
Tumor Radiosensitization by Gene Electrotransfer-Mediated Double Targeting of Tumor Vasculature
 , and
, and 
Abstract
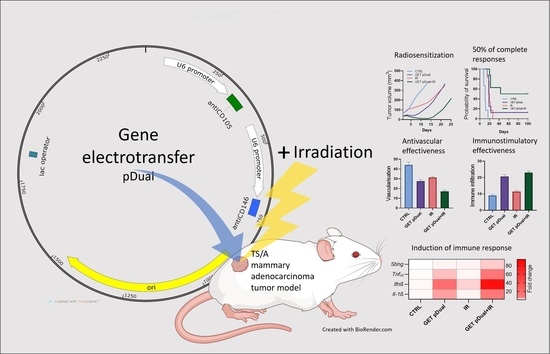
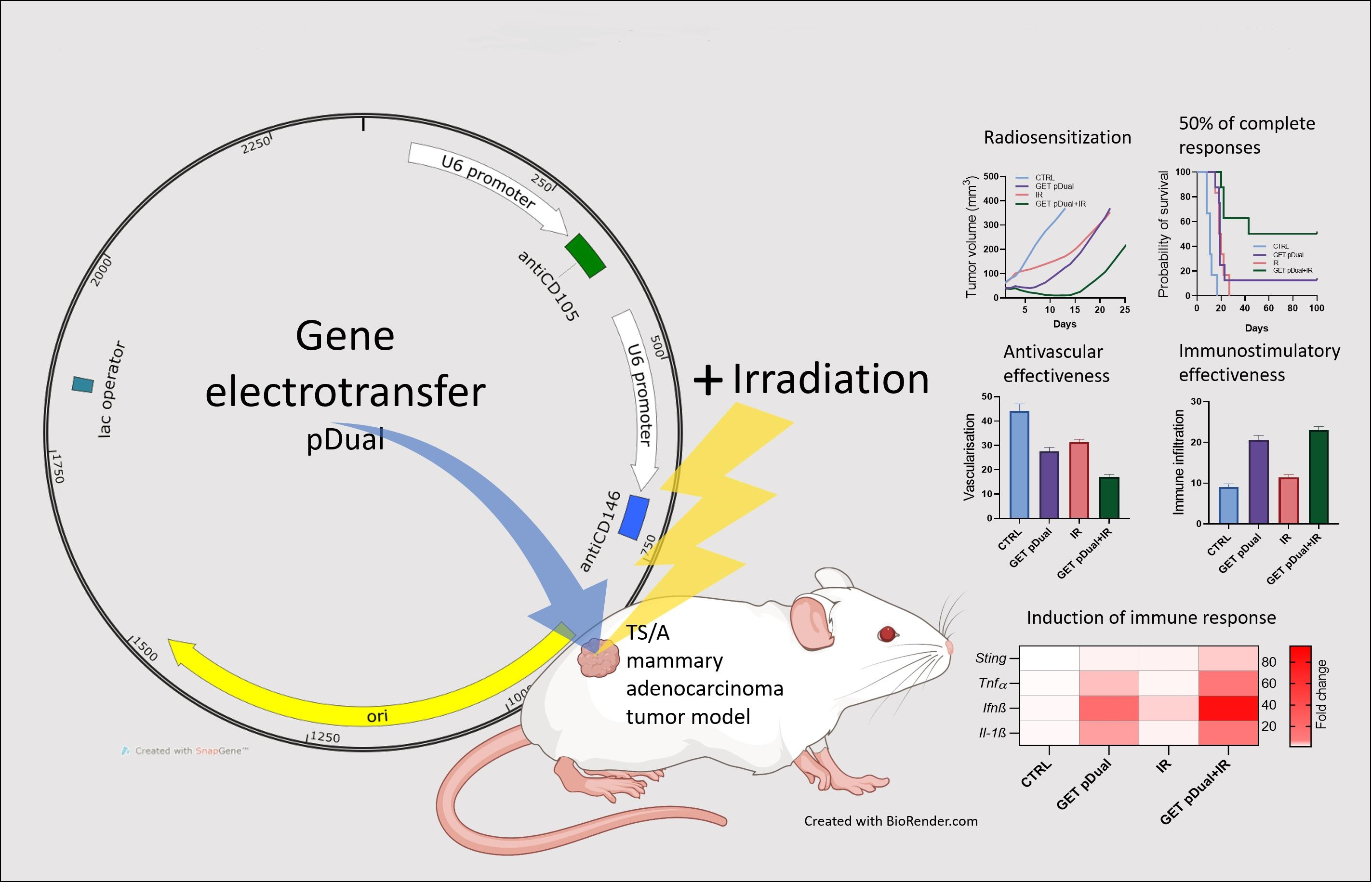
:
1. Introduction
2. Results
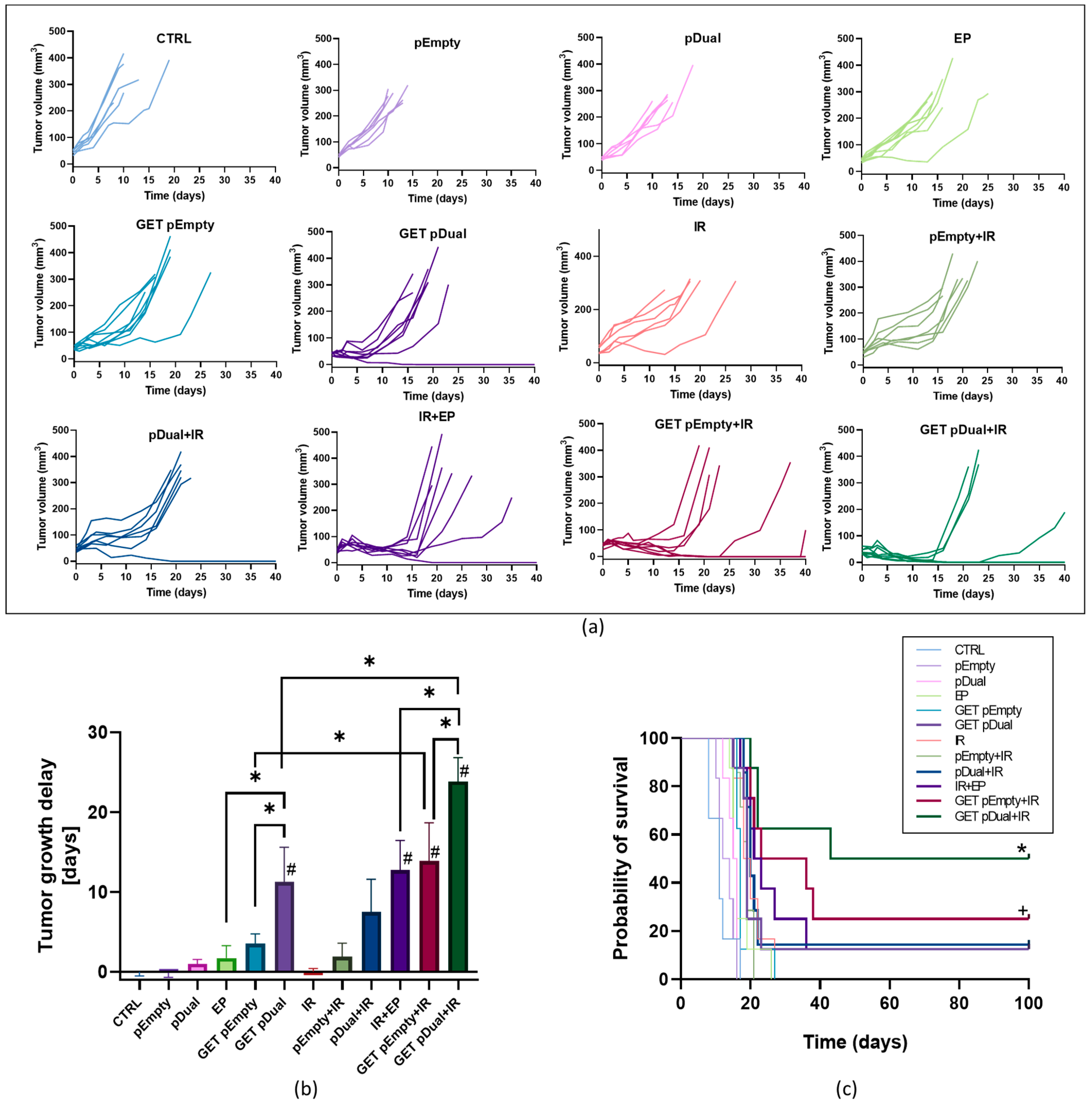
2.1. Dual Silencing of CD105 and CD146 Promotes Radiosensitization of TS/A Tumors
2.2. Histological Analyses Indicate Vascular Targeted Effects Combined with Immune Activation
2.3. DNA Sensor Sting and Proinflammatory Cytokine Levels Indicate Induction of Immune Response
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmids
4.3. Animals
4.4. In Vivo Gene Electrotransfer
4.5. Tumor Irradiation
4.6. The Tumor Growth Delay
4.7. Histology
4.8. Quantitative Reverse Transcription-Polymerase Chain Reaction (RT–qPCR)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the role of angiogenesis in cancer ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Paauwe, M.; Nixon, A.B.; Hawinkels, L.J.A.C. Molecular Sciences Endoglin Targeting: Lessons Learned and Questions That Remain. Int. J. Mol. Sci. 2020, 22, 147. [Google Scholar] [CrossRef]
- Prosen, L.; Markelc, B.; Dolinsek, T.; Music, B.; Cemazar, M.; Sersa, G. Mcam silencing with RNA interference using magnetofection has antitumor effect in murine melanoma. Mol. Ther.-Nucleic Acids 2014, 3, e205. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Wang, F.; Feng, J.; Yang, D.; Yang, X.; Yan, X. Knockdown of CD146 reduces the migration and proliferation of human endothelial cells. Cell Res. 2006, 16, 313–318. [Google Scholar] [CrossRef]
- Zabouo, G.; Imbert, A.M.; Jacquemier, J.; Finetti, P.; Moreau, T.; Esterni, B.; Birnbaum, D.; Bertucci, F.; Chabannon, C. CD146 expression is associated with a poor prognosis in human breast tumors and with enhanced motility in breast cancer cell lines. Breast Cancer Res. 2009, 11, R1. [Google Scholar] [CrossRef]
- Chen, H.X.; Cleck, J.N. Adverse effects of anticancer agents that target the VEGF pathway. Nat. Rev. Clin. Oncol. 2009, 6, 465–477. [Google Scholar] [CrossRef]
- Yan, X.; Lin, Y.; Yang, D.; Shen, Y.; Yuan, M.; Zhang, Z.; Li, P.; Xia, H.; Li, L.; Luo, D.; et al. A novel anti-CD146 monoclonal antibody, AA98, inhibits angiogenesis and tumor growth. Blood 2003, 102, 184–191. [Google Scholar] [CrossRef]
- Heller, R.; Heller, L.C. Gene Electrotransfer Clinical Trials. Adv. Genet. 2015, 89, 235–262. [Google Scholar] [CrossRef]
- Čemažar, M.; Jesenko, T.; Bošnjak, M.; Markelc, B.; Kamenšek, U.; Brezar, S.K.; Kos, Š.; Tratar, U.L.; Žnidar, K.; Renčelj, A.; et al. Gene therapy in oncology, first steps of development in Slovenia. Onkol. Med.-Sci. J. 2022, 26, 12–21. [Google Scholar] [CrossRef]
- Algazi, A.; Bhatia, S.; Agarwala, S.; Molina, M.; Lewis, K.; Faries, M.; Fong, L.; Levine, L.P.; Franco, M.; Oglesby, A.; et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol. 2020, 31, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Canton, D.A.; Shirley, S.; Wright, J.; Connolly, R.; Burkart, C.; Mukhopadhyay, A.; Twitty, C.; Qattan, K.E.; Campbell, J.S.; Le, M.H.; et al. Melanoma treatment with intratumoral electroporation of tavokinogene telseplasmid (pIL-12, tavokinogene telseplasmid). Immunotherapy 2017, 9, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Brault, M.; Olsen, T.M.; Martinez, J.; Stetson, D.B.; Oberst, A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J. Immunol. 2018, 200, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Search of: Interleukin 12 Electroporation|Cancer—List Results—ClinicalTrials.gov, (n.d.). Available online: https://clinicaltrials.gov/ct2/results?cond=Cancer&term=interleukin+12+electroporation&cntry=&state=&city=&dist= (accessed on 12 October 2022).
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef]
- Low, L.; Mander, A.; McCann, K.; Dearnaley, D.; Tjelle, T.; Mathiesen, I.; Stevenson, F.; Ottensmeier, C.H. DNA vaccination with electroporation induces increased antibody responses in patients with prostate cancer. Hum. Gene Ther. 2009, 20, 1269–1278. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Chiappori, A.; Aurisicchio, L.; Bagchi, A.; Clark, J.; Dubey, S.; Fridman, A.; Fabregas, J.C.; Marshall, J.; Scarselli, E.; et al. Phase 1 studies of the safety and immunogenicity of electroporated HER2/CEA DNA vaccine followed by adenoviral boost immunization in patients with solid tumors. J. Transl. Med. 2013, 11, 62. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef]
- McAnuff, M.A.; Rettig, G.R.; Rice, K.G. Potency of siRNA versus shRNA mediated knockdown in vivo. J. Pharm. Sci. 2007, 96, 2922–2930. [Google Scholar] [CrossRef]
- Lambricht, L.; Lopes, A.; Kos, S.; Sersa, G.; Préat, V.; Vandermeulen, G.; Preat, V.; Vandermeulen, G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin. Drug Deliv. 2016, 13, 295–310. [Google Scholar] [CrossRef]
- Scholz, C.; Wagner, E. Therapeutic plasmid DNA versus siRNA delivery: Common and different tasks for synthetic carriers. J. Control. Release 2012, 161, 554–565. [Google Scholar] [CrossRef]
- Gill, D.R.; Pringle, I.A.; Hyde, S.C. Progress and prospects: The design and production of plasmid vectors. Gene Ther. 2009, 16, 165–171. [Google Scholar] [CrossRef]
- Kamensek, U.; Sersa, G.; Cemazar, M. Evaluation of p21 promoter for interleukin 12 radiation induced transcriptional targeting in a mouse tumor model. Mol. Cancer 2013, 12, 136. [Google Scholar] [CrossRef]
- Savarin, M.; Kamensek, U.; Znidar, K.; Todorovic, V.; Sersa, G.; Cemazar, M. Evaluation of a novel plasmid for simultaneous gene electrotransfer-mediated silencing of cd105 and cd146 in combination with irradiation. Int. J. Mol. Sci. 2021, 22, 3069. [Google Scholar] [CrossRef] [PubMed]
- Kamensek, U.; Cemazar, M.; Lampreht Tratar, U.; Ursic, K.; Sersa, G. Antitumor in situ vaccination effect of TNFα and IL-12 plasmid DNA electrotransfer in a murine melanoma model. Cancer Immunol. Immunother. 2018, 67, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Dolinsek, T.; Sersa, G.; Prosen, L.; Bosnjak, M.; Stimac, M.; Razborsek, U.; Cemazar, M. Electrotransfer of plasmid DNA encoding an anti-mouse endoglin (CD105) shRNA to B16 melanoma tumors with low and high metastatic potential results in pronounced anti-tumor effects. Cancers 2015, 8, 3. [Google Scholar] [CrossRef]
- Tesic, N.; Kamensek, U.; Sersa, G.; Kranjc, S.; Stimac, M.; Lampreht, U.; Preat, V.; Vandermeulen, G.; Butinar, M.; Turk, B. Endoglin (CD105) Silencing Mediated by shRNA Under the Control of Endothelin-1 Promoter for Targeted Gene Therapy of Melanoma. Mol. Ther. Acids 2015, 4, e239. [Google Scholar] [CrossRef]
- Stimac, M.; Kamensek, U.; Cemazar, M.; Kranjc, S.; Coer, A.; Sersa, G. Tumor radiosensitization by gene therapy against endoglin. Cancer Gene Ther. 2016, 23, 214–220. [Google Scholar] [CrossRef]
- Savarin, M.; Kamensek, U.; Cemazar, M.; Heller, R.; Sersa, G. Electrotransfer of plasmid DNA radiosensitizes B16F10 tumors through activation of immune response. Radiol. Oncol. 2017, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Prosen, L.; Hudoklin, S.; Cemazar, M.; Stimac, M.; Lampreht Tratar, U.; Ota, M.; Scancar, J.; Romih, R.; Sersa, G. Magnetic field contributes to the cellular uptake for effective therapy with magnetofection using plasmid DNA encoding against Mcam in B16F10 melanoma in vivo. Nanomedicine 2016, 11, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Stimac, M.; Dolinsek, T.; Lampreht, U.; Cemazar, M.; Sersa, G. Gene electrotransfer of plasmid with tissue specific promoter encoding shRNA against endoglin exerts antitumor efficacy against murine TS/A tumors by vascular targeted effects. PLoS ONE 2015, 10, e0124913. [Google Scholar] [CrossRef] [PubMed]
- Brezar, S.K.; Mrak, V.; Bosnjak, M.; Savarin, M.; Sersa, G.; Cemazar, M. Intratumoral gene electrotransfer of plasmid DNA encoding shRNA against melanoma cell adhesion molecule radiosensitizes tumors by antivascular effects and activation of an immune response. Vaccines 2020, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Ciric, E.; Sersa, G. Radiotherapy in combination with vascular-targeted therapies. Radiol. Oncol. 2010, 44, 67–78. [Google Scholar] [CrossRef]
- Mazeron, R.; Anderson, B.; Supiot, S.; Paris, F.; Deutsch, E. Current state of knowledge regarding the use of antiangiogenic agents with radiation therapy. Cancer Treat. Rev. 2011, 37, 476–486. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Cemazar, M.; Heller, L.C. Cytosolic DNA Sensor Upregulation Accompanies DNA Electrotransfer in B16.F10 Melanoma Cells. Mol. Ther. Nucleic Acids 2016, 5, e322. [Google Scholar] [CrossRef]
- Jesenko, T.; Bosnjak, M.; Markelc, B.; Sersa, G.; Znidar, K.; Heller, L.; Cemazar, M. Radiation induced upregulation of dna sensing pathways is cell-type dependent and can mediate the off-target effects. Cancers 2020, 12, 3365. [Google Scholar] [CrossRef] [PubMed]
- Polajzer, T.; Jarm, T.; Miklavcic, D. Analysis of damage-associated molecular pattern molecules due to electroporation of cells in vitro. Radiol. Oncol. 2020, 54, 317–328. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The Impact of Radiation-Induced DNA Damage on cGAS-STING-Mediated Immune Responses to Cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- De Giovanni, C.; Nicoletti, G.; Landuzzi, L.; Palladini, A.; Lollini, P.L.; Nanni, P. Bioprofiling TS/A murine mammary cancer for a functional precision experimental model. Cancers 2019, 11, 1889. [Google Scholar] [CrossRef]
- Kamensek, U.; Ursic, K.; Markelc, B.; Cemazar, M.; Setrajcic Dragos, V.; Sersa, G. Mutational burden, MHC-I expression and immune infiltration as limiting factors for in situ vaccination by TNFα and IL-12 gene electrotransfer. Bioelectrochemistry 2021, 140, 107831. [Google Scholar] [CrossRef]
- Dolinsek, T.; Markelc, B.; Sersa, G.; Coer, A.; Stimac, M.; Lavrencak, J.; Brozic, A.; Kranjc, S.; Cemazar, M. Multiple Delivery of siRNA against Endoglin into Murine Mammary Adenocarcinoma Prevents Angiogenesis and Delays Tumor Growth. PLoS ONE 2013, 8, e58723. [Google Scholar] [CrossRef]
- Dolinsek, T.; Markelc, B.; Bosnjak, M.; Blagus, T.; Prosen, L.; Kranjc, S.; Stimac, M.; Lampreht, U.; Sersa, G.; Cemazar, M. Endoglin Silencing has Significant Antitumor Effect on Murine Mammary Adenocarcinoma Mediated by Vascular Targeted Effect. Curr. Gene Ther. 2015, 15, 228–244. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Sedlar, A.; Kranjc, S.; Dolinsek, T.; Cemazar, M.; Coer, A.; Sersa, G. Radiosensitizing effect of intratumoral interleukin-12 gene electrotransfer in murine sarcoma. BMC Cancer 2013, 13, 38. [Google Scholar] [CrossRef]
- Todorovic, V.; Groselj, B.; Cemazar, M.; Prevc, A.; Zakelj, M.N.; Strojan, P.; Sersa, G. Expression of DNA-damage response and repair genes after exposure to DNA-damaging agents in isogenic head and neck cells with altered radiosensitivity. Radiol. Oncol. 2022, 56, 173–184. [Google Scholar] [CrossRef]
- Desmet, C.J.; Ishii, K.J. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef]
- Znidar, K.; Bosnjak, M.; Semenova, N.; Pakhomova, O.; Heller, L.; Cemazar, M. Tumor cell death after electrotransfer of plasmid DNA is associated with cytosolic DNA sensor upregulation. Oncotarget 2018, 9, 18665–18681. [Google Scholar] [CrossRef] [PubMed]
- Nanni, P.; De Giovanni, C.; Lollini, P.L.; Nicoletti, G.; Prodi, G. TS/A: A new metastasizing cell line from a BALB/c spontaneous mammary adenocarcinoma. Clin. Exp. Metastasis 1983, 1, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Group | TGD (Days) | Complete Responders | Secondary Challenge | |||
| n | AM ± SEM | n | % | n | % | |
| CTRL | 6 | 0.0 ± 0.5 | 0 | 0 | / | / |
| pEmpty | 6 | −0.1 ± 0.6 | 0 | 0 | / | / |
| pDual | 6 | 1.0 ± 0.5 | 0 | 0 | / | / |
| EP | 8 | 1.7 ± 1.5 | 0 | 0 | / | / |
| GET pEmpty | 8 | 3.5 ± 1.2 | 0 | 0 | / | / |
| GET pDual | 8 | 11.3 ± 4.3 | 1 | 12.5 | 0 | 0 |
| IR | 6 | 0.0 ± 0.4 | 0 | 0 | / | / |
| pEmpty + IR | 7 | 1.9 ± 1.7 | 0 | 0 | / | / |
| pDual + IR | 7 | 7.5 ± 4.1 | 1 | 14.3 | 0 | 0 |
| EP + IR | 8 | 12.8 ± 3.7 | 1 | 12.5 | 0 | 0 |
| GET pEmpty + IR | 8 | 13.9 ± 4.8 | 2 | 25 | 0 | 0 |
| GET pDual + IR | 8 | 23.9 ± 3.0 | 4 | 50 | 1 | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savarin, M.; Znidar, K.; Sersa, G.; Komel, T.; Cemazar, M.; Kamensek, U. Tumor Radiosensitization by Gene Electrotransfer-Mediated Double Targeting of Tumor Vasculature. Int. J. Mol. Sci. 2023, 24, 2755. https://doi.org/10.3390/ijms24032755
Savarin M, Znidar K, Sersa G, Komel T, Cemazar M, Kamensek U. Tumor Radiosensitization by Gene Electrotransfer-Mediated Double Targeting of Tumor Vasculature. International Journal of Molecular Sciences. 2023; 24(3):2755. https://doi.org/10.3390/ijms24032755
Chicago/Turabian StyleSavarin, Monika, Katarina Znidar, Gregor Sersa, Tilen Komel, Maja Cemazar, and Urska Kamensek. 2023. "Tumor Radiosensitization by Gene Electrotransfer-Mediated Double Targeting of Tumor Vasculature" International Journal of Molecular Sciences 24, no. 3: 2755. https://doi.org/10.3390/ijms24032755