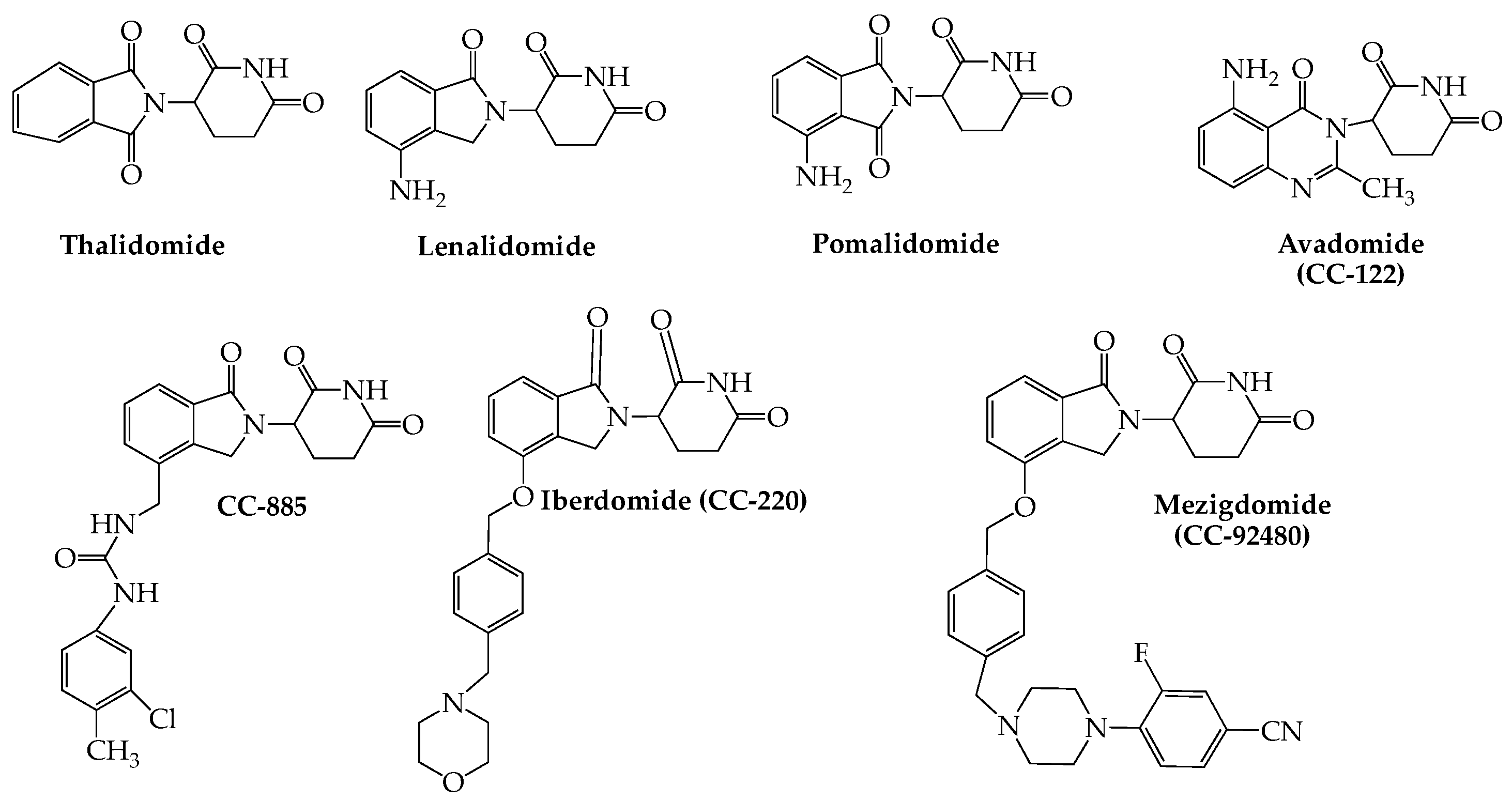
3. Cereblon E3 Ligase Modulators
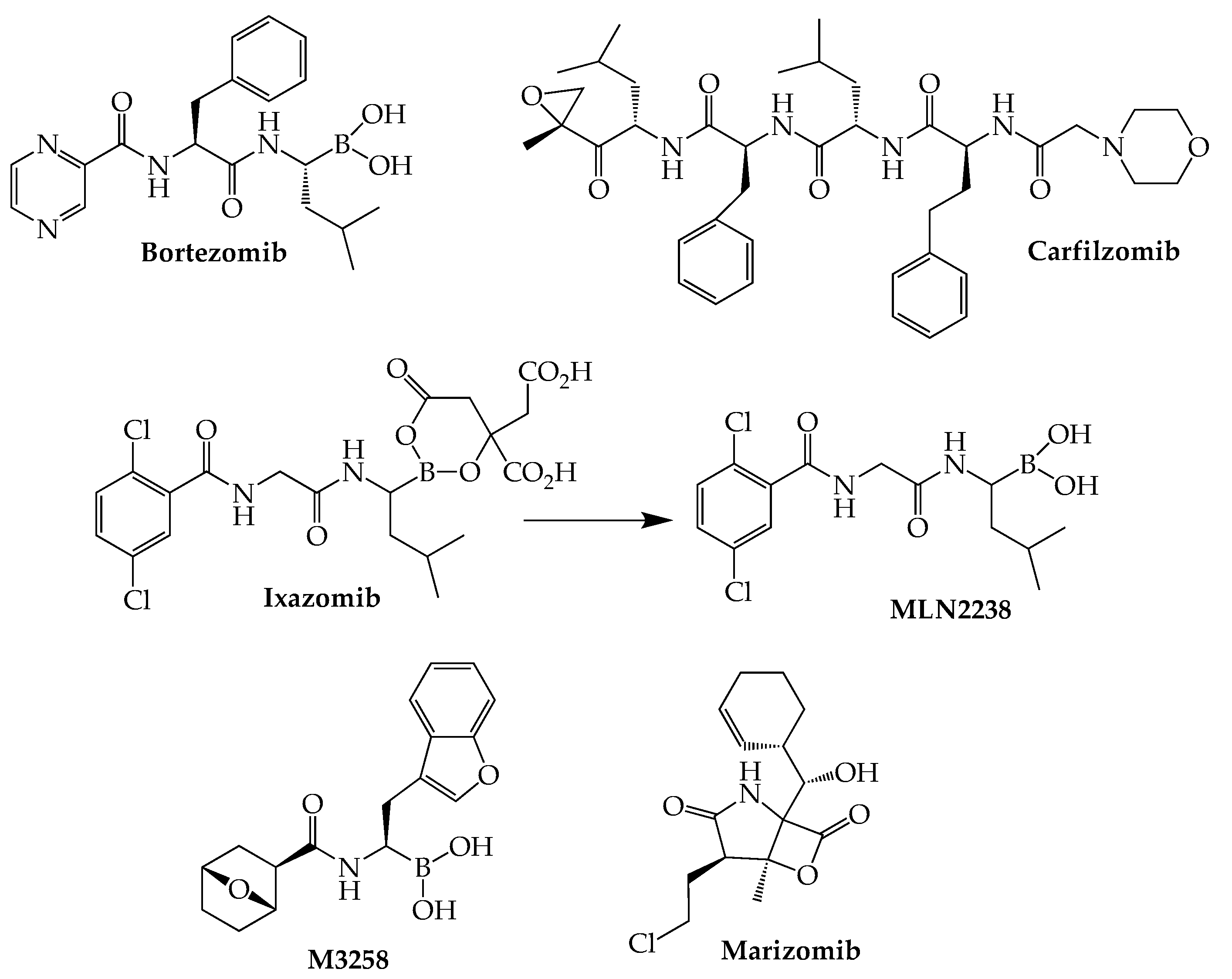
4. Proteasome Inhibitors
5. Deubiquitinase Inhibitors
6. Neddylation Inhibitors
7. HDAC Inhibitors
8. Bromodomain Inhibitors
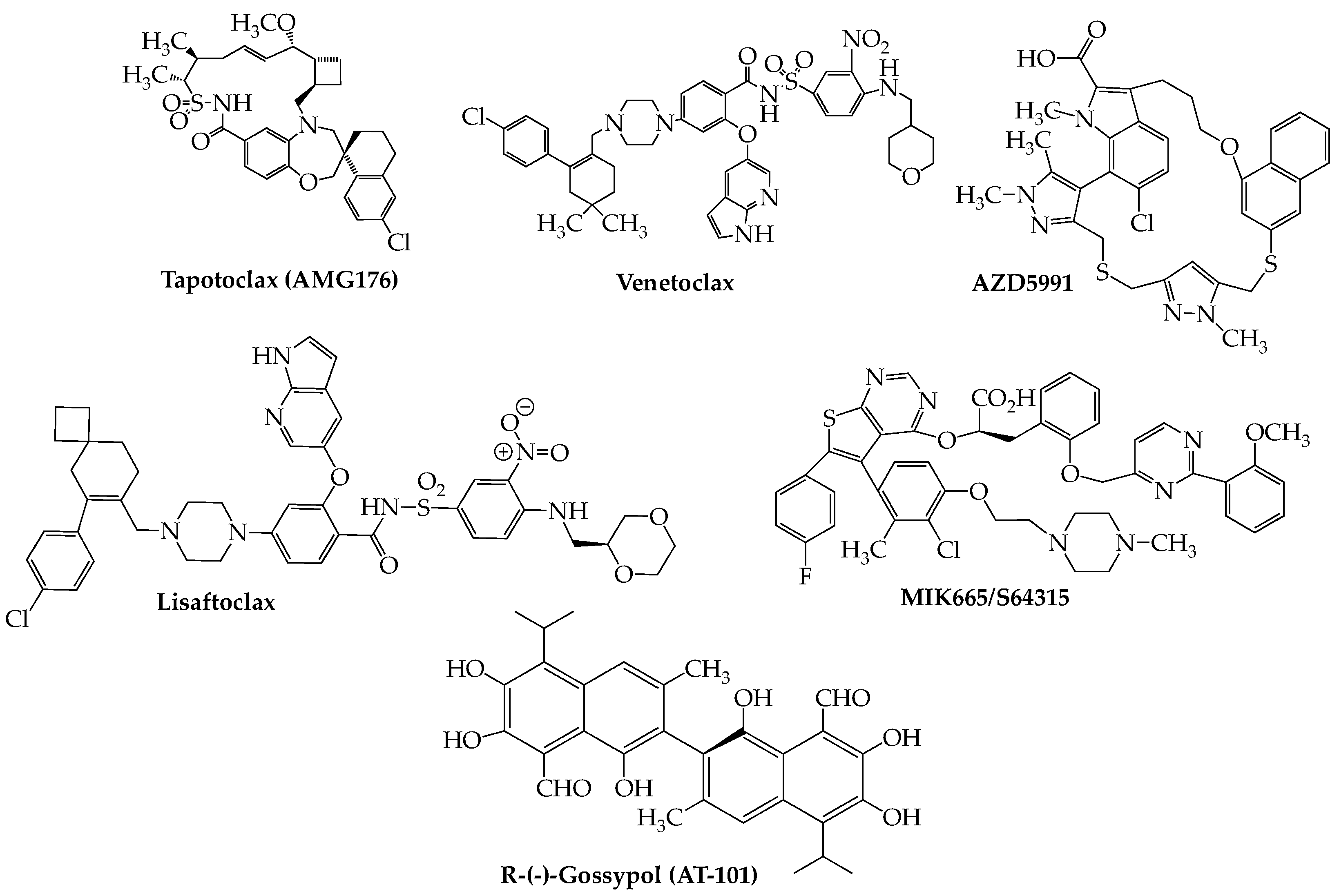
9. Apoptosis Inducers: Bcl2, IAP, and Mcl-1 Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial ID (Reference) | Phase | Drugs | Enrollment (N) | Prior Lines of Therapy (Median) | ORR (%) | PFS (Median in Months) |
|---|---|---|---|---|---|---|
| NCT03314181 [117] | I | (Ven + Dara + Dex) vs. (Ven + Dara + Dex + Bort) | Part 1: [24 with t(11;14)—Ven + Dara + Dex]; Part 2: [6 with t(11;14) + 18 other RRMM— Ven + Dara + Dex + Bort] | Part 1: 2.5; Part 2: 1 | Ven + Dara + Dex: 96; Ven + Dara + Dex + Bort: 92 | NR |
| NCT03314181 [118] | I/Ii | (Ven + Dara + Dex) vs. (Ven + Dara + Dex + Bort) | 34 all t(11;14): 11 Ven + Dara + Dex (12 at 400 mg Ven, 7 at 800 mg. Ven); 16 Ven + Dara + Dex + Bort | Ven + Dara + Dex: 1; Ven + Dara + Dex + Bort: 2 | Ven + Dara + Dex: 72.7 (at 400 mg.) and 100 (at 800 mg.); Ven + Dara + Dex + Bort: 62.5 | NR |
| NCT02899052 [119] | II | Ven + Carf + Dex | 49: 13 t(11;14); 36 non-t(11;14) | 1 | t(11;14): 92; non-t(11;14): 75 | With t(11;14): 24.8; without t(11;14): 22.8 |
| NCT01794520 [120] | I | Ven and Ven + Dex | 66: 30 t(11;14); 36 non-t(11;14) | 5 | t(11;14): 40; non-t(11;14): 6 | t(11;14): 6.6; non-t(11;14): 1.9 |
| NCT01794520 [121] | I/II | Ven + Dex | Phase I: 20; Phase II: 31. All t(11;14) positive | Phase I: 3; Phase II: 5 | Phase I: 60; Phase II: 48 | Phase I: 12.4; Phase II: 10.8 |
| NCT02755597 [122] | III | (Ven + Bort + Dex) vs. (Bort + Dex + Pbo) | 291: 35 with t(11;14); 194 (Ven + Bort + Dex), 97 (Bort + Dex + Pbo) | 1–3 | NR | With t(11;14): Ven + Bort + Dex: 36.8; Bort + Dex + Pbo: 9.3 |
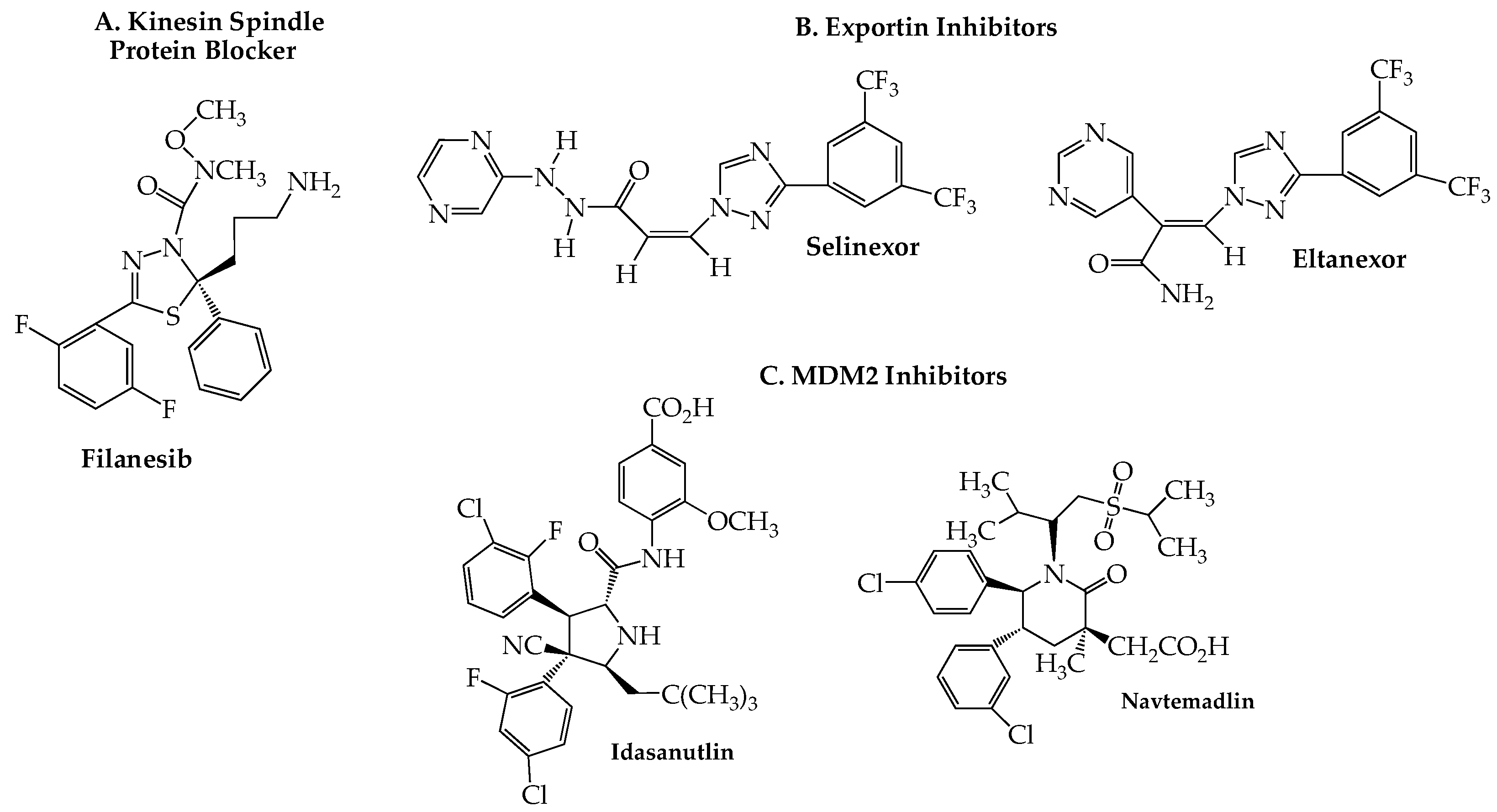
10. Kinesin Spindle Protein Inhibitors
11. Exportin Inhibitors
12. MDM2 Blockers
13. Kinase Inhibitors
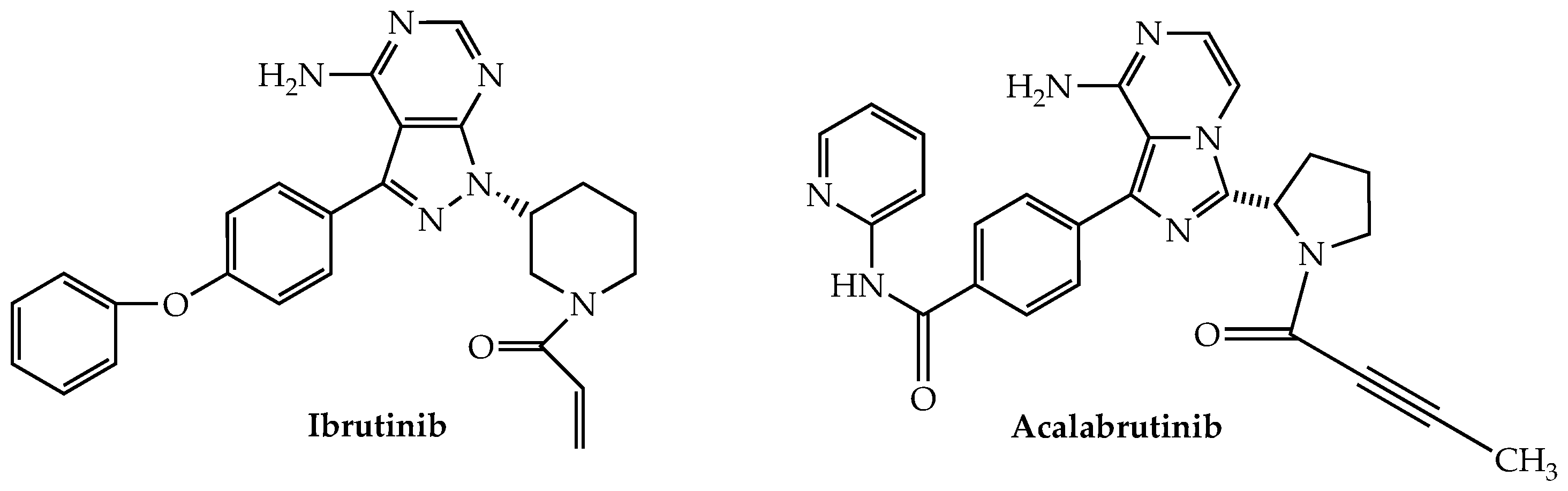
13.1. Bruton’s Tyrosine Kinase Inhibitors
13.2. Transforming Growth Factor Receptor Inhibitors
13.3. Raf-Mek-Erk Pathway Inhibitors
13.4. PI3K-Akt-mTOR Pathway Inhibitors
14. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munshi, N.C.; Longo, D.L.; Anderson, K.C. Plasma cell disorders. In Harrison’s Principles of Internal Medicine, 21st ed.; Loscalzo, J., Fauci, A., Kasper, D., Hauser, S., Longo, D., Jameson, J.L., Eds.; McGraw-Hill Education: New York, NY, USA, 2022; pp. 1–22. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Padala, S.A.; Barsouk, A.; Barsouk, A.; Rawla, P.; Vakiti, A.; Kolhe, R.; Kota, V.; Ajebo, G.H. Epidemiology, staging, and management of multiple myeloma. Med. Sci. 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Cancer statistics for African Americans, 2019. CA Cancer J. Clin. 2019, 69, 211–233. [Google Scholar] [CrossRef] [Green Version]
- Callander, N.S.; Baljevic, M.; Adekola, K.; Anderson, L.D.; Campagnaro, E.; Castillo, J.J.; Costello, C.; Devarakonda, S.; Elsedawy, N.; Faiman, M.; et al. NCCN guidelines® insights: Multiple myeloma, version 3.2022. J. Natl. Compr. Cancer Netw. 2022, 20, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.S.; Chen, C.I.; Reece, D.E. Current review on high-risk multiple myeloma. Curr. Hematol. Malig. Rep. 2017, 12, 96–108. [Google Scholar] [CrossRef]
- National Cancer Institute. SEER Cancer Statistics Review (CSR) 1975–2017: Myeloma; National Cancer Institute: Bethesda, MD, USA, 2020.
- Yamamoto, C.; Minakata, D.; Koyama, S.; Sekiguchi, K.; Fukui, Y.; Murahashi, R.; Nakashima, H.; Matsuoka, S.; Ikeda, T.; Kawaguchi, S.I.; et al. Daratumumab in first-line is cost-effective in transplant-eligible newly diagnosed myeloma patients. Blood 2022, 140, 594–607. [Google Scholar] [CrossRef]
- Blommestein, H.M.; Zweegman, S. Cost-effectiveness: Maximizing impact by meticulous data. Blood 2022, 140, 525–526. [Google Scholar] [CrossRef]
- Blokhin, N.; Larionov, L.; Perevodchikova, N.; Chebotareva, L.; Merkulova, N. Clinical experiences with sarcolysin in neoplastic diseases. Ann. N. Y. Acad. Sci. 1958, 68, 1128–1132. [Google Scholar] [CrossRef]
- Ray, A.; Ravillah, D.; Das, D.S.; Song, Y.; Nordstrom, E.; Gullbo, J.; Richardson, P.G.; Chauhan, D.; Anderson, K.C. A novel alkylating agent melflufen induces irreversible DNA damage and cytotoxicity in multiple myeloma cells. Br. J. Haematol. 2016, 174, 397–409. [Google Scholar] [CrossRef]
- Holstein, S.A.; Hillengass, J.; McCarthy, P.L. Melflufen: A next-generation nitrogen mustard. J. Clin. Oncol. 2021, 39, 836–839. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.H.; Bakker, N.A.; Sonneveld, P. Authors’ reply: Perspective: The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 25, 101528. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.H.; Dimopoulos, M.A.; Delimpasi, S.; Robak, P.; Coriu, D.; Legiec, W.; Pour, L.; Špička, I.; Masszi, T.; Doronin, V.; et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): A randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022, 9, e98–e110. [Google Scholar] [CrossRef] [PubMed]
- Olivier, T.; Prasad, V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 18, 101374. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Hurst, S.N.; Ebert, B.L. Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 2014, 3, e941742. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Handa, H. Molecular mechanisms of thalidomide and its derivatives. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2020, 96, 189–203. [Google Scholar] [CrossRef]
- Barankiewicz, J.; Salomon-Perzyński, A.; Misiewicz-Krzemińska, I.; Lech-Marańda, E. CRL4(CRBN) E3 ligase complex as a therapeutic target in multiple myeloma. Cancers 2022, 14, 4492. [Google Scholar] [CrossRef]
- LaPlante, G.; Zhang, W. Targeting the ubiquitin-proteasome system for cancer therapeutics by small-molecule Inhibitors. Cancers 2021, 13, 3079. [Google Scholar] [CrossRef]
- Faust, T.B.; Donovan, K.A.; Yue, H.; Chamberlain, P.P.; Fischer, E.S. Small-molecule approaches to targeted protein degradation. Annu. Rev. Cancer Biol. 2021, 5, 181–201. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Siegel, D.S.; Schiller, G.J.; Samaras, C.; Sebag, M.; Berdeja, J.; Ganguly, S.; Matous, J.; Song, K.; Seet, C.S.; Talamo, G.; et al. Pomalidomide, dexamethasone, and daratumumab in relapsed refractory multiple myeloma after lenalidomide treatment. Leukemia 2020, 34, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.; Weisel, K.; Moreau, P.; Anderson, L.D., Jr.; White, D.; San-Miguel, J.; Sonneveld, P.; Engelhardt, M.; Jenner, M.; Corso, A.; et al. Pomalidomide, bortezomib, and dexamethasone for multiple myeloma previously treated with lenalidomide (OPTIMISMM): Outcomes by prior treatment at first relapse. Leukemia 2021, 35, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Sperling, A.S.; Ebert, B.L. Cancer therapies based on targeted protein degradation—Lessons learned with lenalidomide. Nat. Rev. Clin. Oncol. 2021, 18, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Matyskiela, M.E.; Zhang, W.; Man, H.W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; LeBrun, L.; Pagarigan, B.; Carmel, G.; et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. [Google Scholar] [CrossRef]
- Lonial, S.; Popat, R.; Hulin, C.; Jagannath, S.; Oriol, A.; Richardson, P.G.; Facon, T.; Weisel, K.; Larsen, J.T.; Minnema, M.C.; et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): A multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematol. 2022, 9, e822–e832. [Google Scholar] [CrossRef]
- Hagner, P.R.; Man, H.W.; Fontanillo, C.; Wang, M.; Couto, S.; Breider, M.; Bjorklund, C.; Havens, C.G.; Lu, G.; Rychak, E.; et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 2015, 126, 779–789. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A first-in-human study of novel cereblon modulator avadomide (CC-122) in advanced malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.D.; Correa, M.; Nagy, M.A.; Alexander, M.; Plantevin, V.; Grant, V.; Whitefield, B.; Huang, D.; Kercher, T.; Harris, R.; et al. Discovery of CRBN E3 ligase modulator CC-92480 for the treatment of relapsed and refractory multiple myeloma. J. Med. Chem. 2020, 63, 6648–6676. [Google Scholar] [CrossRef]
- Wong, L.; Lamba, M.; Nunez, M.D.J.; Bauer, D.; Richardson, P.G.; Bahlis, N.J.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martinez-Lopez, J.; et al. Dose- and schedule-dependent immunomodulatory effects of the novel Celmod agent CC-92480 in patients with relapsed/refractory multiple myeloma. Blood 2020, 136, 47–48. [Google Scholar] [CrossRef]
- Richardson, P.G.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martinez, J.; Mateos, M.V.; Otero, P.R.; Lonial, S.; Popat, R.; Oriol, A.; et al. First-in-human phase I study of the novel CELMoD agent CC-92480 combined with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8500. [Google Scholar] [CrossRef]
- Lonial, S.; Berdeja, J.G.; Dimopoulos, M.A.; Jagannath, S.; Knop, S.; Quach, H.; Rodriguez-Otero, P.; Richardson, P.G.; Sorrell, A.; Chen, M.; et al. EXCALIBER: A phase 3 study comparing iberdomide, daratumumab, and dexamethasone (IberDd) with daratumumab, bortezomib, and dexamethasone (DVd) in patients with relapsed or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, S150. [Google Scholar] [CrossRef]
- Weisel, K.; Knop, S.; Lonial, S.; Richardson, P.G.; Popat, R.; Stadtmauer, E.A.; Larsen, J.T.; Oriol, A.; Jagannath, S.; Cook, G.; et al. Iberdomide (IBER) in combination with dexamethasone (DEX) and daratumumab (DARA), bortezomib (BORT), or carfilzomib (CFZ) in patients with relapsed/refractory multiple myeloma (RRMM). Oncol. Res. Treat. 2021, 44, 86–87. [Google Scholar]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Imtiaz, H.; Khan, M.; Ehsan, H.; Wahab, A.; Rafae, A.; Khan, A.Y.; Jamil, A.; Sana, M.K.; Jamal, A.; Ali, T.J.; et al. Efficacy and toxicity profile of carfilzomib-based regimens for treatment of newly diagnosed multiple myeloma: A systematic review. Onco Targets Ther. 2021, 14, 4941–4960. [Google Scholar] [CrossRef]
- Latif, A.; Kapoor, V.; Lateef, N.; Ahsan, M.J.; Usman, R.M.; Malik, S.U.; Ahmad, N.; Rosko, N.; Rudoni, J.; William, P.; et al. Incidence and management of carfilzomib-induced cardiovascular toxicity; a systematic review and meta-analysis. Cardiovasc. Hematol. Disord. Drug Targets 2021, 21, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Dasgupta, S.; Gong, Y.; Shah, U.A.; Fradley, M.G.; Cheng, R.K.; Roy, B.; Guha, A. Cardiotoxicity as an adverse effect of immunomodulatory drugs and proteasome inhibitors in multiple myeloma: A network meta-analysis of randomized clinical trials. Hematol. Oncol. 2022, 40, 233–242. [Google Scholar] [CrossRef]
- Xie, J.; Wan, N.; Liang, Z.; Zhang, T.; Jiang, J. Ixazomib—The first oral proteasome inhibitor. Leuk. Lymphoma 2019, 60, 610–618. [Google Scholar] [CrossRef]
- Richardson, P.G.; Moreau, P.; Laubach, J.P.; Gupta, N.; Hui, A.M.; Anderson, K.C.; San Miguel, J.F.; Kumar, S. The investigational proteasome inhibitor ixazomib for the treatment of multiple myeloma. Future Oncol. 2015, 11, 1153–1168. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Song, Y.; Richardson, P.; Trikha, M.; Chauhan, D.; Anderson, K.C. Synergistic anti-myeloma activity of the proteasome inhibitor marizomib and the IMiD immunomodulatory drug pomalidomide. Br. J. Haematol. 2015, 171, 798–812. [Google Scholar] [CrossRef] [Green Version]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): Final study results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, Y.; Liang, Q.; Qu, Y.; Zhang, L.; Liu, Y.; Fang, B.; Yun, Z.; Du, X.; Xi, Y.; et al. Phase I study of a novel oral proteasome inhibitor TQB3602 in relapsed/refractory multiple myeloma. Blood 2022, 140, 4396–4397. [Google Scholar] [CrossRef]
- Tang, W.; Li, Y.; Zhong, X.; Liang, Q.; Liu, Y.; Zeng, Y.; Fang, B.; Zheng, L.; Niu, T. Phase I study of TQB3602 capsule, an oral proteasome inhibitor, in relapsed refractory multiple myeloma. Hemasphere 2022, 6, 843–844. [Google Scholar] [CrossRef]
- Lei, H.; Wang, J.; Hu, J.; Zhu, Q.; Wu, Y. Deubiquitinases in hematological malignancies. Biomark. Res. 2021, 9, 66. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef] [Green Version]
- Rowinsky, E.K.; Paner, A.; Berdeja, J.G.; Paba-Prada, C.; Venugopal, P.; Porkka, K.; Gullbo, J.; Linder, S.; Loskog, A.; Richardson, P.G.; et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig. New Drugs 2020, 38, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Zhang, M.; Qiu, X.; Lu, Y. Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemother. Pharmacol. 2018, 81, 797–808. [Google Scholar] [CrossRef]
- Zhou, L.; Jia, L. Targeting protein neddylation for cancer therapy. Adv. Exp. Med. Biol. 2020, 1217, 297–315. [Google Scholar] [CrossRef]
- Yu, Q.; Jiang, Y.; Sun, Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm. Sin. B 2020, 10, 746–765. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Yu, G.; Chen, P.; Li, H.; Wei, D.; Zhu, J.; Xie, L.; Jia, H.; Shi, J.; et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J. Natl. Cancer Inst. 2014, 106, dju083. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Yang, J.P.; Cao, Y.; Peng, L.X.; Zheng, L.S.; Sun, R.; Meng, D.F.; Wang, M.Y.; Mei, Y.; Qiang, Y.Y.; et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017, 8, e2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillin, D.W.; Jacobs, H.M.; Delmore, J.E.; Buon, L.; Hunter, Z.R.; Monrose, V.; Yu, J.; Smith, P.G.; Richardson, P.G.; Anderson, K.C.; et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther. 2012, 11, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faessel, H.M.; Mould, D.R.; Zhou, X.; Faller, D.V.; Sedarati, F.; Venkatakrishnan, K. Population pharmacokinetics of pevonedistat alone or in combination with standard of care in patients with solid tumours or haematological malignancies. Br. J. Clin. Pharmacol. 2019, 85, 2568–2579. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, C.; Muraoka, H.; Ochiiwa, H.; Tsuji, S.; Hashimoto, A.; Kazuno, H.; Nakagawa, F.; Komiya, Y.; Suzuki, S.; Takenaka, T.; et al. TAS4464, a highly potent and selective inhibitor of NEDD8-activating enzyme, suppresses neddylation and shows antitumor activity in diverse cancer models. Mol. Cancer Ther. 2019, 18, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.C.; Young, N.L. Combinations of histone post-translational modifications. Biochem. J. 2021, 478, 511–532. [Google Scholar] [CrossRef]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef]
- Ilango, S.; Paital, B.; Jayachandran, P.; Padma, P.R.; Nirmaladevi, R. Epigenetic alterations in cancer. Front. Biosci. 2020, 25, 1058–1109. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Caprio, C.; Sacco, A.; Giustini, V.; Roccaro, A.M. Epigenetic aberrations in multiple myeloma. Cancers 2020, 12, 2996. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting histone deacetylases: Opportunities for cancer treatment and chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Siegel, D.S.; Lonial, S.; Qi, J.; Hajek, R.; Facon, T.; Rosinol, L.; Williams, C.; Blacklock, H.; Goldschmidt, H.; et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): A multicentre, randomised, double-blind study. Lancet Oncol. 2013, 14, 1129–1140. [Google Scholar] [CrossRef]
- Richardson, P.G.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Guenther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: Outcomes by prior treatment. Blood 2016, 127, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.H.; Hou, J.; Zhang, Y.M.; Zhou, Y.B.; Jia, L.; Nan, F.J. Phase 1 study of bisthianostat, an orally efficacious pan-HDAC inhibitor: Part results of safety, pharmacokinetics and efficacy in patients with relapsed or refractory multiple myeloma. Blood 2019, 134, 5591. [Google Scholar] [CrossRef]
- Zhou, Y.B.; Zhang, Y.M.; Huang, H.H.; Shen, L.J.; Han, X.F.; Hu, X.B.; Yu, S.D.; Gao, A.H.; Sheng, L.; Su, M.B.; et al. Pharmacodynamic, pharmacokinetic, and phase 1a study of bisthianostat, a novel histone deacetylase inhibitor, for the treatment of relapsed or refractory multiple myeloma. Acta Pharmacol. Sin. 2021, 43, 1091–1099. [Google Scholar] [CrossRef]
- Vogl, D.T.; Raje, N.S.; Jagannath, S.; Richardson, P.G.; Hari, P.; Orlowski, R.Z.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Yan, L.; Shang, J.; Jin, S.; Shi, X.; Yan, S.; Wu, D.; Fu, C. Phase 2 Suzhou MM02 study: Chidamide with VRD versus VRD in newly diagnosed high risk transplant eligible multiple myeloma patients. Blood 2021, 138, 4765. [Google Scholar] [CrossRef]
- Sborov, D.W.; Benson, D.M.; Williams, N.; Huang, Y.; Bowers, M.A.; Humphries, K.; Efebera, Y.; Devine, S.; Hofmeister, C.C. Lenalidomide and vorinostat maintenance after autologous transplant in multiple myeloma. Br. J. Haematol. 2015, 171, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.S.; Dimopoulos, M.; Jagannath, S.; Goldschmidt, H.; Durrant, S.; Kaufman, J.L.; Leleu, X.; Nagler, A.; Offner, F.; Graef, T.; et al. VANTAGE 095: An international, multicenter, open-label study of vorinostat (MK-0683) in combination with bortezomib in patients with relapsed and refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2016, 16, 329–334.e1. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Pawlyn, C.; Tillotson, A.L.; Sherratt, D.; Flanagan, L.; Low, E.; Morgan, G.J.; Williams, C.; Kaiser, M.; Davies, F.E.; et al. Bortezomib, vorinostat, and dexamethasone combination therapy in relapsed myeloma: Results of the phase 2 MUK four trial. Clin. Lymphoma Myeloma Leuk. 2021, 21, 154–161.e3. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Mina, R.; Shah, J.J.; Laubach, J.P.; Nooka, A.K.; Lewis, C.; Gleason, C.; Sharp, C.; Harvey, R.D.; Heffner, L.T.; et al. Phase 1 trial evaluating vorinostat plus bortezomib, lenalidomide, and dexamethasone in patients with newly diagnosed multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2020, 20, 797–803. [Google Scholar] [CrossRef]
- Vesole, D.H.; Bilotti, E.; Richter, J.R.; McNeill, A.; McBride, L.; Raucci, L.; Anand, P.; Bednarz, U.; Ivanovski, K.; Smith, J.; et al. Phase I study of carfilzomib, lenalidomide, vorinostat, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Br. J. Haematol. 2015, 171, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Waldschmidt, J.M.; Keller, A.; Ihorst, G.; Grishina, O.; Muller, S.; Wider, D.; Frey, A.V.; King, K.; Simon, R.; May, A.; et al. Safety and efficacy of vorinostat, bortezomib, doxorubicin and dexamethasone in a phase I/II study for relapsed or refractory multiple myeloma (VERUMM study: Vorinostat in elderly, relapsed and unfit multiple myeloma). Haematologica 2018, 103, e473–e479. [Google Scholar] [CrossRef] [Green Version]
- Niesvizky, R.; Richardson, P.G.; Gabrail, N.Y.; Madan, S.; Yee, A.J.; Quayle, S.N.; Almeciga-Pinto, I.; Jones, S.S.; Houston, L.; Hayes, D.; et al. ACY-241, a novel, HDAC6 selective inhibitor: Synergy with immunomodulatory (IMiD (R)) drugs in multiple myeloma (MM) cells and early clinical results (ACE-MM-200 study). Blood 2015, 126, 3. [Google Scholar] [CrossRef]
- Niesvizky, R.; Richardson, P.G.; Yee, A.J.; Nooka, A.K.; Raab, M.S.; Shain, K.H.; Gabrail, N.Y.; Matous, J.; Agarwal, A.B.; Hoffman, J.; et al. Selective HDAC6 inhibitor ACY-241, an oral tablet, combined with pomalidomide and dexamethasone: Safety and efficacy of escalation and expansion cohorts in patients with relapsed or relapsed-and-refractory multiple myeloma (ACE-MM-200 study). Blood 2016, 128, 7. [Google Scholar] [CrossRef]
- North, B.J.; Almeciga-Pinto, I.; Tamang, D.; Yang, M.; Jones, S.S.; Quayle, S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE 2017, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.J.; Yao, W.Q.; Yan, L.Z.; Shi, X.L.; Wang, R.J.; Yan, S.; Liu, Y.; Wu, D.P.; Cheng, F.C. Initial safety and efficacy of dose-escalating HDACs inhibitor chidamide with VRD (Chi-VRD) treatment for newly-diagnosed high-risk transplant eligible multiple myeloma patients. Blood 2019, 134, 1855. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef]
- Gokani, S.; Bhatt, L.K. Bromodomains: A novel target for the anticancer therapy. Eur. J. Pharmacol. 2021, 911, 174523. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016, 106, 1–18. [Google Scholar] [CrossRef]
- Llombart, V.; Mansour, M.R. Therapeutic targeting of “undruggable” MYC. eBioMedicine 2022, 75, 103756. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.W.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 903–916. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Gelato, K.A.; Fernandez-Montalvan, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef]
- Martin, M.P.; Olesen, S.H.; Georg, G.I.; Schonbrunn, E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Aird, F.; Kandela, I.; Mantis, C. Replication study: BET bromodomain inhibition as a therapeutic strategy to target c-Myc. eLife 2017, 6, e21253. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Boi, M.; Vurchio, V.; Ercole, E.; Machiorlatti, R.; Messana, K.; Landra, I.; Urigu, S.; Aliberti, S.; Riveiro, E.; et al. OTX015, a novel BET inhibitor, is a promising anticancer agent for multiple myeloma. Cancer Res. 2014, 74, 5531. [Google Scholar] [CrossRef]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Cote, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R.; et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, K.; Nooka, A.; Quach, H.; Htut, M.; Popat, R.; Liedtke, M.; Tuchman, S.A.; Laubach, J.P.; Gasparetto, C.; Chanan-Khan, A.A.; et al. Open label, multicenter, dose-escalation/ expansion phase Ib study to evaluate safety and activity of BET Inhibitor RO6870810 (RO), given as monotherapy to patients (pts) with advanced multiple myeloma. Blood 2020, 136, 12. [Google Scholar] [CrossRef]
- Siu, K.T.; Eda, H.; Santo, L.; Ramachandran, J.; Koulnis, M.; Mertz, J.; Sims, R.J.; Cooper, M.; Raje, N.S. Effect of the BET inhibitor, CPI-0610, alone and in combination with lenalidomide in multiple myeloma. Blood 2015, 126, 4255. [Google Scholar] [CrossRef]
- Klener, P.; Sovilj, D.; Renesova, N.; Andera, L. BH3 mimetics in hematologic malignancies. Int. J. Mol. Sci. 2021, 22, 10157. [Google Scholar] [CrossRef]
- Yap, J.L.; Chen, L.; Lanning, M.E.; Fletcher, S. Expanding the cancer arsenal with targeted therapies: Disarmament of the anti-apoptotic Bcl-2 proteins by small-molecules. J. Med. Chem. 2016, 60, 821–838. [Google Scholar] [CrossRef]
- Krishna, S.; Kumar, S.B.; Murthy, T.P.K.; Murahari, M. Structure-based design approach of potential BCL-2 inhibitors for cancer chemotherapy. Comput. Biol. Med. 2021, 134, 104455. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Kumar, S.; Harrison, S.J.; Cavo, M.; de La Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Updated results from BELLINI, a phase III study of venetoclax or placebo in combination with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38, 8509. [Google Scholar] [CrossRef]
- Mateos, M.V.; Moreau, P.; Dimopoulos, M.A.; Hong, W.J.; Cooper, S.; Yu, Y.; Jalaluddin, M.; Ross, J.A.; Karve, S.; Coppola, S.; et al. A phase III, randomized, multicenter, open-label study of venetoclax or pomalidomide in combination with dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38, TPS8554. [Google Scholar] [CrossRef]
- Fu, C.C.; Chen, Z.; Li, W.S.; Men, L.C.; Wu, D.P.; Yang, D.J.; Zhai, Y.F. Trial in progress: Phase 1b/2 open-label study of lisaftoclax (APG-2575) monotherapy or in combination with lenalidomide/dexamethasone in patients with relapsed or refractory multiple myeloma (R/R MM). Blood 2021, 138, 4764. [Google Scholar] [CrossRef]
- Hu, N.; Guo, Y.H.; Xue, H.; Liu, Y.; Guo, Y.; Wang, F.; Song, X.M.; Guo, Y.; Chen, S.S.; Xu, H.P.; et al. Preclinical characterization of BGB-11417, a potent and selective Bcl-2 inhibitor with superior antitumor activities in haematological tumor models. Cancer Res. 2020, 80, 3077. [Google Scholar] [CrossRef]
- Quach, H.; Rajagopal, R.; Spencer, A.; Low, M.; Kazandjian, D.; Crescenzo, R.; Du, C.; Patel, S.; Mundra, V.; Cheng, H.; et al. Preliminary safety of a Bcl-2 Inhibitor, Bgb-11417, in patients with relapsed/refractory multiple myeloma harboring t(11,14): A non-randomized, open-label, phase 1b/2 study. Blood 2022, 140, 7269–7271. [Google Scholar] [CrossRef]
- Lu, X.; Liang, H.; Orvig, C.; Chen, Z.F. Peptide and small molecule inhibitors targeting myeloid cell leukemia 1 (Mcl-1) as novel antitumor agents. Curr. Mol. Med. 2021, 21, 426–439. [Google Scholar] [CrossRef]
- Spencer, A.; Rosenberg, A.S.; Jakubowiak, A.; Raje, N.; Chatterjee, M.; Trudel, S.; Bahlis, N.J.; Siegel, D.S.; Wilop, S.; Harrison, S.J.; et al. A phase 1, first-in-human study of AMG 176, a selective MCL-1 Inhibitor, in patients with relapsed or refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, E53–E54. [Google Scholar] [CrossRef]
- Halilovic, E.; Chanrion, M.; Mistry, P.; Wartmann, M.; Qiu, S.M.; Sanghavi, S.; Chen, Y.; Lysiak, G.; Maragno, A.L.; Pfaar, U.; et al. MIK665/S64315, a novel Mcl-1 inhibitor, in combination with Bcl-2 inhibitors exhibits strong synergistic antitumor activity in a range of hematologic malignancies. Cancer Res. 2019, 79, 4477. [Google Scholar] [CrossRef]
- Bhagwat, N.; Grego, A.; Gowen-MacDonald, W.; Wang, M.; Cowart, M.; Wu, X.W.; Zhuo, J.C.; Combs, A.; Ruggeri, B.; Scherle, P.; et al. Preclinical characterization of PRT1419, a potent, selective and orally available inhibitor of MCL1. Cancer Res. 2021, 81, 983. [Google Scholar] [CrossRef]
- Matulis, S.M.; Gupta, V.A.; Brown, I.; Keats, J.J.; Secrist, P.; Cidado, J.; Tron, A.E.; Neri, P.; Bahlis, N.; Kaufman, J.L.; et al. Preclinical activity of novel MCL1 inhibitor AZD5991 in multiple myeloma. Blood 2018, 132, 952. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, S.; Wang, Y.; Mo, Y.; Xiong, F.; Zhang, S.; Zeng, Z.; Xiong, W.; Li, G.; Chen, H.; et al. Gossypol induces apoptosis of multiple myeloma cells through the JUN-JNK pathway. Am. J. Cancer Res. 2020, 10, 870–883. [Google Scholar]
- Ailawadhi, S.; Alegria, V.R.; Ahmed, S.; Laplant, B.; Manna, A.; Parrondo, R.; Roy, V.; Sher, T.; Edwards, B.; Lanier, S.; et al. Phase I Study of a novel Bcl-2 inhibitor, AT-101 in combination with lenalidomide and dexamethasone in patients with relapsed and/or refractory multiple myeloma (RRMM). Blood 2019, 134, 3137. [Google Scholar] [CrossRef]
- Parrondo, R.D.; Paulus, A.; Ailawadhi, S. Updates in the Use of BCL-2-family small molecule inhibitors for the treatment of relapsed/refractory multiple myeloma. Cancers 2022, 14, 3330. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; Baz, R.; Harrison, S.J.; Quach, H.; Ho, S.J.; Vangsted, A.J.; Plesner, T.; Moreau, P.; Gibbs, S.D.; Coppola, S.; et al. Phase I study of venetoclax plus daratumumab and dexamethasone, with or without bortezomib, in patients with relapsed or refractory multiple myeloma with and without t(11;14). J. Clin. Oncol. 2021, 39, 3602–3612. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Quach, H.; Baz, R.; Vangsted, A.J.; Ho, S.J.; Harrison, S.J.; Plesner, T.; Moreau, P.; Gibbs, S.D.; Medvedova, E.; et al. Safety and preliminary efficacy from the expansion cohort of a phase 1/2 study of venetoclax plus daratumumab and dexamethasone vs daratumumab plus bortezomib and dexamethasone in patients with t(11;14) relapsed/refractory multiple myeloma. Blood 2021, 138 (Suppl. S1), 817. [Google Scholar] [CrossRef]
- Costa, L.J.; Davies, F.E.; Monohan, G.P.; Kovacsovics, T.J.; Burwick, N.; Jakubowiak, A.J.; Kaufman, J.L.; Hong, W.J.; Dail, M.; Salem, A.H.; et al. Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 3748–3759. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J.L.; Gasparetto, C.; Schjesvold, F.H.; Moreau, P.; Touzeau, C.; Facon, T.; Boise, L.H.; Jiang, Y.; Yang, X.; Dunbar, F.; et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am. J. Hematol. 2021, 96, 418–427. [Google Scholar] [CrossRef]
- Kumar, S.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.J.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Final overall survival results from BELLINI, a phase 3 study of venetoclax or placebo in combination with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Blood 2021, 138, 84. [Google Scholar] [CrossRef]
- Shahin, R.; Aljamal, S. Kinesin spindle protein inhibitors in cancer: From high throughput screening to novel therapeutic strategies. Future Sci. OA 2022, 8, Fso778. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J.; Anderson, D.; Williams, L.; Rieger, R. ARRY-520 combined with pomalidomide displays enhanced anti-tumor activity in preclinical models of multiple myeloma. Blood 2013, 122, 3167. [Google Scholar] [CrossRef]
- Woessner, R.; Tunquist, B.; Lemieux, C.; Chlipala, E.; Jackinsky, S.; Dewolf, W., Jr.; Voegtli, W.; Cox, A.; Rana, S.; Lee, P.; et al. ARRY-520, a novel KSP inhibitor with potent activity in hematological and taxane-resistant tumor models. Anticancer Res. 2009, 29, 4373–4380. [Google Scholar] [PubMed]
- Shah, J.J.; Kaufman, J.L.; Zonder, J.A.; Cohen, A.D.; Bensinger, W.I.; Hilder, B.W.; Rush, S.A.; Walker, D.H.; Tunquist, B.J.; Litwiler, K.S.; et al. A phase 1 and 2 study of filanesib alone and in combination with low-dose dexamethasone in relapsed/refractory multiple myeloma. Cancer 2017, 123, 4617–4630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, A.; Htut, M.; Zonder, J.A.; Fay, J.W.; Jakubowiak, A.J.; Levy, J.B.; Lau, K.; Burt, S.M.; Tunquist, B.J.; Hilder, B.W.; et al. A phase 1 dose-escalation study of filanesib plus bortezomib and dexamethasone in patients with recurrent/refractory multiple myeloma. Cancer 2016, 122, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Kaufman, J.L.; Htut, M.; Agrawal, M.; Mazumder, A.; Cornell, R.F.; Zonder, J.A.; Fay, J.W.; Modiano, M.R.; Moshier, E.L.; et al. Filanesib plus bortezomib and dexamethasone in relapsed/refractory t(11;14) and 1q21 gain multiple myeloma. Cancer Med. 2022, 11, 358–370. [Google Scholar] [CrossRef]
- Hanamura, I. Gain/amplification of chromosome arm 1q21 in multiple myeloma. Cancers 2021, 13, 256. [Google Scholar] [CrossRef]
- Schmidt, T.M.; Barwick, B.G.; Joseph, N.; Heffner, L.T.; Hofmeister, C.C.; Bernal, L.; Dhodapkar, M.V.; Gupta, V.A.; Jaye, D.L.; Wu, J.; et al. Gain of chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Mohan, M.; Weinhold, N.; Schinke, C.; Thanedrarajan, S.; Rasche, L.; Sawyer, J.R.; Tian, E.; van Rhee, F.; Zangari, M. Daratumumab in high-risk relapsed/refractory multiple myeloma patients: Adverse effect of chromosome 1q21 gain/amplification and GEP70 status on outcome. Br. J. Haematol. 2020, 189, 67–71. [Google Scholar] [CrossRef]
- Lee, H.C.; Shah, J.J.; Feng, L.; Manasanch, E.E.; Lu, R.; Morphey, A.; Crumpton, B.; Patel, K.K.; Wang, M.L.; Alexanian, R.; et al. A phase 1 study of filanesib, carfilzomib, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Blood Cancer J. 2019, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Ocio, E.M.; Motllo, C.; Rodriguez-Otero, P.; Martinez-Lopez, J.; Cejalvo, M.J.; Martin-Sanchez, J.; Blade, J.; Garcia-Malo, M.D.; Dourdil, M.V.; Garcia-Mateo, A.; et al. Filanesib in combination with pomalidomide and dexamethasone in refractory MM patients: Safety and efficacy, and association with alpha 1-acid glycoprotein (AAG) levels. Phase Ib/II Pomdefil clinical trial conducted by the Spanish MM group. Br. J. Haematol. 2021, 192, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Alexandrakis, M.G.; Passam, F.H.; Ganotakis, E.S.; Sfiridaki, K.; Xilouri, I.; Perisinakis, K.; Kyriakou, D.S. The clinical and prognostic significance of erythrocyte sedimentation rate (ESR), serum interleukin-6 (IL-6) and acute phase protein levels in multiple myeloma. Clin. Lab. Haematol. 2003, 25, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Tunquist, B.; Brown, K.; Hingorani, G.; Aitchison, R.; Regensburger, J.; Lonial, S.; Kaufman, J.; Zonder, J.; Cohen, A.; Bensinger, W.; et al. Alpha 1-acid glycoprotein (AAG) is a potential patient selection biomarker for improved clinical activity of ARRY-520 in relapsed and refractory multiple myeloma (MM). Haematologica 2013, 98, 328–329. [Google Scholar]
- Algarin, E.M.; Hernandez-Garcia, S.; Garayoa, M.; Ocio, E.M. Filanesib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs 2020, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zonder, J.A.; Usiinani, S.; Scott, E.C.; Harneister, C.C.; Lendvai, N.; Berdeja, J.C.; Anderson, L.D.; Hari, P.; Singhal, S.; Orloff, G.; et al. Phase 2 study of carfilzomib (CFZ) with or without filanesib (FIL) in patients with advanced multiple myeloma (MM). Blood 2015, 126, 728. [Google Scholar] [CrossRef]
- Lonial, S.; Delforge, M.; Einsele, H.; Moreau, P.; Kaiser, M.; Dimopoulos, M.A.; Oriol, A.; Gyger, M.; Hilder, B.; Ptaszynski, A.M.; et al. The AfFIRM study: A multicenter phase 2 study of single-agent filanesib (ARRY-520) in patients with advanced multiple myeloma. J. Clin. Oncol. 2015, 33, TPS8613. [Google Scholar] [CrossRef]
- Schwartz, T.U. Solving the nuclear pore puzzle. Science 2022, 376, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Raices, M.; D’Angelo, M.A. Structure, maintenance, and regulation of nuclear pore complexes: The gatekeepers of the eukaryotic genome. Cold Spring Harb. Perspect. Biol. 2022, 14, a040691. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Li, S.; Fu, J.; Lu, C.; Mapara, M.Y.; Raza, S.; Hengst, U.; Lentzsch, S. Elevated translation initiation factor eIF4E Is an attractive therapeutic target in multiple myeloma. Mol. Cancer Ther. 2016, 15, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.; Munshi, A.A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Braggio, E.; Kortuem, K.M.; Egan, J.B.; Zhu, Y.X.; Xin, C.S.; Tiedemann, R.E.; Palmer, S.E.; Garbitt, V.M.; McCauley, D.; et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Selinexor: First global approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Restrepo, P.; Bhalla, S.; Ghodke-Puranik, Y.; Aleman, A.; Leshchenko, V.; Melnekoff, D.T.; Agte, S.; Jiang, J.; Madduri, D.; Richter, J.; et al. A three-gene signature predicts response to selinexor in multiple myeloma. JCO Precis. Oncol. 2022, 6, e2200147. [Google Scholar] [CrossRef]
- Mateos, M.V.; Gavriatopoulou, M.; Facon, T.; Auner, H.W.; Leleu, X.; Hájek, R.; Dimopoulos, M.A.; Delimpasi, S.; Simonova, M.; Špička, I.; et al. Effect of prior treatments on selinexor, bortezomib, and dexamethasone in previously treated multiple myeloma. J. Hematol. Oncol. 2021, 14, 59. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Cornell, R.F.; Baz, R.; Richter, J.R.; Rossi, A.; Vogl, D.T.; Chen, C.; Shustik, C.; Alvarez, M.J.; Shen, Y.; Unger, T.J.; et al. A phase 1 clinical trial of oral eltanexor in patients with relapsed or refractory multiple myeloma. Am. J. Hematol. 2022, 97, E54–E58. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.L.; Grant, S.; Shain, K.H.; Dalton, W.S.; Dai, Y.; Meads, M.; Baz, R.; Kauffman, M.; Shacham, S.; et al. Treatment of acquired drug resistance in multiple myeloma by combination therapy with XPO1 and topoisomerase II inhibitors. J. Hematol. Oncol. 2016, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, G.; Daniele, P.; Breeze, J.; Li, L.; Shah, J.; Shacham, S.; Kauffman, M.; Engelhardt, M.; Chari, A.; Nooka, A.; et al. Quality of life analyses in patients with multiple myeloma: Results from the selinexor (KPT-330) treatment of refractory myeloma (STORM) phase 2b study. BMC Cancer 2021, 21, 993. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Florendo, E.; Mancia, I.S.; Cho, H.; Madduri, D.; Parekh, S.; Richter, J.; Dhadwal, A.; Thomas, J.; Jiang, G.; et al. Optimal supportive care with selinexor improves outcomes in patients with relapsed/refractory multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, e975–e984. [Google Scholar] [CrossRef] [PubMed]
- Gasparetto, C.; Lentzsch, S.; Schiller, G.J.; Callander, N.S.; Tuchman, S.; Bahlis, N.J.; White, D.; Chen, C.; Baljevic, M.; Sutherland, H.J.; et al. Selinexor, daratumumab, and dexamethasone in patients with relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2020, 38, 8510. [Google Scholar] [CrossRef]
- Gasparetto, C.; Schiller, G.J.; Tuchman, S.; Callander, N.S.; Baljevic, M.; Lentzsch, S.; Rossi, A.C.; Kotb, R.; White, D.; Bahlis, N.J.; et al. Once weekly selinexor, carfilzomib, and dexamethasone (SKd) in carfilzomib nonrefractory multiple myeloma (MM) patients. J. Clin. Oncol. 2021, 39, 8038. [Google Scholar] [CrossRef]
- Turner, J.G.; Cui, Y.; Bauer, A.A.; Dawson, J.L.; Gomez, J.A.; Kim, J.; Cubitt, C.L.; Nishihori, T.; Dalton, W.S.; Sullivan, D.M. Melphalan and exportin 1 inhibitors exert synergistic anti-tumor effects in preclinical models of human multiple myeloma. Cancer Res. 2020, 80, 5344–5354. [Google Scholar] [CrossRef] [PubMed]
- Jakubowiak, A.J.; Jasielec, J.K.; Rosenbaum, C.A.; Cole, C.E.; Chari, A.; Mikhael, J.; Nam, J.; McIver, A.; Severson, E.; Stephens, L.A.; et al. Phase 1 study of selinexor plus carfilzomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Br. J. Haematol. 2019, 86, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Delimpasi, S.; Mateos, M.V.; Auner, H.W.; Gavriatopoulou, M.; Dimopoulos, M.A.; Quach, H.; Pylypenko, H.; Hájek, R.; Leleu, X.; Dolai, T.K.; et al. Efficacy and tolerability of once-weekly selinexor, bortezomib, and dexamethasone in comparison with standard twice-weekly bortezomib and dexamethasone in previously treated multiple myeloma with renal impairment: Subgroup analysis from the BOSTON study. Am. J. Hematol. 2021, 97, E83–E86. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Gavriatopoulou, M.; Delimpasi, S.; Simonova, M.; Spicka, I.; Pour, L.; Dimopoulos, M.A.; Kriachok, I.; Pylypenko, H.; Leleu, X.; et al. Effect of age and frailty on the efficacy and tolerability of once-weekly selinexor, bortezomib, and dexamethasone in previously treated multiple myeloma. Am. J. Hematol. 2021, 96, 708–718. [Google Scholar] [CrossRef] [PubMed]
- White, D.; Chen, C.; Baljevic, M.; Tuchman, S.; Bahlis, N.J.; Schiller, G.J.; Lipe, B.; Kotb, R.; Sutherland, H.J.; Madan, S.; et al. Oral selinexor, pomalidomide, and dexamethasone (XPd) at recommended phase 2 dose in relapsed refractory multiple myeloma (MM). J. Clin. Oncol. 2021, 39, 8018. [Google Scholar] [CrossRef]
- Gasparetto, C.; Lipe, B.; Tuchman, S.; Callander, N.S.; Lentzsch, S.; Baljevic, M.; Rossi, A.C.; Bahlis, N.J.; White, D.; Chen, C.; et al. Once weekly selinexor, carfilzomib, and dexamethasone (SKd) in patients with relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2020, 38 (Suppl. S15), 8530. [Google Scholar] [CrossRef]
- Rodriguez-Otero, P.; De La Calle, V.G.; Sureda, A.; De Arriba, F.; Segura, M.R.; Ribas, P.; Gonzalez, A.P.; Gonzalez-Montes, Y.; Oriol, A.; Martinez-Lopez, J.; et al. Selinexor in combination with daratumumab-bortezomib and dexamethasone for the treatment of relapse or refractory multiple myeloma: Initial results of the phase 2, open-label, multicenter GEM-Selibordara study. Blood 2021, 138, 1677. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, F.; Li, J.; Zhang, C.; Li, W.; Cheng, H.; Cai, H.; Chen, B.; Guo, J.; Mei, J.; et al. Preliminary results from the launch study-a multicenter, open-label study of selinexor, dexamethasone and chemotherapy drugs in relapsed/refractory multiple myeloma. Blood 2022, 140, 12613. [Google Scholar] [CrossRef]
- Qiu, L.; Xia, Z.; Fu, C.; Chen, W.; Chang, C.; Fang, B.; An, G.; Wei, Y.; Cai, Z.; Gao, S.; et al. Selinexor plus low-dose dexamethasone in Chinese patients with relapsed/refractory multiple myeloma previously treated with an immunomodulatory agent and a proteasome inhibitor (MARCH): A phase II, single-arm study. BMC Med. 2022, 20, 108. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Tan, B.X.; Lane, D. How the other half lives: What p53 does when it is not being a transcription factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double hit events involving tumor suppressor genes underlie relapse from chemotherapy: Myeloma as a model. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Melvin, A.T.; Dumberger, L.D.; Woss, G.S.; Waters, M.L.; Allbritton, N.L. Identification of a p53-based portable degron based on the MDM2-p53 binding region. Analyst 2016, 141, 570–578. [Google Scholar] [CrossRef]
- Raj, N.; Attardi, L.D. The transactivation domains of the p53 protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-Krzeminska, I.; Krzeminski, P.; Gutierrez, N.C. Molecular mechanisms of p53 deregulation in cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Price, S.; Al-Bahou, A. Navtemadlin. Drugs Future 2022, 47, 247–259. [Google Scholar] [CrossRef]
- Gluck, W.L.; Gounder, M.M.; Frank, R.; Eskens, F.; Blay, J.Y.; Cassier, P.A.; Soria, J.C.; Chawla, S.; de Weger, V.; Wagner, A.J.; et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Investig. New Drugs 2020, 38, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Good, L.; Benner, B.; Carson, W.E. Bruton’s tyrosine kinase: An emerging targeted therapy in myeloid cells within the tumor microenvironment. Cancer Immunol. Immunother. 2021, 70, 2439–2451. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Zheng, J.; Han, C.; Jin, J.; Han, H.; Liu, Y.; Lau, Y.L.; Tu, W.; Cao, X. Tyrosine kinase Btk is required for NK cell activation. J. Biol. Chem. 2012, 287, 23769–23778. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, X.; Cao, X.; Xu, S. T-cell expression of Bruton’s tyrosine kinase promotes autoreactive T-cell activation and exacerbates aplastic anemia. Cell. Mol. Immunol. 2020, 17, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dong, Y.; Jiang, Q.L.; Zhang, B.; Hu, A.M. Bruton’s tyrosine kinase: Potential target in human multiple myeloma. Leuk. Lymphoma 2014, 55, 177–181. [Google Scholar] [CrossRef]
- Richardson, P.G.; Bensinger, W.I.; Huff, C.A.; Costello, C.L.; Lendvai, N.; Berdeja, J.G.; Anderson, L.D., Jr.; Siegel, D.S.; Lebovic, D.; Jagannath, S.; et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: Phase 2 trial results. Br. J. Haematol. 2018, 180, 821–830. [Google Scholar] [CrossRef]
- Hajek, R.; Pour, L.; Ozcan, M.; Martin Sánchez, J.; García Sanz, R.; Anagnostopoulos, A.; Oriol, A.; Cascavilla, N.; Terjung, A.; Lee, Y.; et al. A phase 2 study of ibrutinib in combination with bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma. Eur. J. Haematol. 2020, 104, 435–442. [Google Scholar] [CrossRef]
- Woyach, J.A. Ibrutinib and Aspergillus: A Btk-targeted risk. Blood 2018, 132, 1869–1870. [Google Scholar] [CrossRef]
- Rogers, K.A.; Mousa, L.; Zhao, Q.; Bhat, S.A.; Byrd, J.C.; El Boghdadly, Z.; Guerrero, T.; Levine, L.B.; Lucas, F.; Shindiapina, P.; et al. Incidence of opportunistic infections during ibrutinib treatment for B-cell malignancies. Leukemia 2019, 33, 2527–2530. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Cornell, R.F.; Gasparetto, C.; Karanes, C.; Matous, J.V.; Niesvizky, R.; Lunning, M.; Usmani, S.Z.; Anderson, L.D., Jr.; Chhabra, S.; et al. Final analysis of a phase 1/2b study of ibrutinib combined with carfilzomib/dexamethasone in patients with relapsed/refractory multiple myeloma. Hematol. Oncol. 2020, 38, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Ailawadhi, S.; Parrondo, R.D.; Moustafa, M.A.; LaPlant, B.R.; Alegria, V.; Chapin, D.; Roy, V.; Sher, T.; Paulus, A.; Chanan-Khan, A.A. Ibrutinib, lenalidomide and dexamethasone in patients with relapsed and/or refractory multiple myeloma: Phase I trial results. Hematol. Oncol. 2022, 40, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Kyrtsonis, M.C.; Repa, C.; Dedoussis, G.V.; Mouzaki, A.; Simeonidis, A.; Stamatelou, M.; Maniatis, A. Serum transforming growth factor-beta 1 is related to the degree of immunoparesis in patients with multiple myeloma. Med. Oncol. 1998, 15, 124–128. [Google Scholar] [CrossRef]
- Malek, E.; Hwang, S.J.; Caimi, P.F.; Metheny, L.L.; Tomlinson, B.K.; Cooper, B.W.; Boughan, K.M.; Otegbeye, F.; Gallogly, M.; Driscoll, J.J.; et al. Phase Ib trial of vactosertib in combination with pomalidomide in relapsed multiple myeloma: A corticosteroid-free approach by targeting TGF-beta signaling pathway. J. Clin. Oncol. 2021, 39, 8039. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of Ras mutations in cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Md Hanafiah, N.H.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef] [Green Version]
- Schjesvold, F.; Ribrag, V.; Rodriguez-Otero, P.; Robak, P.J.; Hansson, M.; Hajek, R.; Amor, A.A.; Martinez-Lopez, J.; Onishi, M.; Gallo, J.D.; et al. Safety and preliminary efficacy results from a phase Ib/II study of cobimetinib as a single agent and in combination with venetoclax with or without atezolizumab in patients with relapsed/refractory multiple myeloma. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Sharman, J.P.; Chmielecki, J.; Morosini, D.; Palmer, G.A.; Ross, J.S.; Stephens, P.J.; Stafl, J.; Miller, V.A.; Ali, S.M. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, e161–e163. [Google Scholar] [CrossRef]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Chau, I.; Hyman, D.M.; Ribrag, V.; Blay, J.Y.; Tabernero, J.; Elez, E.; Wolf, J.; Yee, A.J.; Kaiser, M.; et al. Vemurafenib in patients with relapsed refractory multiple myeloma harboring BRAF(V600) mutations: A cohort of the histology-independent VE-BASKET study. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Pan, D.; Richter, J. Where we stand with precision therapeutics in myeloma: Prosperity, promises, and pipedreams. Front. Oncol. 2021, 11, 819127. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Nagler, A.; Ben-Yehuda, D.; Badros, A.; Hari, P.N.; Hajek, R.; Spicka, I.; Kaya, H.; LeBlanc, R.; Yoon, S.S.; et al. Randomized, placebo-controlled, phase 3 study of perifosine combined with bortezomib and dexamethasone in patients with relapsed, refractory multiple myeloma previously treated with bortezomib. eJHaem 2020, 1, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Bahlis, N.J.; Venner, C.P.; Hay, A.E. Biomarker driven phase II clinical trial of trametinib in relapsed/refractory multiple myeloma with sequential addition of the AKT inhibitor, GSK2141795 at time of disease progression to overcome treatment failure: A trial of the Princess Margaret phase II consortium. Blood 2016, 128, 4526. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Calimeri, T.; Ferreri, A.J.M. m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 2017, 177, 684–702. [Google Scholar] [CrossRef]
- Gunther, A.; Baumann, P.; Burger, R.; Kellner, C.; Klapper, W.; Schmidmaier, R.; Gramatzki, M. Activity of everolimus (RAD001) in relapsed and/or refractory multiple myeloma: A phase I study. Haematologica 2015, 100, 541–547. [Google Scholar] [CrossRef]







| Trial ID (References) | Drugs | Enrollment (N) | Phase | Trial Title |
|---|---|---|---|---|
| NCT03374085 [32,33] | CC-92480 vs. (CC-92480 + Dex) | 201 | I/II | Multicenter, Open-label Study to Assess the Safety, Pharmacokinetics and Efficacy of CC-92480 Monotherapy and in Combination With Dexamethasone in Subjects With RRMM |
| NCT05519085 | (CC-92480 + Bort + Dex) vs. (Pom + Bort + Dex) | 760 | III | Two-Stage, Randomized, Multicenter, Open-Label Study Comparing CC-92480, Bortezomib and Dexamethasone (480Vd) Versus Pomalidomide, Bortezomib and Dexamethasone (PVd) in Subjects With RRMM (SUCCESSOR-1) |
| NCT05552976 | (CC-92480 + Carf + Dex) vs. (Carf + Dex) | 525 | III | Two-stage, Randomized, Multicenter, Open-label Study Comparing CC-92480 (BMS-986348), Carfilzomib, and Dexamethasone (480Kd) Versus Carfilzomib and Dexamethasone (Kd) in Participants With RRMM (SUCCESSOR-2) |
| NCT01421524 [30] | CC-122 | 271 | I | Multi-center, Open-label, Dose Finding Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Efficacy of the Pleiotropic Pathway Modifier CC-122 Administered Orally to Subjects With Advanced Solid Tumors, NHL, or MM |
| NCT05177536 | Iber | 38 | II | Iberdomide Maintenance Therapy Following ASCT in Patients With MM |
| NCT04998786 | Iber + Ixaz + Dex | 80 | II | A Multi-center Open-label Study of Ixazomib, Iberdomide and Dexamethasone in Elderly Patients With MM at First Relapse |
| NCT04975997 [34] | (Iber + Dara + Dex) vs. (Dara + Bort + Dex) | 864 | III | Two-Stage, Randomized, Multicenter, Open-label Study Comparing Iberdomide, Daratumumab and Dexamethasone (IberDd) Versus Daratumumab, Bortezomib, and Dexamethasone (DVd) in Subjects With RRMM (EXCALIBER) |
| NCT02773030 [28,35] | Iber vs. (Iber + Dex) vs. [Iber + Dex + (Dara or Bort or Carf)] | 449 | I/II | Multicenter, Open-label, Dose-escalation Study to Determine the Maximum Tolerated Dose, Assess the Safety, Tolerability, Pharmacokinetics and Efficacy of CC-220 as Monotherapy and in Combination With Other Treatments in Subjects With MM |
| NCT05392946 | Iber + Dara + Bort + Dex | 18 | I/II | Iberdomide in Combination With Daratumumab, Bortezomib and Dexamethasone in Patients With NDMM (IDEAL) |
| NCT05199311 | Iber + Carf + Dex | 66 | I/II | Carfilzomib, Iberdomide and Dexamethasone (KID) in Patients With Newly Diagnosed Transplant Eligible MM |
| NCT04564703 | Iber | 160 | II | Iberdomide Maintenance After ASCT in NDMM Patients |
| NCT04392037 | Iber + Ctx + Dex | 60 | II | Study of Iberdomide Combined With Low-dose Cyclophosphamide and Dexamethasone in RRMM (ICON) |
| NCT05558319 | (Bort + Isa + Dex +/− Len) vs. (Bort + Isa + Dex + Iber) | 480 | III | Trial for NDMM Patients Who Are Candidates for ASCT Comparing Extended VRD Plus Early Rescue Intervention vs. Isatuximab-VRD vs. Isatuximab-V-Iberdomide-D |
| Trial ID (References) | Drugs | Enrollment (N) | Phase | Trial Title |
|---|---|---|---|---|
| NCT00729118 [76] | Vor + Len | 19 | I | Vorinostat (SAHA) and Lenalidomide After Autologous Transplant for Patients With MM |
| NCT01502085 | Vor + Len + Dex | 25 | I/II | Vorinostat in Combination With Lenalidomide and Dexamethasone in MM Patients Refractory to Previous Lenalidomide Containing Regimens |
| NCT00642954 | Vor + Len + Dex | 31 | I | Vorinostat in Combination With Lenalidomide and Dexamethasone in Patients With RRMM |
| NCT00773747 [69] | (Vor + Bort) vs. (Bort + placebo) | 637 | III | An International, Multicenter, Randomized, Double-Blind Study of Vorinostat (MK-0683) or Placebo in Combination With Bortezomib in Patients With MM (VANTAGE 088) |
| NCT00773838 [77] | Vor + Bort + Dex | 143 | II | An International, Multicenter, Open-Label Study of Vorinostat (MK0683) in Combination With Bortezomib in Patients With RRMM |
| NCT01720875 [78] | Vor + Bort + Dex | 16 | II | Trial of Combination Treatment With Vorinostat, Bortezomib and Dexamethasone in Patients With RRMM |
| NCT01038388 [79] | Vor + Len + Bort + Dex | 30 | I | Trial Evaluating the Safety and Efficacy of Vorinostat + RVD (Lenalidomide + Bortezomib + Dexamethasone) for Patients With NDMM |
| NCT01297764 [80] | Vor + Carf + Len + Dex | 17 | I/II | Study of Carfilzomib, Lenalidomide, Vorinostat, and Dexamethasone in RRMM |
| NCT01394354 [81] | Vor + Bort + Dex + Doxo | 34 | I/II | Safety of Vorinostat in Combination With Bortezomib, Doxorubicin and Dexamethasone (VBDD) in Patients with RRMM |
| Trial Identifier (References) | Drugs | Enrollment (N) | Phase | Trial Title |
|---|---|---|---|---|
| NCT01323751 [73] | Ricol + Bort + Dex | 120 | I/II | Open-Label, Multicenter Study of ACY-1215 Administered Orally as Monotherapy and in Combination With Bortezomib and Dexamethasone for the Treatment of RRMM. |
| NCT01583283 [74] | Ricol + Len + Dex | 38 | I | Open-Label, Multicenter Study of ACY-1215 (Ricolinostat) in Combination With Lenalidomide and Dexamethasone for the Treatment of RRMM |
| NCT01997840 [73] | Ricol + Pom + Dex | 103 | I/II | Multi-Center, Open Label, Dose-Escalation Study to Determine the Maximum Tolerated Dose, Safety, and Efficacy of ACY-1215 (RICOLINOSTAT) in Combination With Pomalidomide and Low-Dose Dexamethasone in Patients With RRMM. |
| NCT02400242 [82,83,84] | Citar + Pom + Dex | 85 | I | Multicenter, Single-Arm, Open-Label, Dose-Escalation Study to Determine the Maximum Tolerated Dose, Safety, and Preliminary Activity of Oral ACY-241 Alone and in Combination With Pomalidomide and Low-Dose Dexamethasone in Patients With RRMM |
| NCT04025450 [75,85] | (Bort + Len + Dex) +/− Chid | 50 | I/II | Comparation of Chidamide Plus VRD (Bortezomib, Lenalidomide, Dexamethasone) With VRD Regimen for Primary High-Risk MM Patients, a Multiple Center, Randomized Clinical Trial. |
| NCT03605056 | Chid + Len + Dex | 25 | II | Chidamide in Combination With Lenalidomide and Dexamethasone for the Treatment of RRMM |
| Trial ID (Reference) | Drugs | Enrollment (N) | Phase | Trial Title |
|---|---|---|---|---|
| NCT00821249 [126] | Fil vs (Fil + Dex) | 55 | I/II | A Study of ARRY-520 in Patients With RRMM |
| NCT01248923 [127,128] | (Fil + Bort) vs. (Fil + Bort + Dex) | 55 | I | A Study of ARRY-520 and Bortezomib Plus Dexamethasone in Patients With RRMM |
| NCT01372540 [132] | Carf + Fil | 76 | I | Filanesib and Carfilzomib in Treating Patients With RRMM or Plasma Cell Leukemia |
| NCT02384083 [133] | Fil + Pom + Dex | 47 | I/II | Filanesib (ARRY-520) in Combination With Pomalidomide and Dexamethasone for RRMM Patients |
| NCT01989325 [137] | (Fil + Carf + Dex) vs. (Carf + Dex) | 77 | II | A Study of Filanesib (ARRY-520) and Carfilzomib in Patients With Advanced MM |
| NCT02092922 [138] | Fil | 154 | II | Trial of Filanesib in RRMM (AfFIRM) |
| Trial ID (Reference) | Drugs | Enrollment (N) | Phase | Trial Title |
|---|---|---|---|---|
| NCT02186834 [152] | Sel + Dox + Dex | 28 | I/II | Investigator-Initiated Trial of Selinexor and Liposomal Doxorubicin for RRMM |
| NCT02336815 [147,153,154] | Sel + Dex | 202 | II | Open-Label, Single-Arm Study of Selinexor Plus Low-Dose Dexamethasone (Sd) in Patients With MM Previously Treated With Lenalidomide, Pomalidomide, Bortezomib, Carfilzomib, and Daratumumab, and Refractory to Prior Treatment With Glucocorticoids, an Immunomodulatory Agent, a Proteasome Inhibitor, and Daratumumab (STORM) |
| NCT05422027 | Sel + Len + Bort + Dex | 42 | I/II | Selinexor Plus Bortezomib, Lenalidomide and Dexamethasone (XVRd) in High Risk NDMM |
| NCT04661137 [155,156] | Sel + Dex + (Pom or Carf or Dara) | 96 | II | Study of Selinexor in Combination With Carfilzomib, Daratumumab or Pomalidomide in Patients With RRMM |
| NCT04414475 | (Sel + Dex) vs. (Sel + Dex + Bort) | 134 | II | Open-label, Multi-arm Trial of Selinexor Plus Low-dose Dexamethasone (Sd) in Patients With Penta-refractory MM or Selinexor and Bortezomib Plus Low-dose Dexamethasone (SVd) in Patients With Triple-class Refractory MM |
| NCT02780609 [157] | Sel + Mel + Dex | 22 | I/II | Investigator Sponsored Study of Selinexor in Combination With High-Dose Melphalan Before ASCT for MM |
| NCT02199665 [158] | Sel + Carf + Dex | 100 | I | Study of the Combination of Selinexor With Carfilzomib and Dexamethasone in Patients With RRMM |
| NCT03110562 [149,150,158,159,160] | (Sel + Bort + Dex) vs. (Bort + Dex) | 402 | III | Randomized, Controlled, Open-label Study of Selinexor, Bortezomib, and Dexamethasone (SVd) Versus Bortezomib and Dexamethasone (Vd) in Patients With RRMM (BOSTON) |
| NCT05028348 | (Sel + Pom + Dex) vs. (Elo + Pom + Dex) | 300 | III | Randomized, Open-label Trial of Selinexor, Pomalidomide, and Dexamethasone (SPd) Versus Elotuzumab, Pomalidomide, and Dexamethasone (EloPd) in Patients With RRMM |
| NCT04764942 [161] | (Sel + Pom + Dex +/- Carf | 81 | I/II | Trial of Selinexor in Combination With Pomalidomide and Dexamethasone ± Carfilzomib for Patients With Proteasome-Inhibitor and Immunomodulatory Drug RRMM (SCOPE) |
| NCT02343042 [155,162] | Sel + Various | 518 | I/II | Study of Selinexor in Combination With Backbone Treatments for RRMM and NDMM |
| NCT03589222 [163] | Sel + Dara + Bort + Dex | 62 | II | Open-label, Multicenter Trial of Selinexor, Bortezomib and Low-dose Dexamethasone Plus Daratumumab (SELIBORDARA) for the Treatment of Patients With RRMM |
| NCT04756401 | Sel + Dara + Carf + Dex | 52 | II | Open Label Single-Arm Study of Selinexor, Daratumumab, Carfilzomib and Dexamethasone for High-Risk, RRMM Patients Who Have Received 1–3 Prior Lines of Therapy |
| NCT04877275 [164] | (Sel + Dex + Dox) vs. (Sel + Ctx + Dex) | 50 | II | Selinexor in Combination With Chemotherapy to Treat RRMM Relapsed/Refractory Multiple Myeloma Patients |
| NCT04782687 | Sel + Dex + Dara + Len | 100 | II | Trial of Daratumumab, Lenalidomide and Dexamethasone (DRd) in Combination With Selinexor for Patients With NDMM |
| NCT04941937 | (Sel + Thal + Len) vs. (Sel + Len + Dex) vs. (Sel + Pom + Dex) | 90 | II | Selinexor in Combination With Immunomodulator to Treat RRMM Patients |
| NCT04717700 | (Sel + Bort + Len + Dex) vs. (Bort + Len + Dex) | 100 | II | Selinexor With Alternating Bortezomib or Lenalidomide Plus Dexamethasone in Transplant Ineligible NDMM Patients (SABLe): An Investigator Sponsored Trial |
| NCT04891744 | Sel + Thal + Dex | 48 | I/II | Study of Selinexor in Combination With Thalidomide and Dexamethasone for RRMM |
| NCT03944057 [165] | Sel + Dex | 82 | II | Open-Label, Single-Arm Study of Selinexor Plus Dexamethasone in Patients With MM Refractory to Prior Treatment With Immunomodulatory Agents and Proteasome Inhibitor |
| NCT04939142 | (Bort + Dex) vs. (Sel + Bort + Dex) | 150 | III | Randomized, Controlled, Multicenter, Open-label Study of Selinexor, Bortezomib, and Dexamethasone (SVd) Versus Bortezomib and Dexamethasone (Vd) in Patients With RRMM |
| NCT05478993 | Sel + Pom + Dex | 21 | II | Study of Selinexor, Pomalidomide, and Dexamethasone For MM With Central Nervous System Involvement |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abramson, H.N. Recent Advances in the Applications of Small Molecules in the Treatment of Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 2645. https://doi.org/10.3390/ijms24032645
Abramson HN. Recent Advances in the Applications of Small Molecules in the Treatment of Multiple Myeloma. International Journal of Molecular Sciences. 2023; 24(3):2645. https://doi.org/10.3390/ijms24032645
Chicago/Turabian StyleAbramson, Hanley N. 2023. "Recent Advances in the Applications of Small Molecules in the Treatment of Multiple Myeloma" International Journal of Molecular Sciences 24, no. 3: 2645. https://doi.org/10.3390/ijms24032645
APA StyleAbramson, H. N. (2023). Recent Advances in the Applications of Small Molecules in the Treatment of Multiple Myeloma. International Journal of Molecular Sciences, 24(3), 2645. https://doi.org/10.3390/ijms24032645