
Exploring AlphaFold2′s Performance on Predicting Amino Acid Side-Chain Conformations and Its Utility in Crystal Structure Determination of B318L Protein

Abstract
:1. Introduction
2. Results
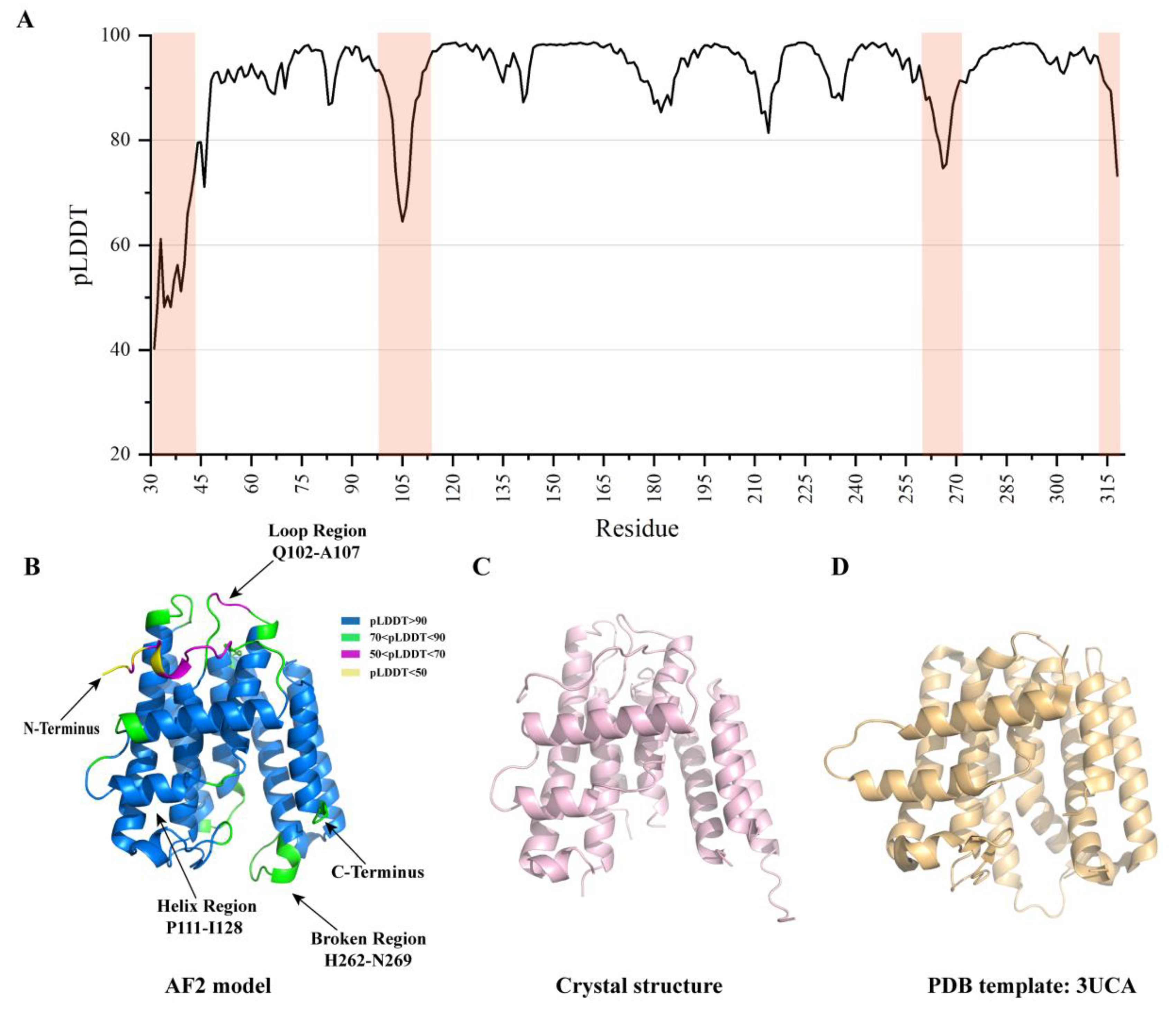
2.1. Overall Analysis of AF2 Model against B318L Crystal Structure
2.2. Assessing Prediction Accuracy of B318L Protein Side-Chains at the Individual Amino Acid Residue Level
2.3. Comparing AF2 Predicted Structure with Other Computational Models
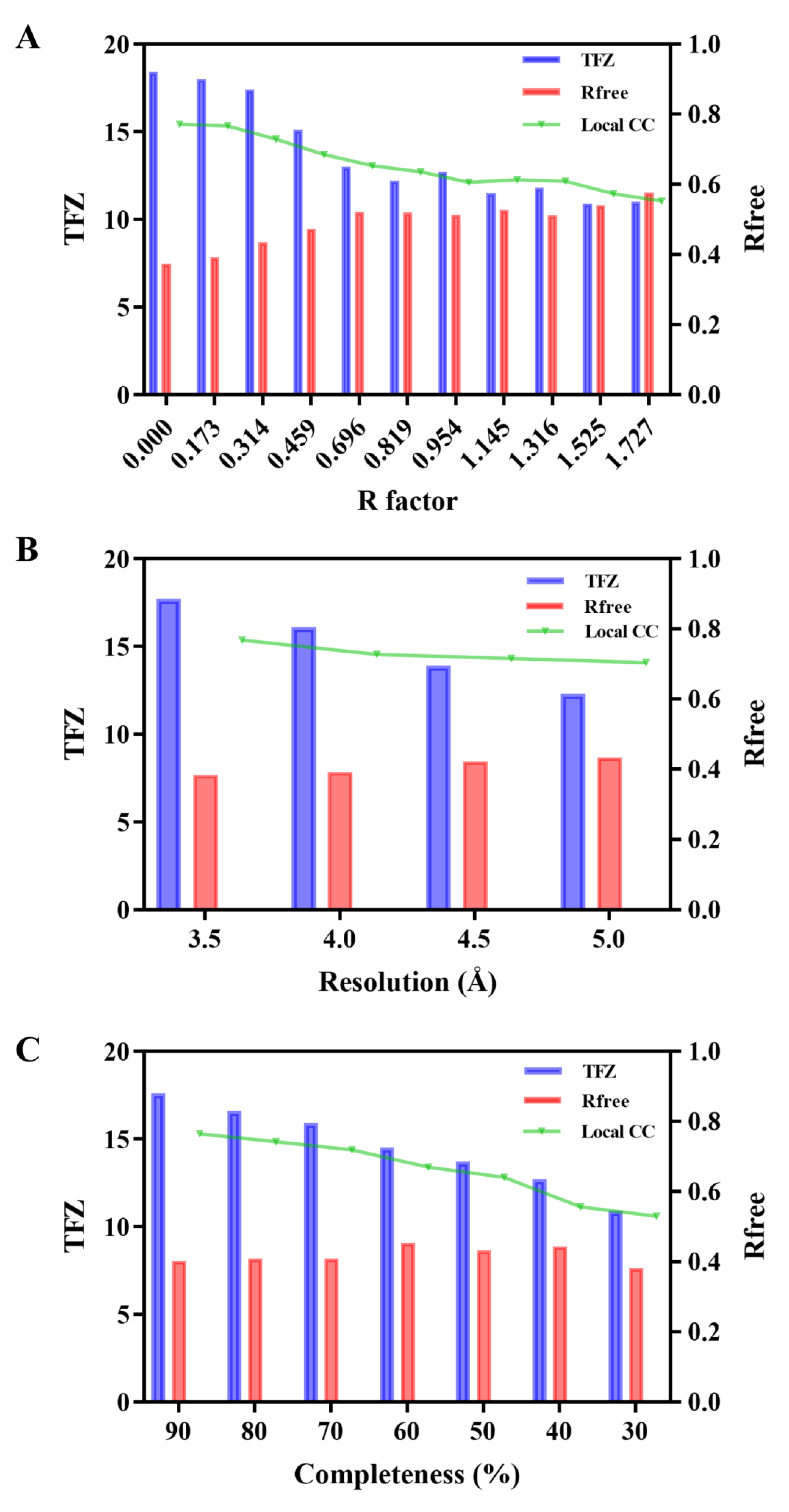
2.4. Effect of Data Quality on AF2-Assistant Crystal Structural Determination
2.5. Crystal Structure of B318L Protein and Comparison with Its Homologs
2.6. Molecular Docking with Crystal Structure and AF2 Model for B318L Protein
3. Discussion
4. Materials and Methods
4.1. In Silico Model Predictions of B318L Protein
4.2. Plasmid Construction
4.3. Protein Expression, Purification, and Crystallization
4.4. Experimental Data Collection and Structural Determination
4.5. Structural Alignment and RMSD Calculations
4.6. Molecular Docking
4.7. Analysis of AF2 Templates Used for Modeling B318L Protein
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.X.; Chen, L.; Di Costanzo, L.; Christie, C.; Duarte, J.M.; Dutta, S.; Feng, Z.K.; et al. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar]
- Lander, E.S.; Consortium, I.H.G.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Dill, K.A.; MacCallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nat. Struct. Mol. Biol. 2021, 28, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins 2021, 89, 1687–1699. [Google Scholar] [CrossRef]
- Alexander, L.T.; Lepore, R.; Kryshtafovych, A.; Adamopoulos, A.; Alahuhta, M.; Arvin, A.M.; Bomble, Y.J.; Bottcher, B.; Breyton, C.; Chiarini, V.; et al. Target highlights in CASP14: Analysis of models by structure providers. Proteins 2021, 89, 1647–1672. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Moult, J.; Albrecht, R.; Chang, G.A.; Chao, K.; Fraser, A.; Greenfield, J.; Hartmann, M.D.; Herzberg, O.; Josts, I.; et al. Computational models in the service of X-ray and cryo-electron microscopy structure determination. Proteins 2021, 89, 1633–1646. [Google Scholar] [CrossRef]
- Masrati, G.; Landau, M.; Ben-Tal, N.; Lupas, A.; Kosloff, M.; Kosinski, J. Integrative Structural Biology in the Era of Accurate Structure Prediction. J. Mol. Biol. 2021, 433, 167127. [Google Scholar] [CrossRef]
- Edich, M.; Briggs, D.C.; Kippes, O.; Gao, Y.; Thorn, A. The impact of AlphaFold2 on experimental structure solution. Faraday Discuss. 2022, 240, 184–195. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Sammito, M.D.; Read, R.J. Implications of AlphaFold2 for crystallographic phasing by molecular replacement. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Salmen, W.; Sankaran, B.; Lasanajak, Y.; Smith, D.F.; Crawford, S.E.; Estes, M.K.; Prasad, B.V.V. Novel fold of rotavirus glycan-binding domain predicted by AlphaFold2 and determined by X-ray crystallography. Commun. Biol. 2022, 5, 419. [Google Scholar] [CrossRef] [PubMed]
- Barbarin-Bocahu, I.; Graille, M. The X-ray crystallography phase problem solved thanks to AlphaFold and RoseTTAFold models: A case-study report. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 517–531. [Google Scholar] [CrossRef]
- Stsiapanava, A.; Xu, C.; Nishio, S.; Han, L.; Yamakawa, N.; Carroni, M.; Tunyasuvunakool, K.; Jumper, J.; de Sanctis, D.; Wu, B.; et al. Structure of the decoy module of human glycoprotein 2 and uromodulin and its interaction with bacterial adhesin FimH. Nat. Struct. Mol. Biol. 2022, 29, 190–193. [Google Scholar] [CrossRef]
- Hryc, C.F.; Baker, M.L. AlphaFold2 and CryoEM: Revisiting CryoEM modeling in near-atomic resolution density maps. iScience 2022, 25, 104496. [Google Scholar] [CrossRef]
- Mosalaganti, S.; Obarska-Kosinska, A.; Siggel, M.; Taniguchi, R.; Turonova, B.; Zimmerli, C.E.; Buczak, K.; Schmidt, F.H.; Margiotta, E.; Mackmull, M.T.; et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 2022, 376, eabm9506. [Google Scholar] [CrossRef]
- Tai, L.; Zhu, Y.; Ren, H.; Huang, X.; Zhang, C.; Sun, F. 8 A structure of the outer rings of the Xenopus laevis nuclear pore complex obtained by cryo-EM and AI. Protein Cell 2022, 13, 760–777. [Google Scholar] [CrossRef]
- Skalidis, I.; Kyrilis, F.L.; Tuting, C.; Hamdi, F.; Chojnowski, G.; Kastritis, P.L. Cryo-EM and artificial intelligence visualize endogenous protein community members. Structure 2022, 30, 575–589.e576. [Google Scholar] [CrossRef]
- Huang, Y.J.; Zhang, N.; Bersch, B.; Fidelis, K.; Inouye, M.; Ishida, Y.; Kryshtafovych, A.; Kobayashi, N.; Kuroda, Y.; Liu, G.; et al. Assessment of prediction methods for protein structures determined by NMR in CASP14: Impact of AlphaFold2. Proteins 2021, 89, 1959–1976. [Google Scholar] [CrossRef]
- Tejero, R.; Huang, Y.J.; Ramelot, T.A.; Montelione, G.T. AlphaFold Models of Small Proteins Rival the Accuracy of Solution NMR Structures. Front. Mol. Biosci. 2022, 9, 877000. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.J.; Williamson, M.P. The accuracy of protein structures in solution determined by AlphaFold and NMR. Structure 2022, 30, 925–933.e922. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Ruiz-Serra, V.; Valentini, S.; Valencia, A. The structural coverage of the human proteome before and after AlphaFold. PLoS Comput. Biol. 2022, 18, e1009818. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Yanez, R.J.; Rodriguez, J.M.; Vinuela, E.; Salas, M.L. African swine fever virus trans-prenyltransferase. J. Biol. Chem. 1997, 272, 9417–9423. [Google Scholar] [CrossRef]
- Alejo, A.; Andres, G.; Vinuela, E.; Salas, M.L. The African swine fever virus prenyltransferase is an integral membrane trans-geranylgeranyl-diphosphate synthase. J. Biol. Chem. 1999, 274, 18033–18039. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Hernaez, B.; Galindo, I.; Munoz-Moreno, R.; Cuesta-Geijo, M.A.; Alonso, C. Small Rho GTPases and Cholesterol Biosynthetic Pathway Intermediates in African Swine Fever Virus Infection. J. Virol. 2012, 86, 1758–1767. [Google Scholar] [CrossRef]
- Baek, K.T.; Kepp, K.P. Assessment of AlphaFold2 for Human Proteins via Residue Solvent Exposure. J. Chem. Inf. Model. 2022, 62, 3391–3400. [Google Scholar] [CrossRef]
- Wu, R.; Ding, F.; Wang, R.; Shen, R.; Zhang, X.; Luo, S.; Su, C.; Wu, Z.; Xie, Q.; Berger, B.; et al. High-resolution de novo structure prediction from primary sequence. BioRxiv 2022. [Google Scholar]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022. [Google Scholar]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Yan, R.X.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.H. Reaction kinetics, catalytic mechanisms, conformational changes, and inhibitor design for prenyltransferases. Biochemistry 2009, 48, 6562–6570. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Cheng, T.H.; Wang, A.H. Structure, catalysis, and inhibition mechanism of prenyltransferase. IUBMB Life 2021, 73, 40–63. [Google Scholar] [CrossRef]
- Wang, K.; Ohnuma, S. Chain-length determination mechanism of isoprenyl diphosphate synthases and implications for molecular evolution. Trends Biochem. Sci. 1999, 24, 445–451. [Google Scholar] [CrossRef]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Chang, T.H.; Hsieh, F.L.; Ko, T.P.; Teng, K.H.; Liang, P.H.; Wang, A.H. Structure of a heterotetrameric geranyl pyrophosphate synthase from mint (Mentha piperita) reveals intersubunit regulation. Plant Cell 2010, 22, 454–467. [Google Scholar] [CrossRef]
- Wallrapp, F.H.; Pan, J.J.; Ramamoorthy, G.; Almonacid, D.E.; Hillerich, B.S.; Seidel, R.; Patskovsky, Y.; Babbitt, P.C.; Almo, S.C.; Jacobson, M.P.; et al. Prediction of function for the polyprenyl transferase subgroup in the isoprenoid synthase superfamily. Proc. Natl. Acad. Sci. USA 2013, 110, E1196–E1202. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stark, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef] [PubMed]
- van Breugel, M.; Rosa, E.S.I.; Andreeva, A. Structural validation and assessment of AlphaFold2 predictions for centrosomal and centriolar proteins and their complexes. Commun. Biol. 2022, 5, 312. [Google Scholar] [CrossRef]
- Paul, B.; Weeratunga, S.; Tillu, V.A.; Hariri, H.; Henne, W.M.; Collins, B.M. Structural Predictions of the SNX-RGS Proteins Suggest They Belong to a New Class of Lipid Transfer Proteins. Front. Cell Dev. Biol. 2022, 10, 826688. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C.; Roussel, A.; Imbert, I. Structure Prediction and Analysis of Hepatitis E Virus Non-Structural Proteins from the Replication and Transcription Machinery by AlphaFold2. Viruses 2022, 14, 1537. [Google Scholar] [CrossRef] [PubMed]
- Schauperl, M.; Denny, R.A. AI-Based Protein Structure Prediction in Drug Discovery: Impacts and Challenges. J. Chem. Inf. Model. 2022, 62, 3142–3156. [Google Scholar] [CrossRef]
- Higgins, M.K. Can We AlphaFold Our Way Out of the Next Pandemic? J. Mol. Biol. 2021, 433, 167093. [Google Scholar] [CrossRef]
- Sen, N.; Anishchenko, I.; Bordin, N.; Sillitoe, I.; Velankar, S.; Baker, D.; Orengo, C. Characterizing and explaining the impact of disease-associated mutations in proteins without known structures or structural homologs. Brief. Bioinform. 2022, 23, bbac187. [Google Scholar] [CrossRef]
- Yao, X.; Kang, T.; Pu, Z.; Zhang, T.; Lin, J.; Yang, L.; Yu, H.; Wu, M. Sequence and Structure-Guided Engineering of Urethanase from Agrobacterium tumefaciens d3 for Improved Catalytic Activity. J. Agric. Food Chem. 2022, 70, 7267–7278. [Google Scholar] [CrossRef]
- Shao, C.; Bittrich, S.; Wang, S.; Burley, S.K. Assessing PDB macromolecular crystal structure confidence at the individual amino acid residue level. Structure 2022, 30, 1385–1394.e1383. [Google Scholar] [CrossRef]
- He, X.H.; You, C.Z.; Jiang, H.L.; Jiang, Y.; Xu, H.E.; Cheng, X. AlphaFold2 versus experimental structures: Evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 2023, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.X.; Kanematsu, Y.; Takano, Y. Structure of Heme-binding Pocket in Heme Protein is Generally Rigid and can be Predicted by AlphaFold2. Chem. Lett. 2022, 51, 704–708. [Google Scholar] [CrossRef]
- Buel, G.R.; Walters, K.J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Chakravarty, D.; Porter, L.L. AlphaFold2 fails to predict protein fold switching. Protein Sci. A Publ. Protein Soc. 2022, 31, e4353. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.B.; Hendrickson, W.A.; Henderson, R.; Brunger, A.T. The protein-folding problem: Not yet solved. Science 2022, 375, 507. [Google Scholar] [CrossRef] [PubMed]
- Martin, J. When Alphafold2 predictions go wrong for protein-protein complexes, is there something to be learnt? Q. Rev. Biophys. 2022, 55, 1–4. [Google Scholar] [CrossRef]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing protein folding neural networks for peptide-protein docking. Nat. Commun. 2022, 13, 176. [Google Scholar] [CrossRef]
- Gao, M.; Nakajima An, D.; Parks, J.M.; Skolnick, J. AF2Complex predicts direct physical interactions in multimeric proteins with deep learning. Nat. Commun. 2022, 13, 1744. [Google Scholar] [CrossRef]
- Bryant, P.; Pozzati, G.; Elofsson, A. Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 2022, 13, 1265. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar]
- Saldano, T.; Escobedo, N.; Marchetti, J.; Zea, D.J.; Mac Donagh, J.; Rueda, A.J.V.; Gonik, E.; Melani, A.G.; Nechcoff, J.N.; Salas, M.N.; et al. Impact of protein conformational diversity on AlphaFold predictions. Bioinformatics 2022, 38, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, G.; Duran, C.; Estevez-Gay, M.; Osuna, S. Estimating conformational heterogeneity of tryptophan synthase with a template-based Alphafold2 approach. Protein Sci. A Publ. Protein Soc. 2022, 31, e4426. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ASFV B318L NΔ30 | |
|---|---|
| Data collection statistics | |
| Data collection | SSRF BL10U2 |
| Wavelength (Å) | 0.9792 |
| Resolution (Å) | 48.2–3.2 (3.28–3.20) |
| Space group | P6522 |
| Unit cell parameters | a = 56.13Å, b = 56.13 Å, c = 376.69 Å, α = β = 90°, γ = 120° |
| No. of unique reflections | 6579 (472) |
| Completeness (%) | 99.7 (99.2) |
| Redundancy | 31.7 (28.3) |
| Mean I/σ (I) | 15.6 (2.2) |
| Molecules in asymmetric unit | 1 |
| Rmerge (%) | 29.7 (251.3) |
| Rmeas (%) | 30.2 (256.0) |
| CC1/2 | 0.998 (0.718) |
| Structure refinement statistics | |
| Rwork/Rfree (%) | 24.6/28.4 |
| Number of atoms | 2058 |
| Protein residues | 261 |
| Root-mean-square deviations | |
| Bond length (Å) | 0.008 |
| Bond angles (°) | 1.421 |
| Ramachandran plot | |
| Favored (%) | 94.5 |
| Allowed (%) | 5.1 |
| Average B-factor (Å) of protein | 99.5 |
| Protein Structure | Binding Affinity (kcal/mol) |
|---|---|
| Crystal structure of B318L | −7.2 |
| AF2 model of B318L | −7.8 |
| Homologous structure (2J1P) | −7.8 |
| Apo | Holo | AF2 Model | Crystal Structure | |
|---|---|---|---|---|
| Mean value (Å2) | 458.56 | 510.84 | 498.8 | 462.6 |
| Standard deviation | 34.83 | 26.4 | 0 | 0 |
| No. of structures | 61 | 43 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Zhang, H.; She, Z.; Gao, Z.; Wang, Q.; Geng, Z.; Dong, Y. Exploring AlphaFold2′s Performance on Predicting Amino Acid Side-Chain Conformations and Its Utility in Crystal Structure Determination of B318L Protein. Int. J. Mol. Sci. 2023, 24, 2740. https://doi.org/10.3390/ijms24032740
Zhao H, Zhang H, She Z, Gao Z, Wang Q, Geng Z, Dong Y. Exploring AlphaFold2′s Performance on Predicting Amino Acid Side-Chain Conformations and Its Utility in Crystal Structure Determination of B318L Protein. International Journal of Molecular Sciences. 2023; 24(3):2740. https://doi.org/10.3390/ijms24032740
Chicago/Turabian StyleZhao, Haifan, Heng Zhang, Zhun She, Zengqiang Gao, Qi Wang, Zhi Geng, and Yuhui Dong. 2023. "Exploring AlphaFold2′s Performance on Predicting Amino Acid Side-Chain Conformations and Its Utility in Crystal Structure Determination of B318L Protein" International Journal of Molecular Sciences 24, no. 3: 2740. https://doi.org/10.3390/ijms24032740
APA StyleZhao, H., Zhang, H., She, Z., Gao, Z., Wang, Q., Geng, Z., & Dong, Y. (2023). Exploring AlphaFold2′s Performance on Predicting Amino Acid Side-Chain Conformations and Its Utility in Crystal Structure Determination of B318L Protein. International Journal of Molecular Sciences, 24(3), 2740. https://doi.org/10.3390/ijms24032740