
Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors

 , , , ,
, , , ,  ,
,  and
and 
Abstract
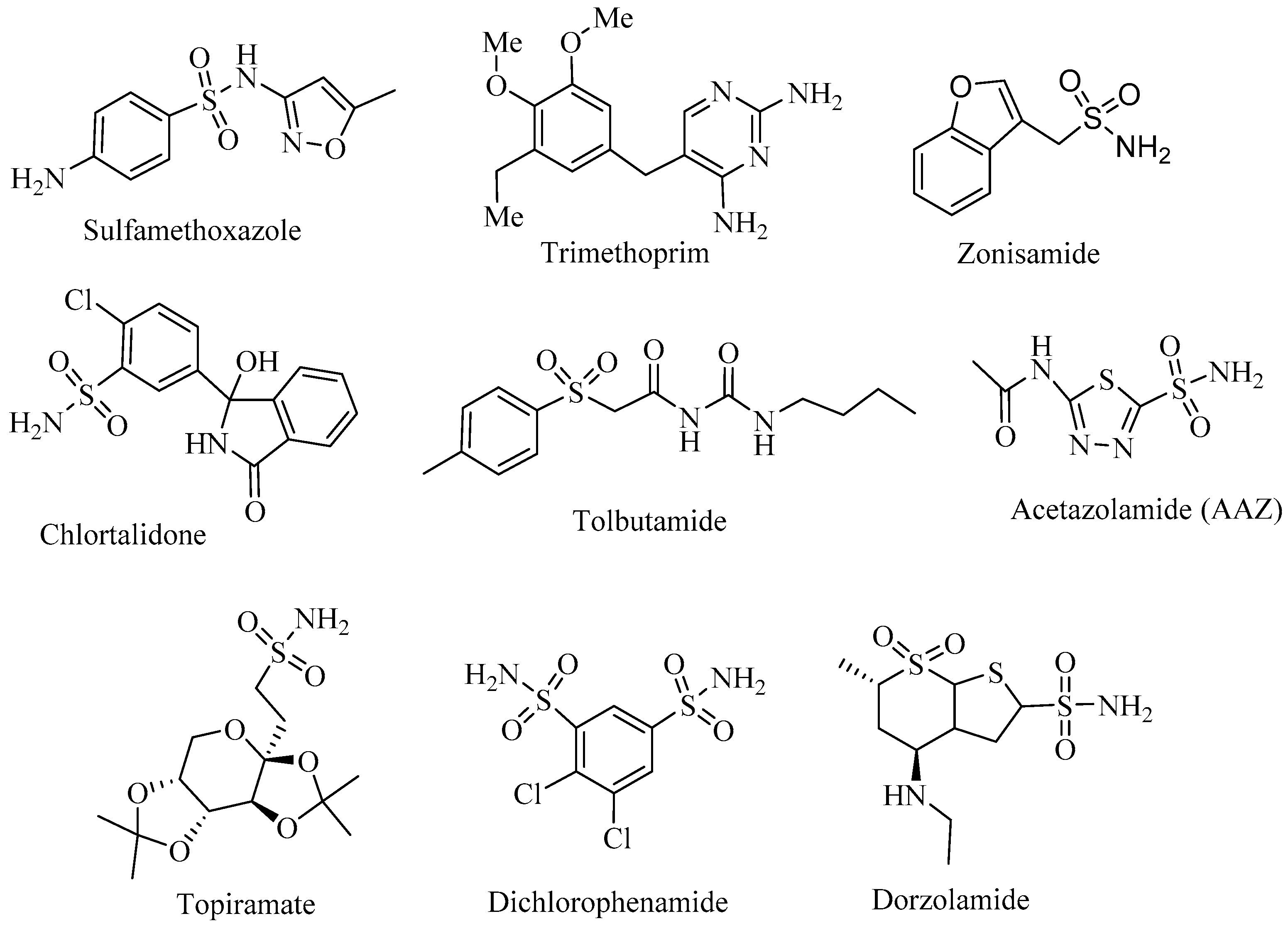
1. Introduction
2. Results and Discussion
2.1. Preliminary Docking Studies
2.2. Prediction of Toxicity
- ▪ Class I: fatal if swallowed (LD50 ≤ 5)
- ▪ Class II: fatal if swallowed (5 < LD50 ≤ 50)
- ▪ Class III: toxic if swallowed (50 < LD50 ≤ 300)
- ▪ Class IV: harmful if swallowed (300 < LD50 ≤ 2000)
- ▪ Class V: may be harmful if swallowed (2000 < LD50 ≤ 5000)
- ▪ Class VI: non-toxic (LD50 > 5000)
2.3. Chemistry
2.4. Evaluation of CA Inhibitory Activity
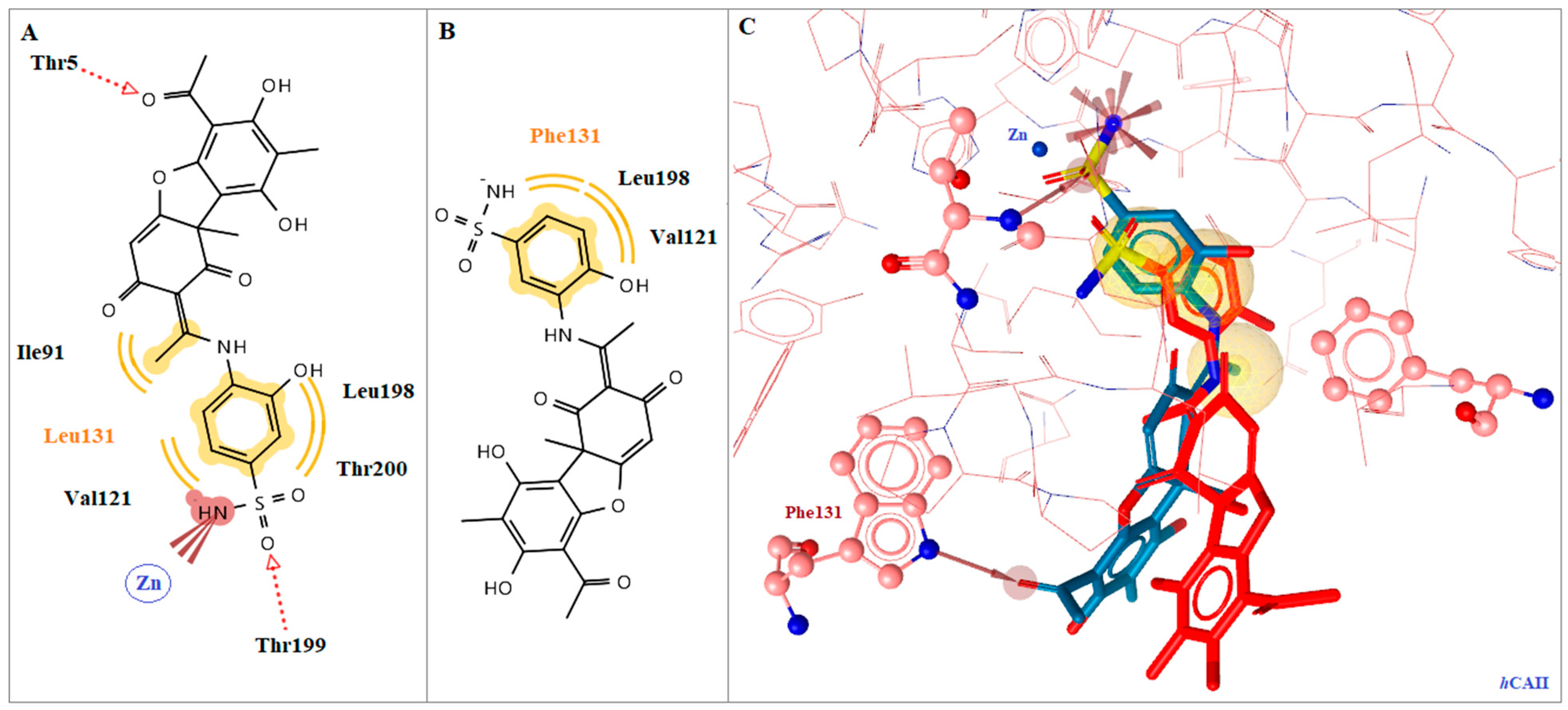
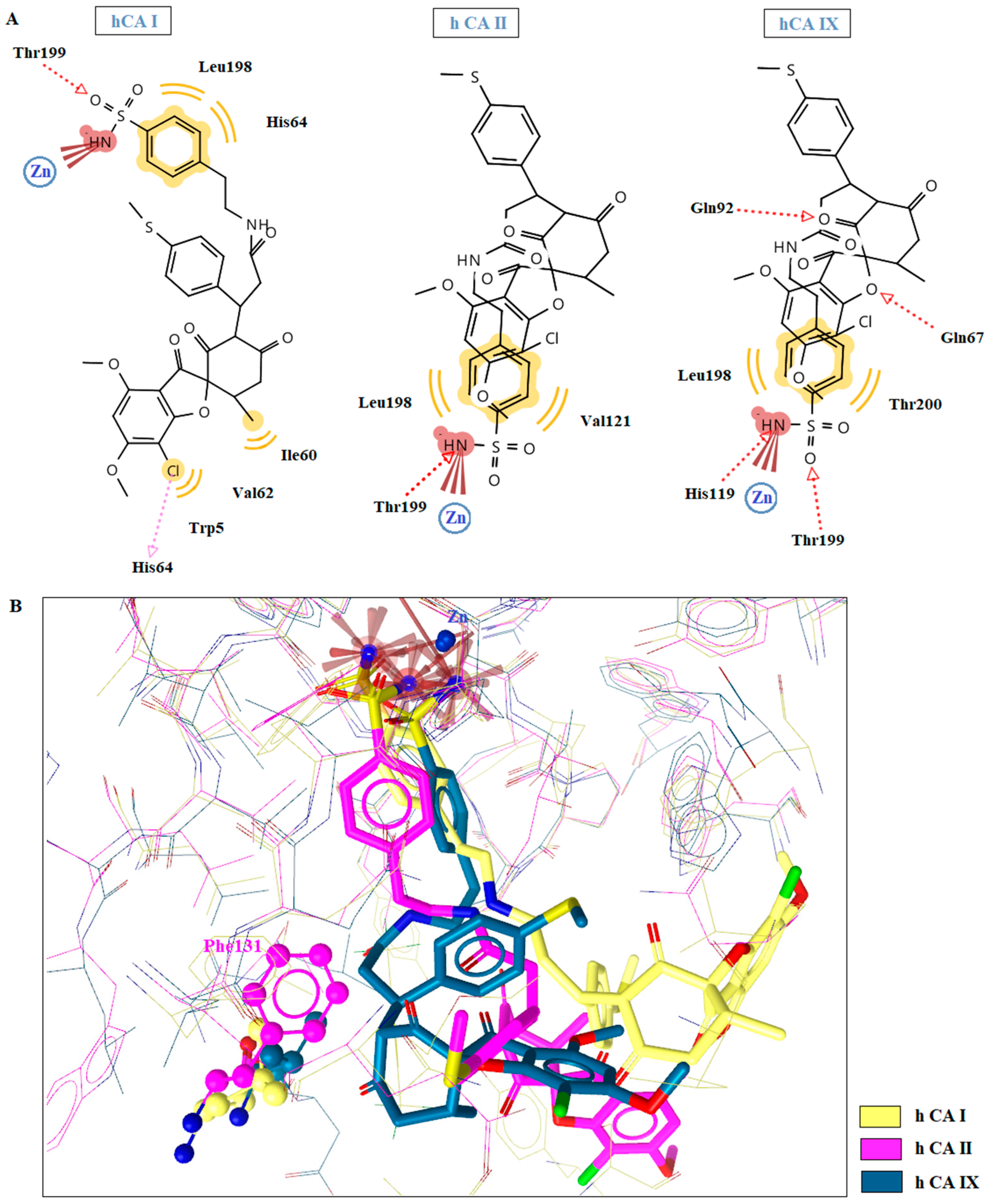
2.5. Molecular Docking Studies in Human CAs Isoforms
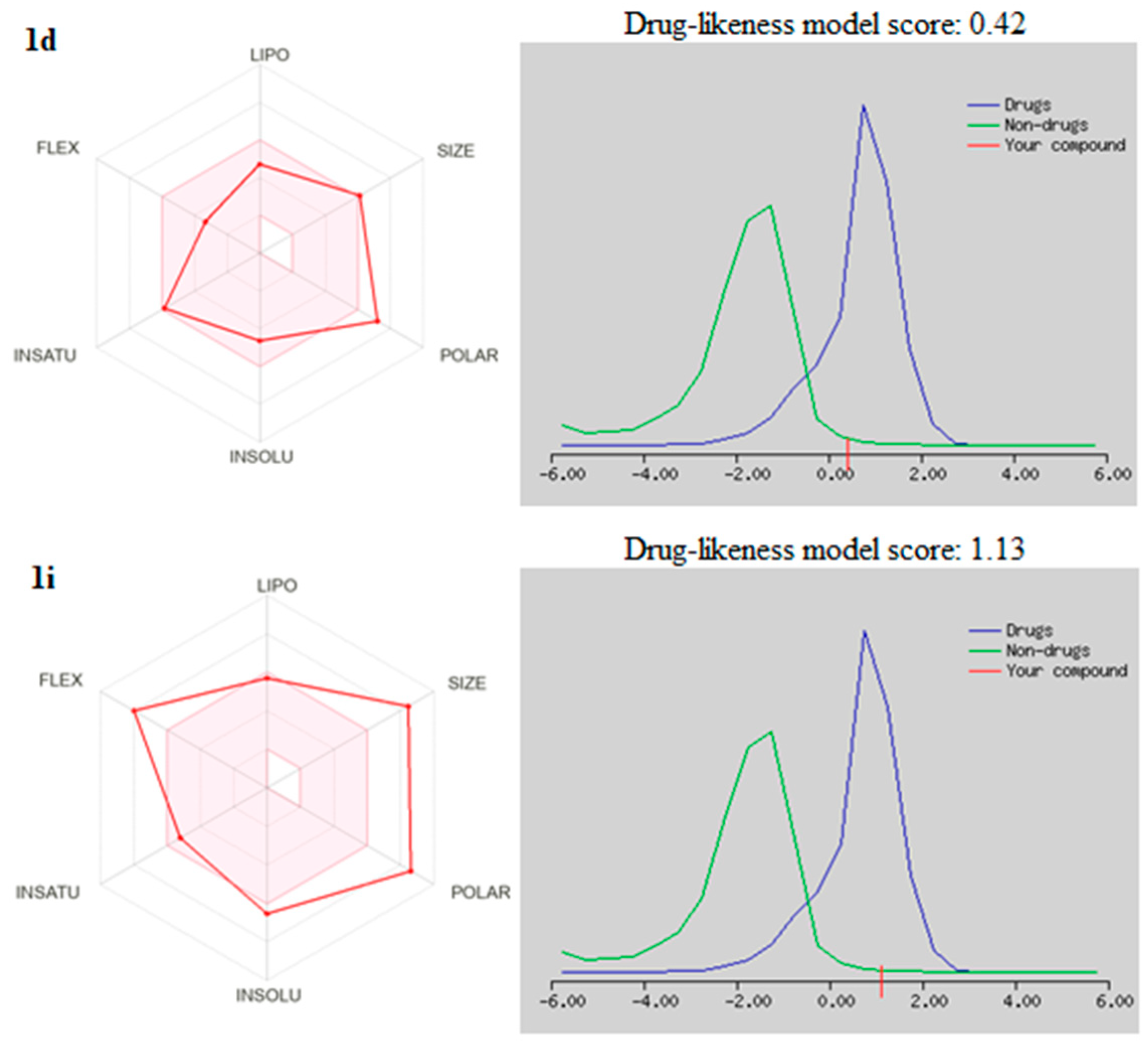
2.6. Drug Likeneess
3. Materials and Methods
3.1. Chemistry
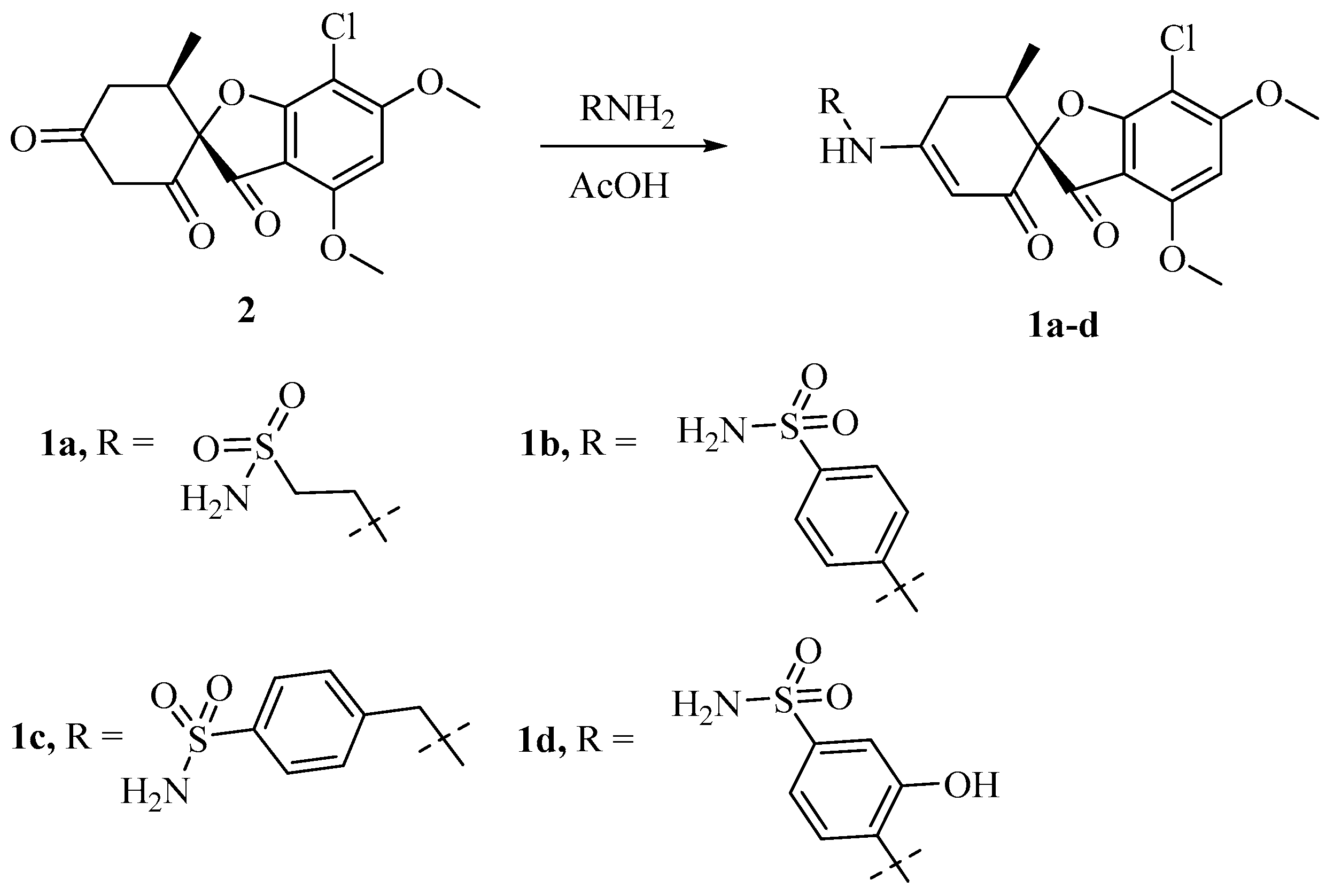
3.1.1. Synthesis of Griseofulvin Derivatives 1a–d
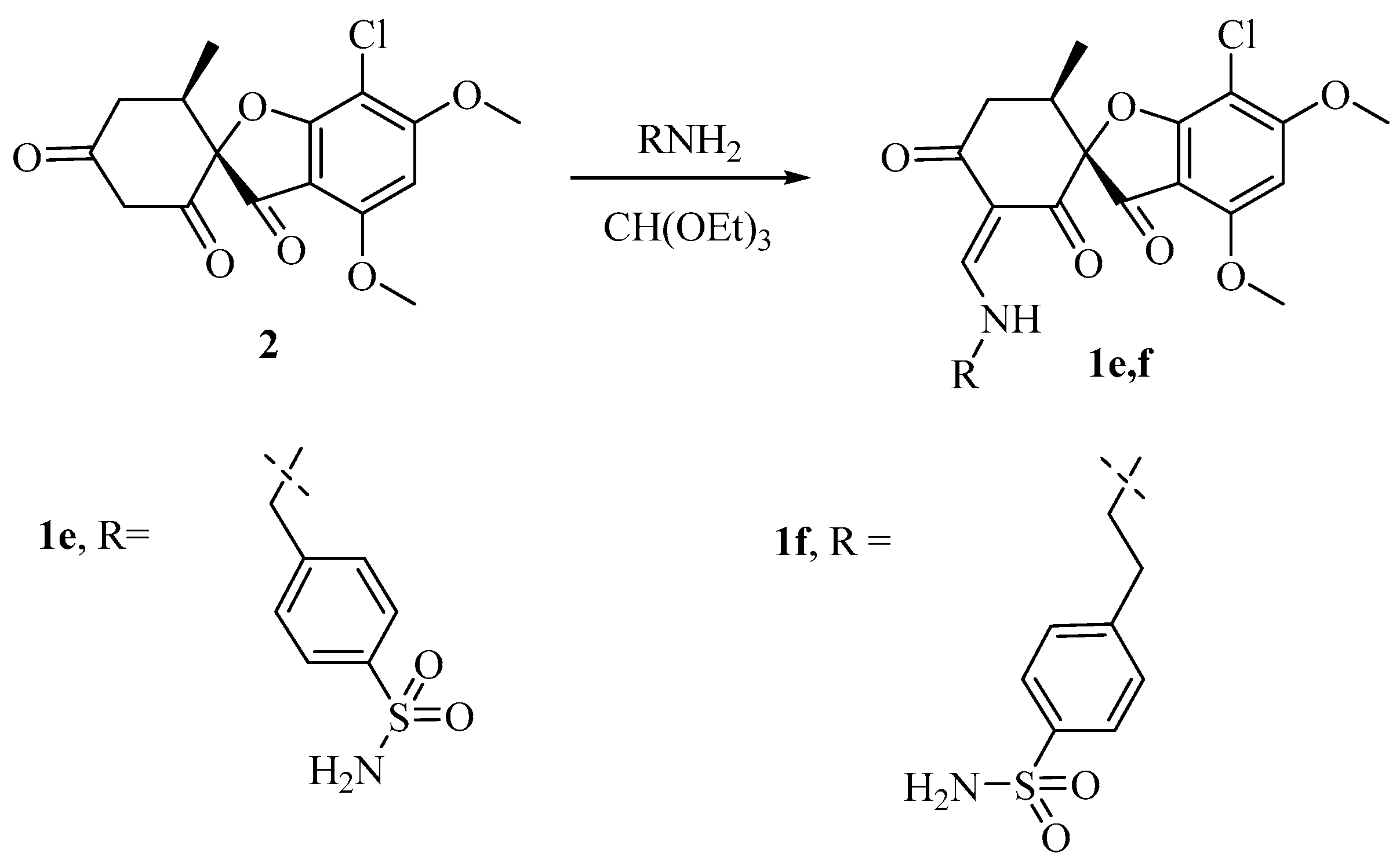
3.1.2. Synthesis of Griseofulvin Derivatives 1e,f
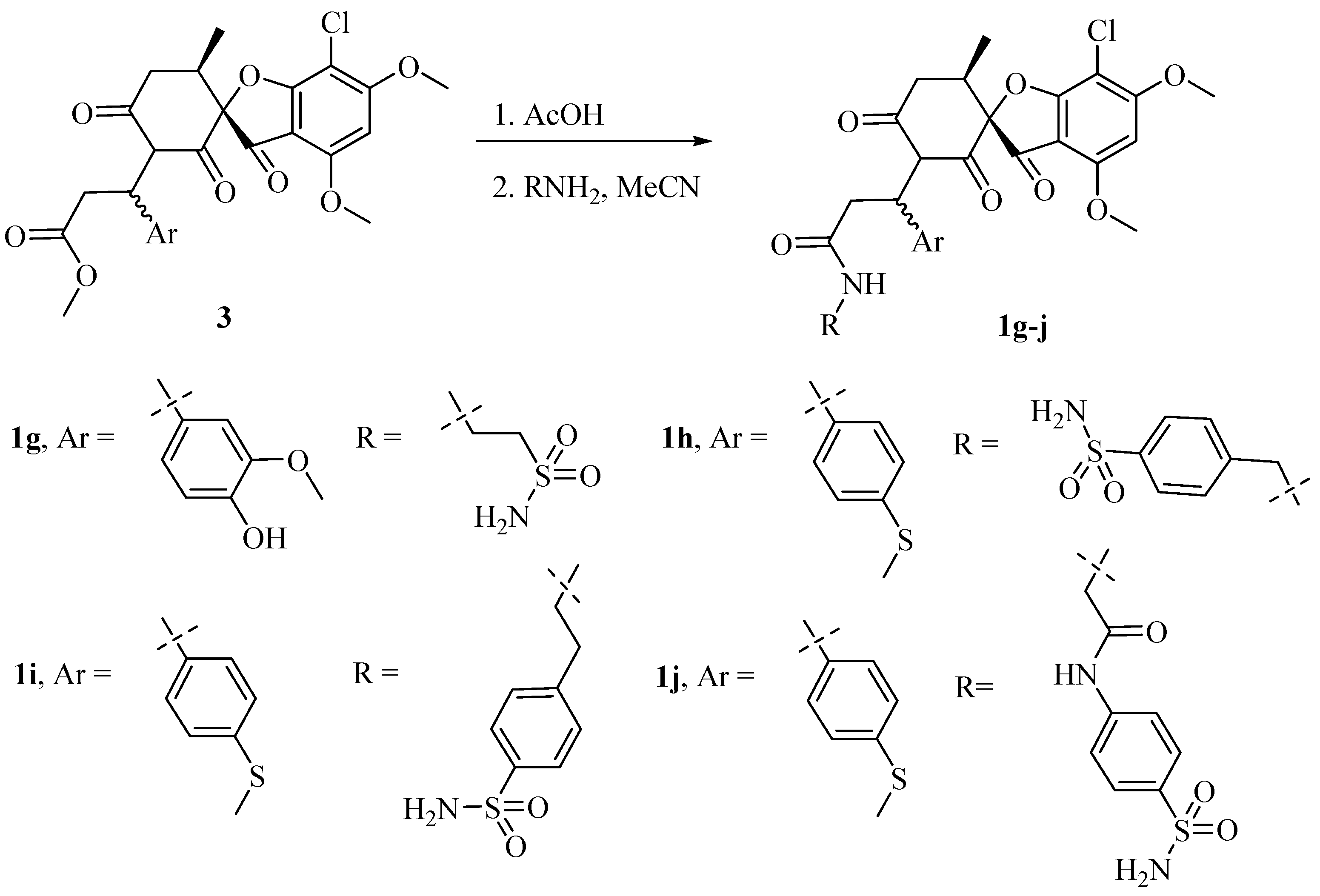
3.1.3. Synthesis of Griseofulvin Derivatives 1g–j
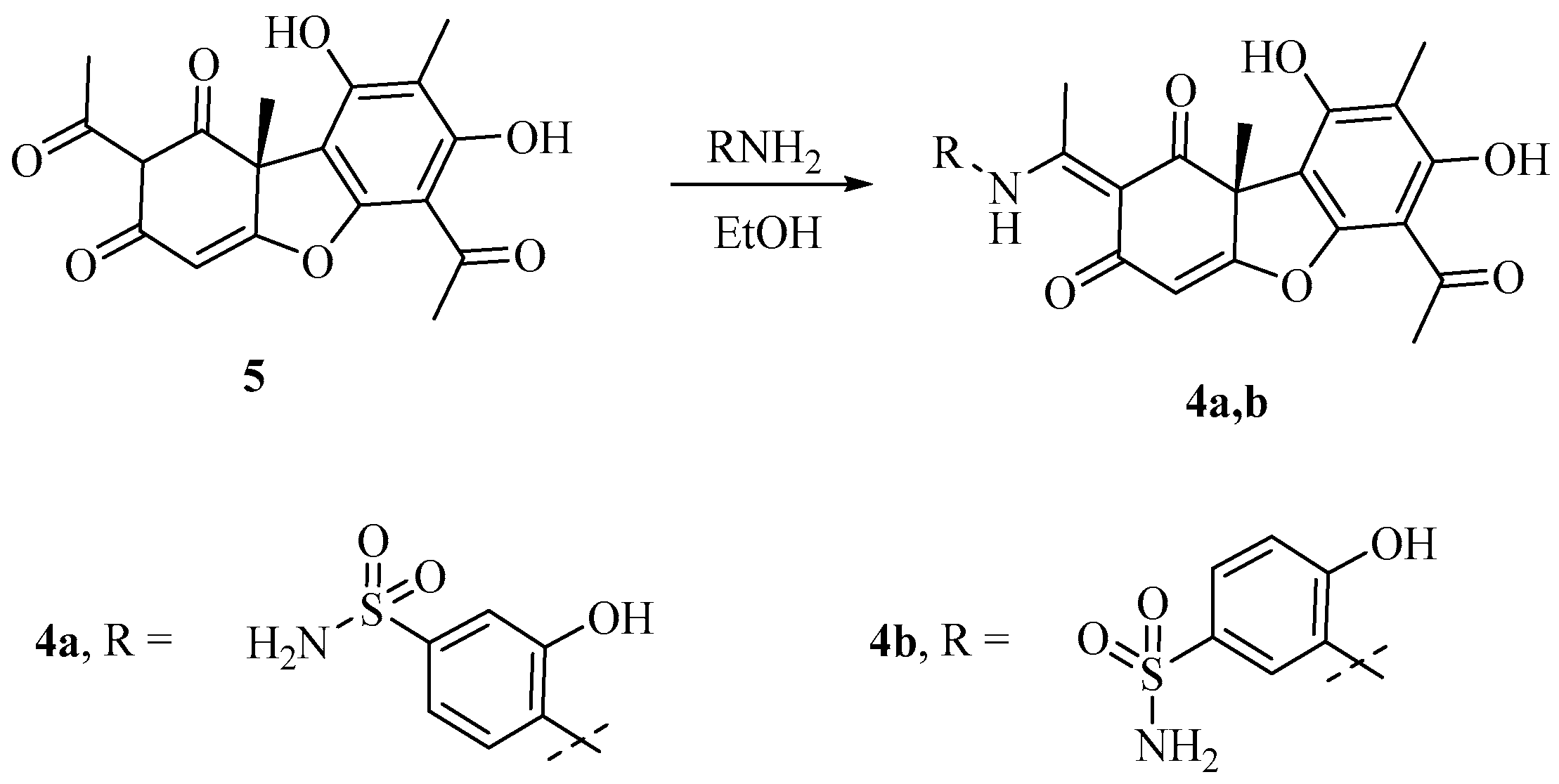
3.1.4. Synthesis of Usnic Acid Derivatives 4a,b
3.2. CA Inhibition Assay
3.3. Molecular Docking Studies
3.4. Drug Likeness
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Supuran, C.T. Carbonic anhydrases: From biomedical applications of the inhibitors and activators to biotechnological use for CO2 capture. J. Enzym. Inhib. Med. Chem. 2013, 28, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; McKenna, R. Carbonic anhydrase inhibitors drug design. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; McKenna, R., Frost, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 291–323. [Google Scholar]
- Esbaugh, A.J.; Tufts, B.L. The structure and function of carbonic anhydrase isozymes in the respiratory system of vertebrates. Respir. Physiol. Neurobiol. 2006, 154, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, I.; Minakuchi, T.; Onishi, S. Carbonic anhydrase inhibitors: Cloning, characterization, and inhibition studies of the cytosolic isozyme III with sulfonamides. Bioorg. Med. Chem. 2007, 15, 7229–7236. [Google Scholar] [CrossRef] [PubMed]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase activators. Future Med. Chem. 2018, 10, 561–573. [Google Scholar] [CrossRef]
- Zamanova, S.; Shabana, A.M.; Mondal, U.K.; Ilies, M.A. Carbonic anhydrases as disease markers. Expert Opin. Ther. Pat. 2019, 29, 509–533. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin. Drug Discov. 2019, 14, 1175–1197. [Google Scholar] [CrossRef]
- Kocak, S.; Lolak, N.; Bua, S. α-Carbonic anhydrases are strongly activated by spinaceamine deriva-tives. Bioorg. Med. Chem. 2019, 27, 800–804. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abdel-Aziz, H.A.; Abou-Seri, S.M.; Supuran, C.T. Novel synthesized SLC-0111 thiazole and thiadiazole analogues:Determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg. Chem. 2019, 87, 794–802. [Google Scholar] [CrossRef]
- Lolak, N.; Akocak, S.; Bua, S.; Koca, M.; Supuran, C.T. Design and synthesis of novel 1,3-diaryltriazene-substituted sulfonamides as potent and selective carbonic anhydrase II inhibitors. Bioorg. Chem. 2018, 77, 542–547. [Google Scholar] [CrossRef]
- Lolak, N.; Akocak, S.; Bua, S.; Sanku, R.K.K.; Supuran, C.T. Discovery of new ureido benzenesulfonamides incorporating 1,3,5-triazine moieties as carbonic anhydrase I, II, IX and inhibitors. Bioorg. Med. Chem. 2019, 27, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Suthar, S.K.; Bansal, S.; Lohan, S.; Modak, V.; Chaudhary, A.; Tiwari, A. Design and synthesis of novel 4-(4-oxo-2-arylthiazolidin-3-yl) benzenesulfonamides as selective inhibitors of carbonic anhydrase IX over I and II with potential anticancer activity. Eur. J. Med. Chem. 2013, 66, 372–379. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. [Google Scholar] [CrossRef]
- Kartsev, V.; Geronikaki, A.; Petrou, A.; Lichitsky, B.; Kostic, M.; Smiljkovic, M.; Soković, M.; Sirakanyan, S. Griseofulvin Derivatives: Synthesis, Molecular Docking and Biological Evaluation. Curr. Top Med. Chem. 2019, 19, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Geronikaki, A.; Kartsev, V.; Petrou, A.; Akrivou, M.G.; Vizirianakis, I.S.; Chatzopoulou, F.M.; Lichitsky, B.; Sirakanyan, S.; Kostic, M.; Smiljkovic, M.; et al. Antibacterial activity of griseofulvin analogues as an example of drug repurposing. Int. J. Antimicrob Agents 2020, 55, 105884. [Google Scholar] [CrossRef]
- Kartsev, V.; Lichisky, B.; Geronikaki, A.; Petrou, A.; Smiljkovic, M.; Kostic, M.; Radanovic, O.; Soković, M. Design, synthesis and antimicrobial activity of usnic acid derivatives. Med. Chem. Commun. 2019, 10, 180. [Google Scholar] [CrossRef]
- Yu, X.; Guo, Q.; Su, G.; Yang, A.; Hu, Z.; Qu, C.; Wan, Z.; Li, R.; Tu, P.; Chai, X. Usnic Acid Derivatives with Cytotoxic and Antifungal Activities from the Lichen Usnea longissima. J. Nat. Prod. 2016, 79, 1373–1380. [Google Scholar] [CrossRef]
- Guzow-Krzemińska, B.; Guzow, K.; Herman-Antosiewicz, A. Usnic Acid Derivatives as Cytotoxic Agents against Cancer Cells and the Mechanisms of Their Activity. Curr. Pharm. Rep. 2019, 5, 429–439. [Google Scholar] [CrossRef]
- Pyrczak-Felczykowska, A.; Narlawar, R.; Pawlik, A.; Guzow-Krzemińska, B.; Artymiuk, D.; Hać, A.; Ryś, K.; Rendina, L.M.; Reekie, T.A.; Herman-Antosiewicz, A.; et al. Synthesis of Usnic Acid Derivatives and Evaluation of Their Antiproliferative Activity against Cancer Cells. J. Nat. Prod. 2019, 82, 1768–1778. [Google Scholar] [CrossRef]
- Sokolov, D.N.; Zarubaev, V.V.; Shtro, A.A.; Polovinka, M.P.; Luzina, O.A.; Komarova, N.I.; Salakhutdinov, N.F.; Kiselev, O.I. Anti-viral activity of (−)- and (+)-usnic acids and their derivatives against influenza virus A (H1N1) 2009. Bioorg. Med. Chem. Lett. 2012, 22, 7060–7064. [Google Scholar] [CrossRef]
- Aris, P.; Mohamadzadeh, M.; Wei, Y.; Xia, X. In Silico Molecular Dynamics of Griseofulvin and Its Derivatives Revealed Potential Therapeutic Applications for COVID-19. Int. J. Mol. Sci. 2022, 23, 6889. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Wang, W.; Park, K.H. (+)-Usnic acid and its salts, inhibitors of SARS-CoV-2,identified by using in silico methods and in vitro assay. Sci. Rep. 2022, 12, 13118. [Google Scholar] [CrossRef] [PubMed]
- Borisov, S.A.; Luzina, O.; Khvostov, M.; Tolstikova, M.; Salak, N. Synthesis and Pharmacological Evaluation of (+)-Usnic Acid Derivatives as Hypoglycemic Agents. Molbank 2022, 2022, M1459. [Google Scholar] [CrossRef]
- Cetin Cakmak, K.; Gülçin, İ. Anticholinergic and antioxidant activities of usnic acid-an activity-structure insight. Toxicol. Rep. 2019, 6, 1273–1280. [Google Scholar] [CrossRef]
- Guo, H.Y.; Jin, C.; Zhang, H.M.; Jin, C.M.; Shen, Q.K.; Quan, Z.S. Synthesis and Biological Evaluation of (+)-Usnic Acid Derivatives as Potential Anti-Toxoplasma gondii Agents. J. Agric. Food Chem. 2019, 67, 9630–9642. [Google Scholar] [CrossRef]
- Ovung, A.; Bhattacharyya, J. Sulfonamide drugs: Structure, antibacterial property, toxicity, and biophysical interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar]
- Mohamed, H.; Haiba, M.; Mohamed, N.; Awad, G.; Ahmed, N. New hydronaphthalene-sulfonamide derivatives: Synthesis, antimicrobial evaluation and QSAR study. J. Mol. Str. 2021, 1246, 131108. [Google Scholar] [CrossRef]
- Shahzad, S.; Qadir, M.A.; Ahmed, M.; Ahmad, S.; Khan, M.J.; Gulzar, A.; Muddassar, M. Folic acid-sulfonamide conjugates as antibacterial agents: Design, synthesis and molecular docking studies. RSC Adv. 2020, 10, 42983–42992. [Google Scholar] [CrossRef]
- Wan, Y.; Fang, G.; Chen, H.; Deng, X. Sulfonamide derivatives as potential anti-cancer agents and their SARs elucidation. Eur. J. Med. Chem. 2021, 226, 113837. [Google Scholar] [CrossRef]
- Pingaew, R.; Mandi, P.; Prachayasittikul, V.; Thongnum, A.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Investigations on Anticancer and Antimalarial Activities of Indole-Sulfonamide Derivatives and In Silico Studies. ACS Omega 2021, 6, 31854–31868. [Google Scholar] [CrossRef]
- Firdous, F.; Ibrahim, R.; Furqan, M.; Khan, N.; Raza, H.; Singh, U.; Emwas, A.-H.; Jaremko, M.; Chotana, G.A.; Faisal, A.; et al. Synthesisand Characterization of Griseofulvin Derivatives as Microtubule-Stabilizing Agents. ChemistrySelect 2022, 7, e20220283. [Google Scholar] [CrossRef]
- Akgul, O.; Di Cesare Mannelli, L.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Altamimi, A.S.A.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of Novel Nonsteroidal Anti-Inflammatory Drugs and Carbonic Anhydrase Inhibitors Hybrids (NSAIDs–CAIs) for the Management of Rheumatoid Arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; Mohamed Hassan, R.; Abdel-Sattar El-Azzouny, A.; Nabil Aboul-Enein, M.; Oh, C.H. Anticancer profile and anti-inflammatory effect of new N-(2-((4-(1,3-diphenyl-1H-pyrazol-4-yl) pyridine sulfonamide derivatives. Bioorg. Chem. 2021, 117, 105424. [Google Scholar] [CrossRef] [PubMed]
- Güngör, S.A.; Şahin, İ.; Güngör, Ö.; Tok, T.T.; Köse, M. Synthesis, Biological Evaluation and Docking Study of Mono- and Di-Sulfonamide Derivatives as Antioxidant Agents and Acetylcholinesterase Inhibitors. Chem. Biodivers. 2022, 19, e202200325. [Google Scholar] [CrossRef]
- Durgapal, S.; Soman, S. Evaluation of novel coumarin-proline sulfonamide hybrids as anticancer and antidiabetic agents. Synth. Commun. 2019, 49, 2869–2883. [Google Scholar] [CrossRef]
- Khalid, Z.; Alnuwaiser, M.A.; Ahmad, H.A.; Shafqat, S.S.; Munawar, M.A.; Kamran, K.; Abbas, M.M.; Kalam, M.A.; Ewida, M.A. Experimental and Computational Analysis of Newly Synthesized Benzotriazinone Sulfonamides as Alpha-Glucosidase Inhibitors. Molecules 2022, 27, 6783–6799. [Google Scholar] [CrossRef]
- Ekoh, O.C.; Okoro, U.; Ugwu, D.; Ali, R.; Okafor, S.; Ugwuja, D.; Attah, S. Novel Dipeptides Bearing Sulfonamide as Antimalarial and Antitrypanosomal Agents: Synthesis and Molecular Docking. Med. Chem. 2022, 18, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Azzam, R.A.; Elsayed, R.E.; Elgemeie, G.H. Design, Synthesis, and Antimicrobial Evaluation of a New Series of N-Sulfonamide 2-Pyridones as Dual Inhibitors of DHPS and DHFR Enzymes. ACS Omega 2020, 5, 10401–10414. [Google Scholar] [CrossRef] [PubMed]
- Xuan, G.S.; Zhan, J.H.; Zhang, A.M.; Li, W.; Zheng, K. Inhibition of carbonic anhydrase II by sulfonamide derivatives. Pharmazie 2021, 76, 412–415. [Google Scholar]
- Angeli, A.; Kartsev, V.; Petrou, A.; Pinteala, M.; Vydzhak, R.M.; Panchishin, S.Y.; Brovarets, V.; De Luca, V.; Capasso, C.; Geronikaki, A.; et al. New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors. Pharmaceuticals 2021, 14, 828–847. [Google Scholar] [CrossRef]
- Angeli, A.; Kartsev, V.; Petrou, A.; Lichitsky, B.; Komogortsev, A.; Pinteala, M.; Geronikaki, A.; Supuran, C.T. Pyrazolo [4, 3-c] pyridine Sulfonamides as Carbonic Anhydrase Inhibitors: Synthesis, Biological and In Silico Studies. Pharmaceuticals 2022, 15, 316–339. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J. Enzym. Inhib. Med. Chem. 2018, 33, 485–495. [Google Scholar] [CrossRef]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.Y.; Qin, H.L. Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- GHS-unece. Available online: http://www.unece.org/trans/danger/publi/ghs/ghs_welcome_e.html (accessed on 5 January 2023).
- Ronnest, M.H.; Harris, P.; Gotfredsen, C.; Larsen, T.; Clausen, M. Synthesis and single crystal X-ray analysis of two griseofulvin metabolites. Tetrahedron Lett. 2010, 51, 5881–5882. [Google Scholar] [CrossRef]
- Hewitson, K.S.; Vullo, D.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Molecular cloning, characterization, and inhibition studies of a β-carbonic anhydrase from Malassezia globosa, a potential antidandruff target. J. Med. Chem. 2012, 55, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; De Luca, V.; Vullo, D.; Scozzafava, A.; Carginale, V.; Supuran, C.T.; Capasso, C. Biochemical characterization of the γ-carbonic anhydrase from the oral pathogen Porphyromonas gingivalis, PgiCA. J. Enzym. Inhib. Med. Chem. 2014, 29, 532–537. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; De Luca, V.; AlOthman, Z.; Osman, S.M.; Supuran, C.T.; Capasso, C. Biochemical characterization of recombinant β-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J. Enzym. Inhib. Med. Chem. 2015, 30, 366–370. [Google Scholar] [CrossRef]
- Vullo, D.; Del Prete, S.; Osman, S.M.; Alasmary, F.A.S.; AlOthman, Z.; Donald, W.A.; Capasso, C.; Supuran, C.T. Comparison of the amine/amino acid activation profiles of the β- and γ-carbonic anhydrases from the pathogenic bacterium Burkholderia pseudomallei. J. Enzym. Inhib. Med. Chem. 2018, 33, 25–30. [Google Scholar] [CrossRef]
- Dedeoglu, N.; De Luca, V.; Isik, S.; Yildirim, H.; Kockar, F.; Capasso, C.; Supuran, C.T. Cloning, characterization and anion inhibition study of a β-class carbonic anhydrase from the caries producing pathogen Streptococcus mutans. Bioorg. Med. Chem. 2015, 23, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- De Luca, V.; Vullo, D.; Del Prete, S.; Carginale, V.; Osman, S.M.; Al Othman, Z.; Supuran, C.T.; Capasso, C. Cloning, characterization and anion inhibition studies of a γ-carbonic anhydrase from the Antarctic bacterium Colwellia psychrerythraea. Bioorg. Med. Chem. 2016, 24, 835–840. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Scozzafava, A. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, A.; Truppo, E.; Supuran, C.T.; Alterio, V.; Dathan, N.; Bootorabi, F.; Parkkila, S.; Monti, S.M.; De Simone, G. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorganic Med. Chem. Lett. 2010, 20, 5023–5026. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef]
- Stefanucci, A.; Angeli, A.; Dimmito, M.P.; Luisi, G.; Del Prete, S.; Capasso, C.; Donald, W.A.; Mollica, A.; Supuran, C.T. Activation of β- and γ-carbonic anhydrases from pathogenic bacteria with tripeptides. J Enzym. Inhib. Med. Chem. 2018, 33, 945–950. [Google Scholar] [CrossRef]
- Angeli, A.; di Cesare Mannelli, L.; Lucarini, E.; Peat, T.S.; Ghelardini, C.; Supuran, C.T. Design, synthesis and X-ray crystallography of selenides bearing benzenesulfonamide moiety with neuropathic pain modulating effects. Eur. J. Med. Chem. 2018, 154, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Angeli, A.; Pinteala, M.; Maier, S.S.; Simionescu, B.C.; Milaneschi, A.; Abbas, G.; Del Prete, S.; Capasso, C.; Capperucci, A.; Tanini, D.; et al. Evaluation of Thio- and Seleno-Acetamides Bearing Benzenesulfonamide as Inhibitor of Carbonic Anhydrases from Different Pathogenic Bacteria. Int. J. Mol. Sci. 2020, 21, 598–606. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.rcsb.org/ (accessed on 19 November 2022).
- Huey, R.; Morris, G.; Olson, A.; Goodsell, D. A semiempirical free energy force field with charge-based desolvation. J. Comp. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Structure | No | Structure |
|---|---|---|---|
| 1a |  | 1g |  |
| 1b |  | 1h |  |
| 1c |  | 1i |  |
| 1d |  | 1j |  |
| 1e |  | 4a |  |
| 1f |  | 4b |  |
| KI (nM) * | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmp | hCA I | hCA II | hCAIX | MgCA | PgiCAβ | PgiCAγ | SmuCA | BpsCAγ | CpsCAγ |
| 1a | 562.0 | 653.4 | 31.2 | 544.0 | 778.6 | 92.1 | 1629 | 448.2 | 3149 |
| 1b | 233.6 | 64.4 | 203.1 | 475.6 | 818.9 | 820.3 | 293.7 | 945.0 | 938.9 |
| 1c | 74.6 | 29.1 | 102.0 | 568.5 | 896.2 | 660.6 | 3997 | 833.0 | 899.9 |
| 1d | 28.8 | 4.9 | 29.4 | 1589 | 1285 | 584.2 | 897.0 | 877.5 | 591.4 |
| 1e | 320.2 | 165.1 | 29.2 | 1826 | 939.6 | 1273 | 1638 | 1061 | 219.6 |
| 1f | 347.6 | 70.8 | 30.6 | 4000 | 8361 | 601.5 | 2468 | 2282 | 726.3 |
| 1g | 2986 | 4052 | 709.6 | 3148 | 5593 | 2219 | 1218 | 2334 | 301.0 |
| 1h | 41.1 | 7.5 | 73.7 | 4346 | 5754 | 853.4 | 579.8 | 1401 | 625.0 |
| 1i | 15.3 | 8.1 | 23.3 | 1912 | 506.2 | 853.9 | 889.4 | 1038 | 804.4 |
| 1j | 27.7 | 5.7 | 22.5 | 2047 | 2998 | 2320 | 1234 | 710.5 | 176.8 |
| 4a | 369.5 | 8.4 | 66.9 | 3430 | 888.2 | 896.7 | 841.3 | 472.9 | 723.2 |
| 4b | 1692 | 755.3 | 305.0 | 2497 | 766.3 | 1841 | 1696 | 1000 | 809.3 |
| AAZ | 250.0 | 12.8 | 25.6 | 40,000 | 214 | 324 | 344 | 149 | 502 |
| No | hCA Isoform | Estimated Free Binding Energy (Kcal/mol) | Chelating the Zn (II) Ion | Residues Involved in H-Bond Interactions | Residues Involved in Hydrophobic Interactions |
|---|---|---|---|---|---|
| 1d | hCA I | −10.35 | Yes | Thr199 | Ala121,Leu198, Thr199, His200 |
| hCA II | −12.02 | Yes | Thr199, Thr200 | Ile91,Val121, Phe131, Leu198, Trp209 | |
| hCA IX | −8.53 | Yes | Thr199 | Val121, Val131, Leu198 | |
| 1i | hCA I | −12.45 | Yes | Thr199 | Trp5, Ile60, Val62, His64, Leu198 |
| hCA II | −11.64 | Yes | Thr199 | Val121, Leu198 | |
| hCA IX | −12.74 | Yes | Glu67, Gln92, Thr199, His199 | Leu198, Thr200 | |
| 4a | hCA I | −6.72 | No | - | Ala121, Leu198 |
| hCA II | −10.23 | Yes | Thr199 | Ile91, Val121, Leu198, Thr200 | |
| hCA IX | −6.57 | No | - | Val121, Leu198, Trp209 | |
| 4b | hCA I | −5.11 | No | - | Leu198 |
| hCA II | −4.89 | No | - | Val121, Phe131, Leu198 | |
| hCA IX | −6.26 | No | - | Val121, Leu198 | |
| AAZ | hCA I | −8.28 | Yes | Gln92 | Leu198, Thr199, His200, Pro201, Trp209 |
| hCA II | −8.87 | Yes | Thr199, Thr200 | Val121, Phe131, Leu198, Trp209 | |
| hCA IX | −9.02 | Yes | Thr199, Thr200 | Val121, Val143, Val131, Leu198, Trp209 |
| No | MW | Number of HBA a | Number of HBD b | Log Po/w (iLOGP) c | Log S d | TPSA e | Lipinski | Bioavailability Score | Drug-Likeness Model Score |
|---|---|---|---|---|---|---|---|---|---|
| 1a | 444.89 | 8 | 2 | 1.19 | Moderately soluble | 142.40 | 0 | 0.55 | 0.70 |
| 1b | 492.93 | 8 | 2 | 2.56 | Moderately soluble | 142.40 | 0 | 0.55 | 0.58 |
| 1c | 506.96 | 8 | 2 | 2.36 | Moderately soluble | 142.40 | 1 violation: MW > 500 | 0.55 | 0.75 |
| 1d | 508.93 | 9 | 3 | 2.33 | Moderately soluble | 162.63 | 1 violation: MW > 500 | 0.55 | 0.42 |
| 1e | 534.97 | 9 | 2 | 2.78 | Moderately soluble | 159.47 | 1 violation: MW > 500 | 0.55 | 0.45 |
| 1f | 548.99 | 9 | 2 | 2.68 | Moderately soluble | 159.47 | 1 violation: MW > 500 | 0.55 | 0.47 |
| 1g | 639.07 | 12 | 3 | 192 | Poorly Soluble | 206.00 | 2 violations: MW > 500, NorO > 10 | 0.17 | 1.24 |
| 1h | 701.21 | 10 | 2 | 3.58 | Poorly soluble | 201.84 | 2 violations: MW > 500, NorO > 10 | 0.17 | 1.19 |
| 1i | 715.23 | 9 | 2 | 3.40 | Poorly soluble | 201.84 | 1 violation: MW > 500 | 0.55 | 1.13 |
| 1j | 744.23 | 11 | 3 | 3.44 | Poorly soluble | 230.94 | 2 violations: MW > 500, NorO > 10 | 0.17 | 1.12 |
| 4a | 514.50 | 10 | 5 | 1.10 | Moderately soluble | 201.70 | 2 violations: MW > 500, NorO > 10 | 0.17 | 0.20 |
| 4b | 514.50 | 10 | 5 | 1.15 | Moderately soluble | 201.70 | 2 violations: MW > 500, NorO > 10 | 0.17 | 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeli, A.; Petrou, A.; Kartsev, V.; Lichitsky, B.; Komogortsev, A.; Capasso, C.; Geronikaki, A.; Supuran, C.T. Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors. Int. J. Mol. Sci. 2023, 24, 2802. https://doi.org/10.3390/ijms24032802
Angeli A, Petrou A, Kartsev V, Lichitsky B, Komogortsev A, Capasso C, Geronikaki A, Supuran CT. Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors. International Journal of Molecular Sciences. 2023; 24(3):2802. https://doi.org/10.3390/ijms24032802
Chicago/Turabian StyleAngeli, Andrea, Anthi Petrou, Victor Kartsev, Boris Lichitsky, Andrey Komogortsev, Clemente Capasso, Athina Geronikaki, and Claudiu T. Supuran. 2023. "Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors" International Journal of Molecular Sciences 24, no. 3: 2802. https://doi.org/10.3390/ijms24032802
APA StyleAngeli, A., Petrou, A., Kartsev, V., Lichitsky, B., Komogortsev, A., Capasso, C., Geronikaki, A., & Supuran, C. T. (2023). Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors. International Journal of Molecular Sciences, 24(3), 2802. https://doi.org/10.3390/ijms24032802