
Mechanisms of Prostate Cancer Cells Survival and Their Therapeutic Targeting


Abstract
:1. Introduction
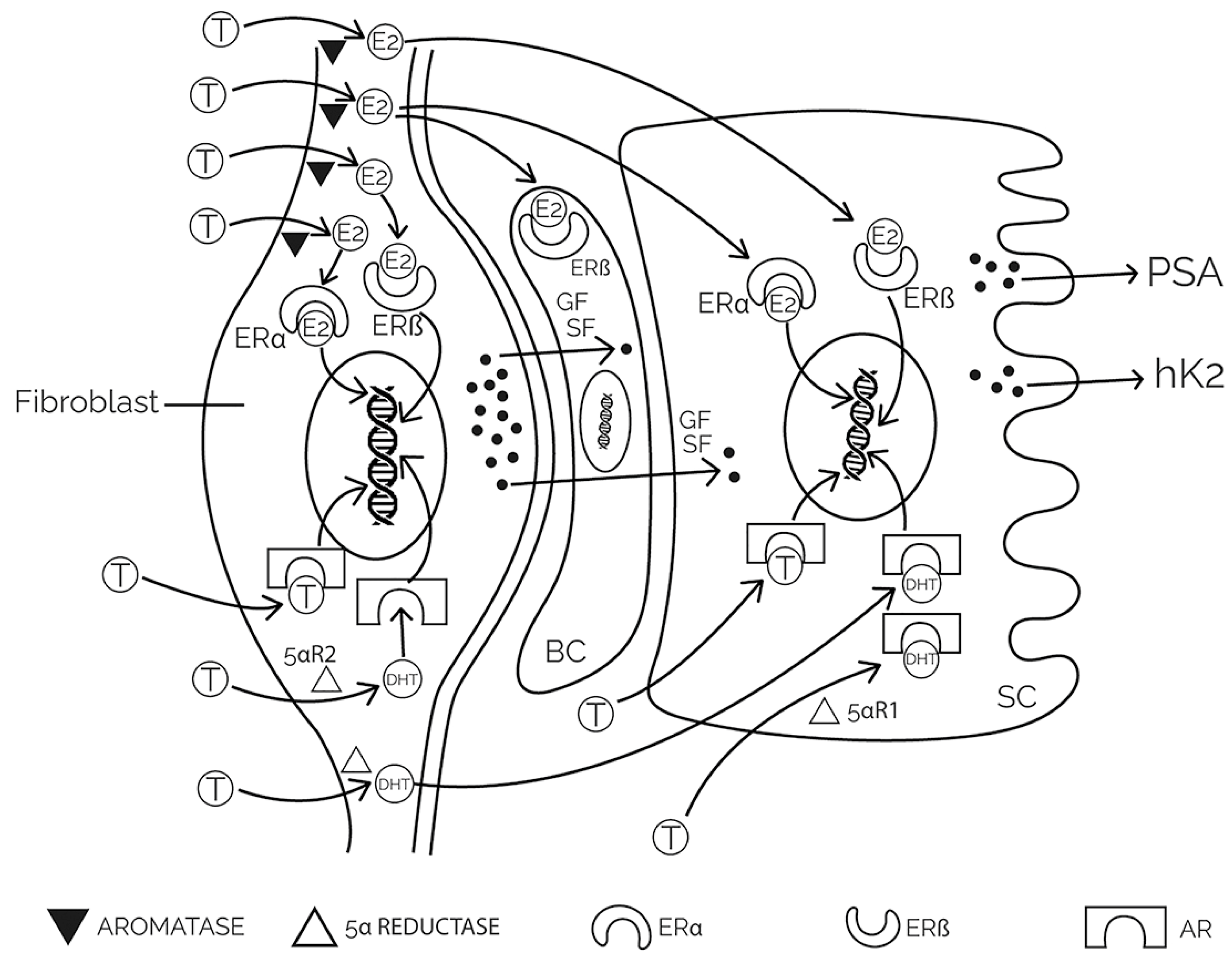
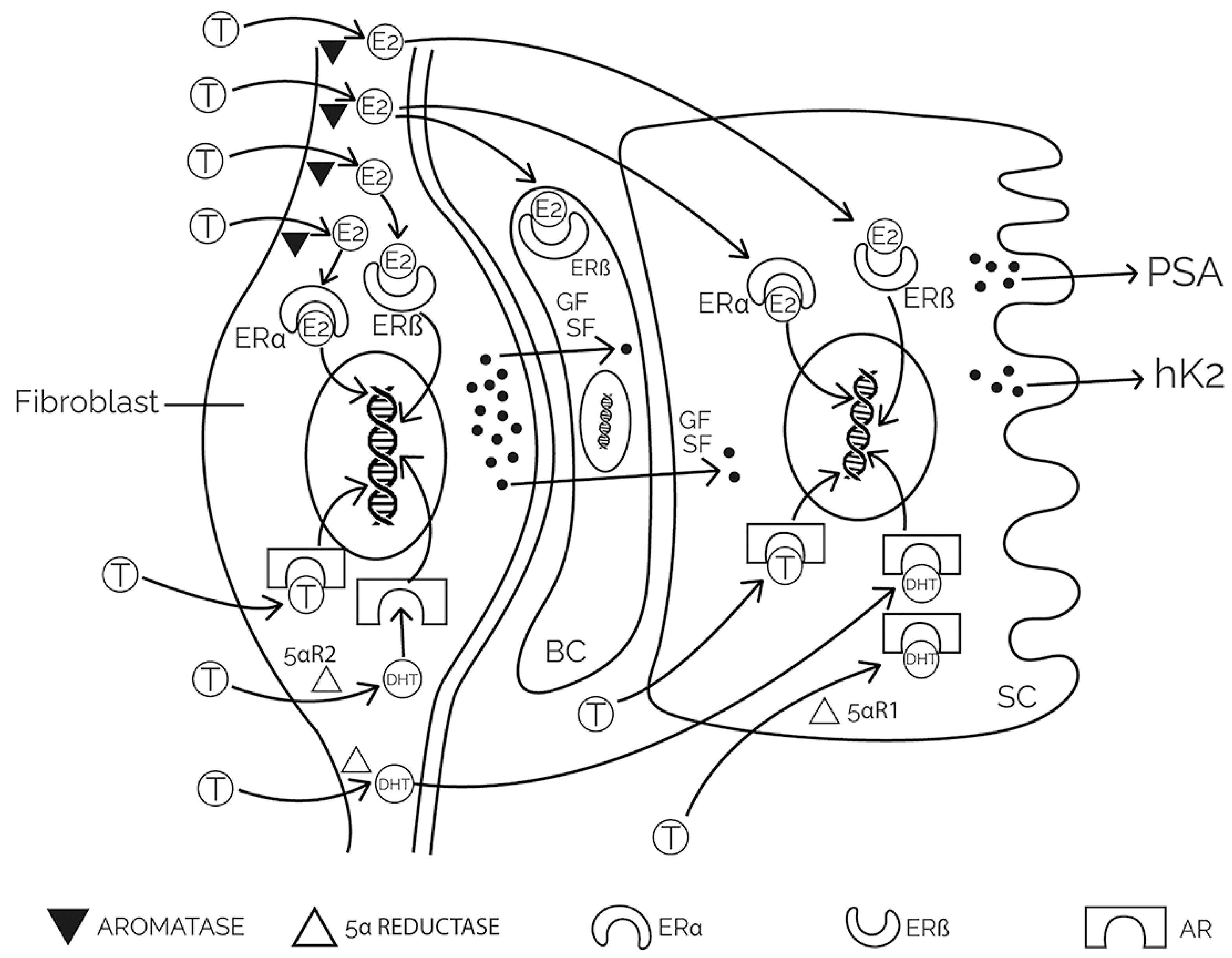
2. Physiology and Pathophysiology of the Prostate
3. PCa Etiology
3.1. Lifestyle Influence
3.2. Hormonal Influence
3.3. Metabolic Influence
3.3.1. Glucose Metabolism Remodulation
3.3.2. Lipid Metabolism Remodulation
3.3.3. Amino Acid Metabolism Remodulation
3.3.4. The Role of Autophagy
3.3.5. Metabolic Crosstalk in PCa Microenvironment
3.4. Microbiome Influence
4. Resistance to Antiandrogens in PCa Therapy
4.1. The Mechanism of Antiandrogens Action in PCa Treatment
- Inhibition of testosterone secretion;
- Inhibition of androgen action.
4.1.1. Inhibition of Testosterone Secretion
4.1.2. Inhibition of Androgen Action
4.2. Resistance to Antiandrogens
- Point mutations of the AR gene;
- Amplification of AR and CYP17;
- Changes in androgen biosynthesis;
- Changes in cofactors for AR;
- Creation of different variants of AR.
4.2.1. Point Mutations of the AR Gene
4.2.2. Amplification of Genes for AR and CYP17
4.2.3. Changes in Androgen Biosynthesis
- The EGF/IGF signaling pathway that is mediated by phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt/PKB) and depends on the level of phosphatidylinositol 3,4,5-triphosphate (PIP3) and PTEN in the cell;
- The Ras signaling pathway (Ras family of proto-oncogenes) that occurs via Cdc42-linked kinase (Ack), Ras/Raf mitogen-activated protein kinase/ERK kinase (MEK), and the proto-oncogene tyrosine protein kinase Src;
- IL6 cytokine signaling pathway that activates AR through Janus kinase signal transducer and activator of transcription (JAK1), signal transducer and activator of transcription 3 (STAT3), and intermediate histone acetyltransferasep300.
4.2.4. Changes in Cofactors for AR
4.2.5. Creation of Different Variants of AR
5. Treatment of Metastatic Prostate Cancer
5.1. Treatment of Metastatic Castration-Sensitive Prostate Cancer
5.2. Treatment of Metastatic Castration-Resistant Prostate Cancer
5.3. Treatment of Non-Metastatic Castration-Resistant Prostate Cancer
5.4. Treatment of Castration-Resistant Prostate Cancer and Autophagy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef]
- Fabiani, R.; Minelli, L.; Bertarelli, G.; Bacci, S. A Western Dietary Pattern Increases Prostate Cancer Risk: A Systematic Review and Meta-Analysis. Nutrients 2016, 8, 626. [Google Scholar] [CrossRef]
- Masola, V.; Franchi, M.; Zaza, G.; Atsina, F.M.; Gambaro, G.; Onisto, M. Heparanase regulates EMT and cancer stem cell properties in prostate tumors. Front. Oncol. 2022, 12, 918419. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.O.; Eiro, N.; Fraile, M.; Beridze, N.; Escaf, A.R.; Escaf, S.; Fernandez-Gomez, J.M.; Vizoso, F.J. Prostate Cancer Tumor Stroma: Responsibility in Tumor Biology, Diagnosis and Treatment. Cancers 2022, 14, 4412. [Google Scholar] [CrossRef]
- Hoeh, B.; Wenzel, M.; Hohenhorst, L.; Kollermann, J.; Graefen, M.; Haese, A.; Tilki, D.; Walz, J.; Kosiba, M.; Becker, A.; et al. Anatomical Fundamentals and Current Surgical Knowledge of Prostate Anatomy Related to Functional and Oncological Outcomes for Robotic-Assisted Radical Prostatectomy. Front. Surg. 2021, 8, 825183. [Google Scholar] [CrossRef] [PubMed]
- Rye, M.B.; Krossa, S.; Hall, M.; van Mourik, C.; Bathen, T.F.; Drablos, F.; Tessem, M.B.; Bertilsson, H. The genes controlling normal function of citrate and spermine secretion are lost in aggressive prostate cancer and prostate model systems. iScience 2022, 25, 104451. [Google Scholar] [CrossRef]
- Deluce, J.E.; Cardenas, L.; Lalani, A.K.; Maleki Vareki, S.; Fernandes, R. Emerging Biomarker-Guided Therapies in Prostate Cancer. Curr. Oncol. 2022, 29, 5054–5076. [Google Scholar] [CrossRef]
- Li, J.; Cao, D.; Huang, Y.; Chen, B.; Chen, Z.; Wang, R.; Dong, Q.; Wei, Q.; Liu, L. Zinc Intakes and Health Outcomes: An Umbrella Review. Front. Nutr. 2022, 9, 798078. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, M.; Mucci, L.A.; Giovannucci, E.L. Zinc supplement use and risk of aggressive prostate cancer: A 30-year follow-up study. Eur. J. Epidemiol. 2022, 37, 1251–1260. [Google Scholar] [CrossRef]
- Torres-Estay, V.; Mastri, M.; Rosario, S.; Fuenzalida, P.; Echeverria, C.E.; Flores, E.; Watts, A.; Cerda-Infante, J.; Montecinos, V.P.; Sotomayor, P.C.; et al. The Differential Paracrine Role of the Endothelium in Prostate Cancer Cells. Cancers 2022, 14, 4750. [Google Scholar] [CrossRef]
- Huhtaniemi, R.; Sipila, P.; Junnila, A.; Oksala, R.; Knuuttila, M.; Mehmood, A.; Aho, E.; Laajala, T.D.; Aittokallio, T.; Laiho, A.; et al. High intratumoral dihydrotestosterone is associated with antiandrogen resistance in VCaP prostate cancer xenografts in castrated mice. iScience 2022, 25, 104287. [Google Scholar] [CrossRef]
- Xu, T.; Liu, Y.; Schulga, A.; Konovalova, E.; Deyev, S.M.; Tolmachev, V.; Vorobyeva, A. Epithelial cell adhesion moleculetargeting designed ankyrin repeat proteintoxin fusion Ec1LoPE exhibits potent cytotoxic action in prostate cancer cells. Oncol. Rep. 2022, 47, 94. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.H.; Funston, G.; Jones, D.; Watson, J. Diagnosing prostate cancer in asymptomatic patients. BMJ 2022, 377, e071076. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J.; Marzouk, K.; Finelli, A.; Saad, F.; So, A.I.; Violette, P.D.; Breau, R.H.; Rendon, R.A. UPDATE—2022 Canadian Urological Association recommendations on prostate cancer screening and early diagnosis Endorsement of the 2021 Cancer Care Ontario guidelines on prostate multiparametric magnetic resonance imaging. Can. Urol. Assoc. J. 2022, 16, E184–E196. [Google Scholar] [CrossRef] [PubMed]
- Pejcic, T.; Dimitrijevic, V.; Hadzi-Djokic, J. Urinary PSA in monitoring of patients with prostate cancer. Acta Chir. Iugosl. 2012, 59, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Pejcic, T.; Hadzi-Djokic, J.; Acimovic, M.; Topuzovic, C.; Milkovic, B.; Janjic, A. Urinary prostate specific antigen: Is the clinical use likely? Acta Chir. Iugosl. 2005, 52, 69–74. [Google Scholar] [CrossRef]
- Pejcic, T.; Hadzi-Djokic, J.; Markovic, B.; Dragicevic, D.; Glisic, B.; Lalic, N.; Acimovic, M.; Dzamic, Z.; Radosavljevic, R. Urinary PSA level and relative tumor volume after prostate biopsy. Acta Chir. Iugosl. 2009, 56, 17–21. [Google Scholar] [CrossRef]
- Pejcic, T.; Hadzi-Djokic, J.; Markovic, B.; Lalic, N.; Glisic, B. What are the possible reasons for urethral PSA varieties after radical prostatectomy? Acta Chir. Iugosl. 2010, 57, 31–35. [Google Scholar] [CrossRef]
- Pejcic, T.; Hadzi-Djokic, J.; Topuzovic, C.; Basic, D.; Marjanovic, A.; Djurasic, L. The analysis of some factors that influence on serum PSA level in localized prostate cancer patients: Mathematical model. Acta Chir. Iugosl. 2011, 58, 81–87. [Google Scholar] [CrossRef]
- Pejcic, T.P.; Tulic, C.; Lalic, N.V.; Glisic, B.D.; Ignjatovic, S.D.; Markovic, B.B.; Hadzi-Djokic, J.B. Urinary prostate-specific antigen: Predictor of benign prostatic hyperplasia progression? Can. J. Urol. 2013, 20, 6707–6713. [Google Scholar]
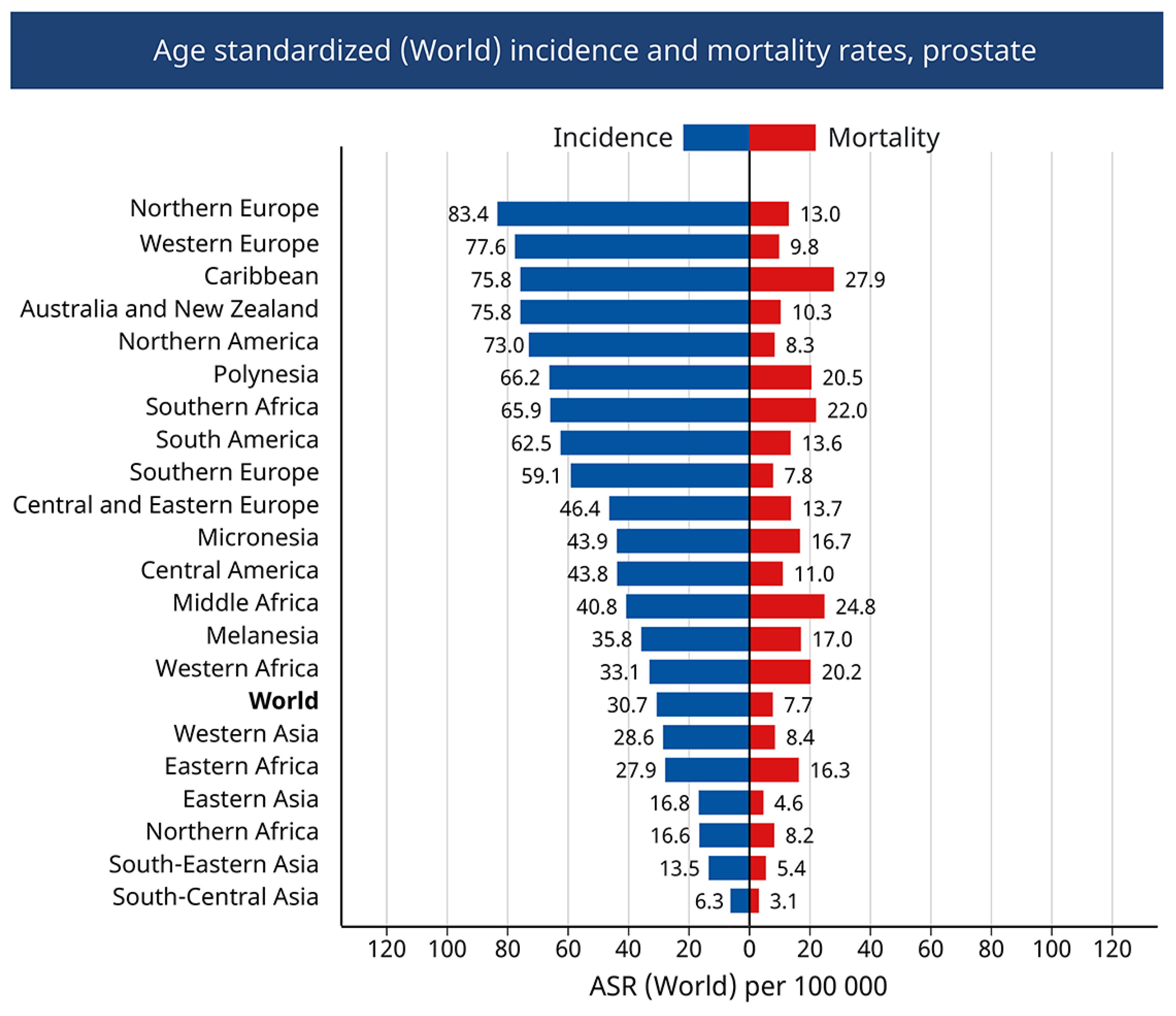
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Force, U.S.P.S.T.; Grossman, D.C.; Curry, S.J.; Owens, D.K.; Bibbins-Domingo, K.; Caughey, A.B.; Davidson, K.W.; Doubeni, C.A.; Ebell, M.; Epling, J.W., Jr.; et al. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2018, 319, 1901–1913. [Google Scholar] [CrossRef]
- Lu-Yao, G.L.; Albertsen, P.C.; Moore, D.F.; Shih, W.; Lin, Y.; DiPaola, R.S.; Barry, M.J.; Zietman, A.; O’Leary, M.; Walker-Corkery, E.; et al. Outcomes of localized prostate cancer following conservative management. JAMA 2009, 302, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.S.; Beaty, T.H.; Steinberg, G.D.; Childs, B.; Walsh, P.C. Mendelian inheritance of familial prostate cancer. Proc. Natl. Acad. Sci. USA 1992, 89, 3367–3371. [Google Scholar] [CrossRef]
- Conti, D.V.; Darst, B.F.; Moss, L.C.; Saunders, E.J.; Sheng, X.; Chou, A.; Schumacher, F.R.; Olama, A.A.A.; Benlloch, S.; Dadaev, T.; et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 2021, 53, 65–75. [Google Scholar] [CrossRef]
- Fujita, K.; Matsushita, M.; Banno, E.; De Velasco, M.A.; Hatano, K.; Nonomura, N.; Uemura, H. Gut microbiome and prostate cancer. Int. J. Urol. 2022, 29, 793–798. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Matsushita, M.; Nonomura, N. Main Inflammatory Cells and Potentials of Anti-Inflammatory Agents in Prostate Cancer. Cancers 2019, 11, 1153. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Bleeker, J.; Wang, Z.A. Applications of Vertebrate Models in Studying Prostatitis and Inflammation-Associated Prostatic Diseases. Front. Mol. Biosci. 2022, 9, 898871. [Google Scholar] [CrossRef]
- de Bono, J.S.; Guo, C.; Gurel, B.; De Marzo, A.M.; Sfanos, K.S.; Mani, R.S.; Gil, J.; Drake, C.G.; Alimonti, A. Prostate carcinogenesis: Inflammatory storms. Nat. Rev. Cancer 2020, 20, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Baio, R.; Napodano, G.; Caruana, C.; Molisso, G.; Di Mauro, U.; Intilla, O.; Pane, U.; D’Angelo, C.; Francavilla, A.B.; Guarnaccia, C.; et al. Association between obesity and frequency of high-grade prostate cancer on biopsy in men: A single-center retrospective study. Mol. Clin. Oncol. 2022, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cornago, A.; Dunneram, Y.; Watts, E.L.; Key, T.J.; Travis, R.C. Adiposity and risk of prostate cancer death: A prospective analysis in UK Biobank and meta-analysis of published studies. BMC Med. 2022, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Lou, K.; Luo, C.; Zou, J.; Zou, X.; Zhang, G. Obesity-Related Cross-Talk between Prostate Cancer and Peripheral Fat: Potential Role of Exosomes. Cancers 2022, 14, 5077. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Vázquez, F.; Pellitero, S.; Puig-Domingo, M. ENDOCRINE OBESITY: Pituitary dysfunction in obesity. Eur. J. Endocrinol. 2022, 186, R79–R92. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Haque, M. Obesity: A Doorway to a Molecular Path Leading to Infertility. Cureus 2022, 14, e30770. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, K.; Hatano, K.; De Velasco, M.A.; Uemura, H.; Nonomura, N. Connecting the Dots Between the Gut-IGF-1-Prostate Axis: A Role of IGF-1 in Prostate Carcinogenesis. Front. Endocrinol. 2022, 13, 852382. [Google Scholar] [CrossRef]
- Siech, C.; Rutz, J.; Maxeiner, S.; Grein, T.; Sonnenburg, M.; Tsaur, I.; Chun, F.K.; Blaheta, R.A. Insulin-like Growth Factor-1 Influences Prostate Cancer Cell Growth and Invasion through an Integrin alpha3, alpha5, alphaV, and beta1 Dependent Mechanism. Cancers 2022, 14, 363. [Google Scholar] [CrossRef]
- Khamit-Kush, K.K.; Powell, J.B.; Handy, J.A.; Bowen, N.; Wu, D.; Odero-Marah, V. Abstract 394: Elevated aryl hydrocarbon receptor and leptin receptor expression correlates with prostate cancer progression and may be associated with obesity-associated leptin-mediated chemoresistance. Cancer Res. 2022, 82, 394. [Google Scholar] [CrossRef]
- Kamel, H.F.M.; Nassir, A.M.; Al Refai, A.A. Assessment of expression levels of leptin and leptin receptor as potential biomarkers for risk of prostate cancer development and aggressiveness. Cancer Med. 2020, 9, 5687–5696. [Google Scholar] [CrossRef]
- Birzniece, V.; Lam, T.; McLean, M.; Reddy, N.; Shahidipour, H.; Hayden, A.; Gurney, H.; Stone, G.; Hjortebjerg, R.; Frystyk, J. Insulin-like growth factor role in determining the anti-cancer effect of metformin: RCT in prostate cancer patients. Endocr. Connect. 2022, 11, e210375. [Google Scholar] [CrossRef] [PubMed]
- Watts, E.L.; Perez-Cornago, A.; Fensom, G.K.; Smith-Byrne, K.; Noor, U.; Andrews, C.D.; Gunter, M.J.; Holmes, M.V.; Martin, R.M.; Tsilidis, K.K.; et al. Circulating insulin-like growth factors and risks of overall, aggressive and early-onset prostate cancer: A collaborative analysis of 20 prospective studies and Mendelian randomization analysis. Int. J. Epidemiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Coffey, D.S. Similarities of prostate and breast cancer: Evolution, diet, and estrogens. Urology 2001, 57, 31–38. [Google Scholar] [CrossRef]
- Baade, P.D.; Youlden, D.R.; Cramb, S.M.; Dunn, J.; Gardiner, R.A. Epidemiology of prostate cancer in the Asia-Pacific region. Prostate Int. 2013, 1, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.C.; Jo, C.; Lee, M. Meat products and consumption culture in the East. Meat Sci. 2010, 86, 95–102. [Google Scholar] [CrossRef]
- Chen, R.; Ren, S.; Chinese Prostate Cancer, C.; Yiu, M.K.; Fai, N.C.; Cheng, W.S.; Ian, L.H.; Naito, S.; Matsuda, T.; Kehinde, E.; et al. Prostate cancer in Asia: A collaborative report. Asian J. Urol. 2014, 1, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, T.; Devesa, S.; Mangtani, P.; Mathew, A.; Cooper, N.; Kao, R.; Sinha, R. Cancer incidence rates among South Asians in four geographic regions: India, Singapore, UK and US. Int. J. Epidemiol. 2008, 37, 147–160. [Google Scholar] [CrossRef]
- Shimizu, H.; Ross, R.K.; Bernstein, L.; Yatani, R.; Henderson, B.E.; Mack, T.M. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br. J. Cancer 1991, 63, 963–966. [Google Scholar] [CrossRef]
- Shin, S.; Saito, E.; Sawada, N.; Ishihara, J.; Takachi, R.; Nanri, A.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Sasazuki, S.; et al. Dietary patterns and prostate cancer risk in Japanese: The Japan Public Health Center-based Prospective Study (JPHC Study). Cancer Causes Control. 2018, 29, 589–600. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Ferro, M.; Terracciano, D.; Tataru, O.S.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Emerging Hallmarks of Metabolic Reprogramming in Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 910. [Google Scholar] [CrossRef]
- Li, D.; Stovall, D.B.; Wang, W.; Sui, G. Advances of Zinc Signaling Studies in Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.K.; Desouki, M.M.; Franklin, R.B.; Costello, L.C. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol. Cancer 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Fregeau-Proulx, L.; Lacouture, A.; Berthiaume, L.; Weidmann, C.; Harvey, M.; Gonthier, K.; Pelletier, J.F.; Neveu, B.; Jobin, C.; Bastien, D.; et al. Multiple metabolic pathways fuel the truncated tricarboxylic acid cycle of the prostate to sustain constant citrate production and secretion. Mol. Metab. 2022, 62, 101516. [Google Scholar] [CrossRef]
- Liang, J.Y.; Liu, Y.Y.; Zou, J.; Franklin, R.B.; Costello, L.C.; Feng, P. Inhibitory effect of zinc on human prostatic carcinoma cell growth. Prostate 1999, 40, 200–207. [Google Scholar] [CrossRef]
- Golovine, K.; Uzzo, R.G.; Makhov, P.; Crispen, P.L.; Kunkle, D.; Kolenko, V.M. Depletion of intracellular zinc increases expression of tumorigenic cytokines VEGF, IL-6 and IL-8 in prostate cancer cells via NF-kappaB-dependent pathway. Prostate 2008, 68, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Han, C.T.; Schoene, N.W.; Lei, K.Y. Influence of zinc deficiency on Akt-Mdm2-p53 and Akt-p21 signaling axes in normal and malignant human prostate cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1188–C1199. [Google Scholar] [CrossRef]
- Braczkowski, R.S.; Kwiatkowski, R.; Danikiewicz, A.; Gorczynska-Kosiorz, S.; Trautsolt, W.; Braczkowska, B.; Grzeszczak, W. Vitamin D receptor gene polymorphisms and prostate cancer. J. Biol. Regul. Homeost. Agents 2018, 32, 1245–1248. [Google Scholar]
- Campbell, M.J.; Trump, D.L. Vitamin D Receptor Signaling and Cancer. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1009–1038. [Google Scholar] [CrossRef]
- Bhoora, S.; Punchoo, R. Policing Cancer: Vitamin D Arrests the Cell Cycle. Int. J. Mol. Sci. 2020, 21, 9296. [Google Scholar] [CrossRef]
- Negri, M.; Gentile, A.; de Angelis, C.; Monto, T.; Patalano, R.; Colao, A.; Pivonello, R.; Pivonello, C. Vitamin D-Induced Molecular Mechanisms to Potentiate Cancer Therapy and to Reverse Drug-Resistance in Cancer Cells. Nutrients 2020, 12, 1798. [Google Scholar] [CrossRef]
- Carlberg, C.; Munoz, A. An update on vitamin D signaling and cancer. Semin. Cancer Biol. 2022, 79, 217–230. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Y.; Liu, Z.; Pei, Y.; Xu, P.; Chong, W.; Hai, Y.; He, L.; He, Y.; Yu, J.; et al. Association between Vitamin D Supplementation and Cancer Mortality: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 3717. [Google Scholar] [CrossRef]
- Xie, D.D.; Chen, Y.H.; Xu, S.; Zhang, C.; Wang, D.M.; Wang, H.; Chen, L.; Zhang, Z.H.; Xia, M.Z.; Xu, D.X.; et al. Low vitamin D status is associated with inflammation in patients with prostate cancer. Oncotarget 2017, 8, 22076–22085. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.; Martin-Gorgojo, V.; Martin-Moreno, J.M. Vitamin D in Cancer Prevention: Gaps in Current Knowledge and Room for Hope. Nutrients 2022, 14, 4512. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Figg, W.D. Revisiting 5alpha-reductase inhibitors and the risk of prostate cancer. Nat. Rev. Urol. 2018, 15, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sheng, J.; Hu, S.; Cui, Y.; Xiao, J.; Yu, W.; Peng, J.; Han, W.; He, Q.; Fan, Y.; et al. Estrogen and G protein-coupled estrogen receptor accelerate the progression of benign prostatic hyperplasia by inducing prostatic fibrosis. Cell Death Dis. 2022, 13, 533. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-de-Arellano, A.; Pereira-Suarez, A.L.; Rico-Fuentes, C.; Lopez-Pulido, E.I.; Villegas-Pineda, J.C.; Sierra-Diaz, E. Distribution and Effects of Estrogen Receptors in Prostate Cancer: Associated Molecular Mechanisms. Front. Endocrinol. 2021, 12, 811578. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, B.; Ou-Yang, L. Role of estrogen receptors in health and disease. Front. Endocrinol. 2022, 13, 839005. [Google Scholar] [CrossRef]
- Li, J.; Liu, Q.; Jiang, C. Signal Crosstalk and the Role of Estrogen Receptor beta (ERbeta) in Prostate Cancer. Med. Sci. Monit. 2022, 28, e935599. [Google Scholar] [CrossRef]
- Tong, D. Selective estrogen receptor modulators contribute to prostate cancer treatment by regulating the tumor immune microenvironment. J. Immunother. Cancer 2022, 10, e002944. [Google Scholar] [CrossRef]
- Lafront, C.; Germain, L.; Weidmann, C.; Audet-Walsh, É. A Systematic Study of the Impact of Estrogens and Selective Estrogen Receptor Modulators on Prostate Cancer Cell Proliferation. Sci. Rep. 2020, 10, 4024. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Tai, Q.; Gu, X.; Schmitz, J.; Poullard, A.; Fajardo, R.J.; Mahalingam, D.; Chen, X.; Zhu, X.; Sun, L.Z. Estrogen and estrogen receptor alpha promotes malignancy and osteoblastic tumorigenesis in prostate cancer. Oncotarget 2015, 6, 44388–44402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; De Luca, A.; Avena, P.; De Amicis, F.; Casaburi, I.; Sirianni, R.; Pezzi, V. Estrogen Receptors-Mediated Apoptosis in Hormone-Dependent Cancers. Int. J. Mol. Sci. 2022, 23, 1242. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, P.; Christopoulos, P.F.; Koutsilieris, M. The role of estrogen receptor beta in prostate cancer. Mol. Med. 2014, 20, 427–434. [Google Scholar] [CrossRef]
- Ricke, W.A.; McPherson, S.J.; Bianco, J.J.; Cunha, G.R.; Wang, Y.; Risbridger, G.P. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling. FASEB J. 2008, 22, 1512–1520. [Google Scholar] [CrossRef]
- Hiroto, A.; Kim, W.K.; Pineda, A.; He, Y.; Lee, D.-H.; Le, V.; Olson, A.W.; Aldahl, J.; Nenninger, C.H.; Buckley, A.J.; et al. Stromal androgen signaling acts as tumor niches to drive prostatic basal epithelial progenitor-initiated oncogenesis. Nat. Commun. 2022, 13, 6552. [Google Scholar] [CrossRef]
- Stone, W.L.; Leavitt, L.; Varacallo, M. Physiology, Growth Factor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Isali, I.; Al-Sadawi, M.A.A.; Qureshi, A.; Khalifa, A.O.; Agrawal, M.K.; Shukla, S. Growth factors involve in cellular proliferation, differentiation and migration during prostate cancer metastasis. Int. J. Cell Biol. Physiol. 2019, 2, 1–13. [Google Scholar]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-beta) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 791272. [Google Scholar] [CrossRef]
- Mirzaei, S.; Paskeh, M.D.A.; Saghari, Y.; Zarrabi, A.; Hamblin, M.R.; Entezari, M.; Hashemi, M.; Aref, A.R.; Hushmandi, K.; Kumar, A.P.; et al. Transforming growth factor-beta (TGF-beta) in prostate cancer: A dual function mediator? Int. J. Biol. Macromol. 2022, 206, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, G.; Liu, Z.; Liu, S.; Cai, Z.; You, P.; Ke, Y.; Lai, L.; Huang, Y.; Gao, H.; et al. Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res. 2018, 78, 4459–4470. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; Hartig, S.M.; Putluri, V.; Foley, C.; Hamilton, M.P.; Smith, E.A.; Saha, P.K.; Panigrahi, A.; Walker, C.; Zong, L.; et al. Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 2019, 1, 70–85. [Google Scholar] [CrossRef]
- Tsouko, E.; Khan, A.S.; White, M.A.; Han, J.J.; Shi, Y.; Merchant, F.A.; Sharpe, M.A.; Xin, L.; Frigo, D.E. Regulation of the pentose phosphate pathway by an androgen receptor-mTOR-mediated mechanism and its role in prostate cancer cell growth. Oncogenesis 2014, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Gorodetska, I.; Klusa, D.; Shi, Q.; Alves, T.C.; Pantel, K.; Dubrovska, A. Metabolic regulation of prostate cancer heterogeneity and plasticity. Semin. Cancer Biol. 2022, 82, 94–119. [Google Scholar] [CrossRef]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1alpha-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Ros, S.; Santos, C.R.; Moco, S.; Baenke, F.; Kelly, G.; Howell, M.; Zamboni, N.; Schulze, A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012, 2, 328–343. [Google Scholar] [CrossRef]
- Pearson, H.B.; McCarthy, A.; Collins, C.M.; Ashworth, A.; Clarke, A.R. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008, 68, 2223–2232. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar] [PubMed]
- Larsen, L.J.; Moller, L.B. Crosstalk of Hedgehog and mTORC1 Pathways. Cells 2020, 9, 2316. [Google Scholar] [CrossRef]
- Chen, K.S.; Fustino, N.J.; Shukla, A.A.; Stroup, E.K.; Budhipramono, A.; Ateek, C.; Stuart, S.H.; Yamaguchi, K.; Kapur, P.; Frazier, A.L.; et al. EGF Receptor and mTORC1 Are Novel Therapeutic Targets in Nonseminomatous Germ Cell Tumors. Mol. Cancer Ther. 2018, 17, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClellan, B.; Gries, P.; Harlow, B.; Tiziani, S.; Jolly, C.; deGraffenried, L. An IGF-1R-mTORC1-SRPK2 signaling Axis contributes to FASN regulation in breast cancer. BMC Cancer 2022, 22, 976. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.N. Editorial: Metabolism and Cell Adhesion in Cancer. Front. Cell Dev. Biol. 2022, 10, 871471. [Google Scholar] [CrossRef]
- Wan, L.; Wang, Y.; Li, J.; Wang, Y.; Zhang, H. Inhibition of the AKT/mTOR pathway negatively regulates PTEN expression via miRNAs. Acta Biochim. Biophys. Sin. 2022, 54, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate 2022, 82 (Suppl. 1), S60–S72. [Google Scholar] [CrossRef]
- Serebriiskii, I.G.; Pavlov, V.; Tricarico, R.; Andrianov, G.; Nicolas, E.; Parker, M.I.; Newberg, J.; Frampton, G.; Meyer, J.E.; Golemis, E.A. Comprehensive characterization of PTEN mutational profile in a series of 34,129 colorectal cancers. Nat. Commun. 2022, 13, 1618. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef]
- Roudsari, N.M.; Lashgari, N.A.; Momtaz, S.; Abaft, S.; Jamali, F.; Safaiepour, P.; Narimisa, K.; Jackson, G.; Bishayee, A.; Rezaei, N.; et al. Inhibitors of the PI3K/Akt/mTOR Pathway in Prostate Cancer Chemoprevention and Intervention. Pharmaceutics 2021, 13, 1195. [Google Scholar] [CrossRef]
- Cham, J.; Venkateswaran, A.R.; Bhangoo, M. Targeting the PI3K-AKT-mTOR Pathway in Castration Resistant Prostate Cancer: A Review Article. Clin. Genitourin. Cancer 2021, 19, 563.e1–563.e7. [Google Scholar] [CrossRef]
- Mao, N.; Zhang, Z.; Lee, Y.S.; Choi, D.; Rivera, A.A.; Li, D.; Lee, C.; Haywood, S.; Chen, X.; Chang, Q.; et al. Defining the therapeutic selective dependencies for distinct subtypes of PI3K pathway-altered prostate cancers. Nat. Commun. 2021, 12, 5053. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Akamatsu, S.; Tsukahara, S.; Nagakawa, S.; Matsumoto, T.; Eto, M. Androgen receptor mutations for precision medicine in prostate cancer. Endocr. Relat. Cancer 2022, 29, R143–R155. [Google Scholar] [CrossRef] [PubMed]
- Morova, T.; McNeill, D.R.; Lallous, N.; Gönen, M.; Dalal, K.; Wilson, D.M.; Gürsoy, A.; Keskin, Ö.; Lack, N.A. Androgen receptor-binding sites are highly mutated in prostate cancer. Nat. Commun. 2020, 11, 832. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Liu, R.Z.; Choi, W.S.; Jain, S.; Dinakaran, D.; Xu, X.; Han, W.H.; Yang, X.H.; Glubrecht, D.D.; Moore, R.B.; Lemieux, H.; et al. The FABP12/PPARgamma pathway promotes metastatic transformation by inducing epithelial-to-mesenchymal transition and lipid-derived energy production in prostate cancer cells. Mol. Oncol. 2020, 14, 3100–3120. [Google Scholar] [CrossRef]
- Amiri, M.; Yousefnia, S.; Seyed Forootan, F.; Peymani, M.; Ghaedi, K.; Nasr Esfahani, M.H. Diverse roles of fatty acid binding proteins (FABPs) in development and pathogenesis of cancers. Gene 2018, 676, 171–183. [Google Scholar] [CrossRef]
- Siltari, A.; Syvala, H.; Lou, Y.R.; Gao, Y.; Murtola, T.J. Role of Lipids and Lipid Metabolism in Prostate Cancer Progression and the Tumor’s Immune Environment. Cancers 2022, 14, 4293. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding protein family as global regulators of lipid synthetic genes in energy metabolism. Vitam. Horm. 2002, 65, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2012, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar] [PubMed]
- Hoque, A.; Chen, H.; Xu, X.C. Statin induces apoptosis and cell growth arrest in prostate cancer cells. Cancer Epidemiol. Biomark. Prev. 2008, 17, 88–94. [Google Scholar] [CrossRef]
- Murtola, T.J.; Syvala, H.; Pennanen, P.; Blauer, M.; Solakivi, T.; Ylikomi, T.; Tammela, T.L. Comparative effects of high and low-dose simvastatin on prostate epithelial cells: The role of LDL. Eur. J. Pharmacol. 2011, 673, 96–100. [Google Scholar] [CrossRef]
- Murtola, T.J.; Pennanen, P.; Syvala, H.; Blauer, M.; Ylikomi, T.; Tammela, T.L. Effects of simvastatin, acetylsalicylic acid, and rosiglitazone on proliferation of normal and cancerous prostate epithelial cells at therapeutic concentrations. Prostate 2009, 69, 1017–1023. [Google Scholar] [CrossRef]
- Kochuparambil, S.T.; Al-Husein, B.; Goc, A.; Soliman, S.; Somanath, P.R. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J. Pharmacol. Exp. Ther. 2011, 336, 496–505. [Google Scholar] [CrossRef]
- White, M.A.; Lin, C.; Rajapakshe, K.; Dong, J.; Shi, Y.; Tsouko, E.; Mukhopadhyay, R.; Jasso, D.; Dawood, W.; Coarfa, C.; et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol. Cancer Res. 2017, 15, 1017–1028. [Google Scholar] [CrossRef]
- Scalise, M.; Console, L.; Rovella, F.; Galluccio, M.; Pochini, L.; Indiveri, C. Membrane Transporters for Amino Acids as Players of Cancer Metabolic Rewiring. Cells 2020, 9, 2028. [Google Scholar] [CrossRef]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Jang, S.K.; Kim, Y.J.; Jin, H.O.; Bae, S.; Hong, J.; Park, I.C.; Lee, J.H. Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8761. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kirsch, B.J.; Asaka, R.; Nabi, K.; Quinones, A.; Tan, J.; Antonio, M.J.; Camelo, F.; Li, T.; Nguyen, S.; et al. Uncovering the Role of N-Acetyl-Aspartyl-Glutamate as a Glutamate Reservoir in Cancer. Cell Rep. 2019, 27, 491–501. [Google Scholar] [CrossRef]
- Asaka, R.; Le, A. Dual role of N-acetyl-aspartyl-glutamate metabolism in cancer monitor and therapy. Mol. Cell Oncol. 2019, 6, e1627273. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. 2017, 35, 461.e15–461.e27. [Google Scholar] [CrossRef]
- Penney, K.L.; Tyekucheva, S.; Rosenthal, J.; El Fandy, H.; Carelli, R.; Borgstein, S.; Zadra, G.; Fanelli, G.N.; Stefanizzi, L.; Giunchi, F.; et al. Metabolomics of Prostate Cancer Gleason Score in Tumor Tissue and Serum. Mol. Cancer Res. 2021, 19, 475–484. [Google Scholar] [CrossRef]
- Song, Y.H.; Shiota, M.; Kuroiwa, K.; Naito, S.; Oda, Y. The important role of glycine N-methyltransferase in the carcinogenesis and progression of prostate cancer. Mod. Pathol. 2011, 24, 1272–1280. [Google Scholar] [CrossRef]
- Lucarelli, G.; Fanelli, M.; Larocca, A.M.; Germinario, C.A.; Rutigliano, M.; Vavallo, A.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/ml. Prostate 2012, 72, 1611–1621. [Google Scholar] [CrossRef]
- Lucarelli, G.; Ditonno, P.; Bettocchi, C.; Spilotros, M.; Rutigliano, M.; Vavallo, A.; Galleggiante, V.; Fanelli, M.; Larocca, A.M.; Germinario, C.A.; et al. Serum sarcosine is a risk factor for progression and survival in patients with metastatic castration-resistant prostate cancer. Future Oncol. 2013, 9, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, D.; Pandolfo, S.D.; Rogers, D.; Cerrato, C.; di Meo, N.A.; Autorino, R.; Mirone, V.; Ferro, M.; Porta, C.; Stella, A.; et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 23, 3826. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; White, E. Autophagy, Metabolism, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 73–78. [Google Scholar] [CrossRef]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Padman, B.S.; Bach, M.; Lucarelli, G.; Prescott, M.; Ramm, G. The protonophore CCCP interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 2013, 9, 1862–1875. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Stewart, L.M.; Gerner, L.; Rettel, M.; Stein, F.; Burrows, J.F.; Mills, I.G.; Evergren, E. CaMKK2 facilitates Golgi-associated vesicle trafficking to sustain cancer cell proliferation. Cell Death Dis. 2021, 12, 1040. [Google Scholar] [CrossRef]
- Lin, C.; Blessing, A.M.; Pulliam, T.L.; Shi, Y.; Wilkenfeld, S.R.; Han, J.J.; Murray, M.M.; Pham, A.H.; Duong, K.; Brun, S.N.; et al. Inhibition of CAMKK2 impairs autophagy and castration-resistant prostate cancer via suppression of AMPK-ULK1 signaling. Oncogene 2021, 40, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 expression is an early event in prostate carcinogenesis and predicts therapeutic response to targeted therapy against MAPK/p38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Nazim, U.M.; Yin, H.; Park, S.Y. Neferine treatment enhances the TRAIL-induced apoptosis of human prostate cancer cells via autophagic flux and the JNK pathway. Int. J. Oncol. 2020, 56, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Qi, F.; Li, L.; Qin, Z.; Li, X.; Wang, X. Autophagy-related genes are potential diagnostic and prognostic biomarkers in prostate cancer. Transl. Androl. Urol. 2020, 9, 2616–2628. [Google Scholar] [CrossRef]
- Hu, D.; Jiang, L.; Luo, S.; Zhao, X.; Hu, H.; Zhao, G.; Tang, W. Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J. Transl. Med. 2020, 18, 160. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Mendrinos, S.; Koutsopoulos, A.V.; Koukourakis, M.I. Autophagy proteins in prostate cancer: Relation with anaerobic metabolism and Gleason score. Urol. Oncol. 2014, 32, 39.e11–39.e18. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Mishra, R.; Haldar, S.; Placencio, V.; Madhav, A.; Rohena-Rivera, K.; Agarwal, P.; Duong, F.; Angara, B.; Tripathi, M.; Liu, Z.; et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J. Clin. Investig. 2018, 128, 4472–4484. [Google Scholar] [CrossRef]
- Keto, C.J.; Aronson, W.J.; Terris, M.K.; Presti, J.C.; Kane, C.J.; Amling, C.L.; Freedland, S.J. Obesity is associated with castration-resistant disease and metastasis in men treated with androgen deprivation therapy after radical prostatectomy: Results from the SEARCH database. BJU Int. 2012, 110, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Toulet, A.; Attane, C.; Milhas, D.; Dauvillier, S.; Zaidi, F.; Clement, E.; Cinato, M.; Le Gonidec, S.; Guerard, A.; et al. Periprostatic Adipose Tissue Favors Prostate Cancer Cell Invasion in an Obesity-Dependent Manner: Role of Oxidative Stress. Mol. Cancer Res. 2019, 17, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, J.; Namdarian, B.; Pedersen, J.; Hovens, C.; Moon, D.; Peters, J.; Costello, A.J.; Ruljancich, P.; Corcoran, N.M. Extraprostatic extension into periprostatic fat is a more important determinant of prostate cancer recurrence than an invasive phenotype. J. Urol. 2013, 190, 2061–2066. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; Valsamakis, G.; Mastorakos, G.; Hanson, P.; Kyrou, I.; Randeva, H.S.; Weickert, M.O. Dietary Influences on the Microbiota-Gut-Brain Axis. Int. J. Mol. Sci. 2021, 22, 3502. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Cintoni, M.; Raoul, P.; Lopetuso, L.R.; Scaldaferri, F.; Pulcini, G.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. Food Components and Dietary Habits: Keys for a Healthy Gut Microbiota Composition. Nutrients 2019, 11, 2393. [Google Scholar] [CrossRef]
- Drasar, B.S.; Crowther, J.S.; Goddard, P.; Hawksworth, G.; Hill, M.J.; Peach, S.; Williams, R.E.; Renwick, A. The relation between diet and the gut microflora in man. Proc. Nutr. Soc. 1973, 32, 49–52. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Zhong, W.; Wu, K.; Long, Z.; Zhou, X.; Zhong, C.; Wang, S.; Lai, H.; Guo, Y.; Lv, D.; Lu, J.; et al. Gut dysbiosis promotes prostate cancer progression and docetaxel resistance via activating NF-kappaB-IL6-STAT3 axis. Microbiome 2022, 10, 94. [Google Scholar] [CrossRef]
- Javier-DesLoges, J.; McKay, R.R.; Swafford, A.D.; Sepich-Poore, G.D.; Knight, R.; Parsons, J.K. The microbiome and prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 159–164. [Google Scholar] [CrossRef]
- Katongole, P.; Sande, O.J.; Joloba, M.; Reynolds, S.J.; Niyonzima, N. The human microbiome and its link in prostate cancer risk and pathogenesis. Infect. Agent. Cancer 2020, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.J.; Shannon, B.A.; McNeal, J.E.; Shannon, T.; Garrett, K.L. Propionibacterium acnes associated with inflammation in radical prostatectomy specimens: A possible link to cancer evolution? J. Urol. 2005, 173, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Luciano, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The Microbiome of the Prostate Tumor Microenvironment. Eur. Urol. 2017, 72, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Ohnishi, K.; Hori, S.; Nakano, A.; Nakano, R.; Yano, H.; Ohnishi, S.; Owari, T.; Morizawa, Y.; Itami, Y.; et al. Mycoplasma genitalium Infection and Chronic Inflammation in Human Prostate Cancer: Detection Using Prostatectomy and Needle Biopsy Specimens. Cells 2019, 8, 212. [Google Scholar] [CrossRef]
- Yu, H.; Meng, H.; Zhou, F.; Ni, X.; Shen, S.; Das, U.N. Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Arch. Med. Sci. 2015, 11, 385–394. [Google Scholar] [CrossRef]
- Alanee, S.; El-Zawahry, A.; Dynda, D.; Dabaja, A.; McVary, K.; Karr, M.; Braundmeier-Fleming, A. A prospective study to examine the association of the urinary and fecal microbiota with prostate cancer diagnosis after transrectal biopsy of the prostate using 16sRNA gene analysis. Prostate 2019, 79, 81–87. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Li, S.; Sun, F.; Wang, G.; Wei, D.; Yang, T.; Gu, S. Androgen deprivationinduced OPHN1 amplification promotes castrationresistant prostate cancer. Oncol. Rep. 2022, 47, 3. [Google Scholar] [CrossRef]
- Morote, J.; Aguilar, A.; Planas, J.; Trilla, E. Definition of Castrate Resistant Prostate Cancer: New Insights. Biomedicines 2022, 10, 689. [Google Scholar] [CrossRef]
- LeVee, A.; Lin, C.Y.; Posadas, E.; Figlin, R.; Bhowmick, N.A.; Di Vizio, D.; Ellis, L.; Rosser, C.J.; Freeman, M.R.; Theodorescu, D.; et al. Clinical Utility of Olaparib in the Treatment of Metastatic Castration-Resistant Prostate Cancer: A Review of Current Evidence and Patient Selection. Onco Targets Ther. 2021, 14, 4819–4832. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, S.; Bjelogrlic, S.; Todorovic, Z.; Prostran, M. Chemosensitisation by poly(ADP-ribose) polymerase-1 (PARP-1) inhitor 5-aminoisoquinoline (5-AIQ) on various melanoma cell lines. J. Clin. Oncol. 2006, 24, 12019. [Google Scholar] [CrossRef]
- Todorovic, Z.; Durasevic, S.; Stojkovic, M.; Grigorov, I.; Pavlovic, S.; Jasnic, N.; Tosti, T.; Macut, J.B.; Thiemermann, C.; Dordevic, J. Lipidomics Provides New Insight into Pathogenesis and Therapeutic Targets of the Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 2798. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens Health 2019, 37, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Liu, Y.F.; Fu, S.Q.; Yan, Y.C.; Gong, B.B.; Xie, W.J.; Yang, X.R.; Sun, T.; Ma, M. Progress in Clinical Research on Gonadotropin-Releasing Hormone Receptor Antagonists for the Treatment of Prostate Cancer. Drug Des. Devel. Ther. 2021, 15, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Olsson, H.; Petri, N.; Erichsen, L.; Malmberg, A.; Grundemar, L. Effect of Degarelix, a Gonadotropin-Releasing Hormone Receptor Antagonist for the Treatment of Prostate Cancer, on Cardiac Repolarisation in a Randomised, Placebo and Active Comparator Controlled Thorough QT/QTc Trial in Healthy Men. Clin. Drug Investig. 2017, 37, 873–879. [Google Scholar] [CrossRef]
- Mostaghel, E.A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag. Res. 2014, 6, 39–51. [Google Scholar] [CrossRef]
- Yin, L.; Hu, Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2014, 11, 32–42. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef]
- Rajakyla, E.K.; Vartiainen, M.K. Rho, nuclear actin, and actin-binding proteins in the regulation of transcription and gene expression. Small GTPases 2014, 5, e27539. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef]
- Salvi, S.; Casadio, V.; Conteduca, V.; Burgio, S.L.; Menna, C.; Bianchi, E.; Rossi, L.; Carretta, E.; Masini, C.; Amadori, D.; et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br. J. Cancer 2015, 112, 1717–1724. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Zhang, A.; Hernandez, S.; Marck, B.T.; Zhang, X.; Tamae, D.; Biehl, H.E.; Tretiakova, M.; Bartlett, J.; Burns, J.; et al. Contribution of Adrenal Glands to Intratumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 426–439. [Google Scholar] [CrossRef]
- Maitland, N.J. Resistance to Antiandrogens in Prostate Cancer: Is It Inevitable, Intrinsic or Induced? Cancers 2021, 13, 327. [Google Scholar] [CrossRef]
- Shore, N.D.; Morgans, A.K.; Ryan, C.J. Resetting the Bar of Castration Resistance—Understanding Androgen Dynamics in Therapy Resistance and Treatment Choice in Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; McDonnell, D.P. Androgen receptor-cofactor interactions as targets for new drug discovery. Trends Pharmacol. Sci. 2005, 26, 225–228. [Google Scholar] [CrossRef]
- Li, P.; Yu, X.; Ge, K.; Melamed, J.; Roeder, R.G.; Wang, Z. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am. J. Pathol. 2002, 161, 1467–1474. [Google Scholar] [CrossRef]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Chmelar, R.; Buchanan, G.; Need, E.F.; Tilley, W.; Greenberg, N.M. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int. J. Cancer 2007, 120, 719–733. [Google Scholar] [CrossRef]
- Tien, J.C.; Liu, Z.; Liao, L.; Wang, F.; Xu, Y.; Wu, Y.L.; Zhou, N.; Ittmann, M.; Xu, J. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res. 2013, 73, 3997–4008. [Google Scholar] [CrossRef]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic Targeting of the CBP/p300 Bromodomain Blocks the Growth of Castration-Resistant Prostate Cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef]
- Sobhani, N.; Neeli, P.K.; D’Angelo, A.; Pittacolo, M.; Sirico, M.; Galli, I.C.; Roviello, G.; Nesi, G. AR-V7 in Metastatic Prostate Cancer: A Strategy beyond Redemption. Int. J. Mol. Sci. 2021, 22, 5515. [Google Scholar] [CrossRef]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Huggins, C. How Charles Huggins made his Nobel prize winning discovery--in his own words: An historic audio recording. Interviewed by Willard Goodwin and Elmer Bell. Prostate 2012, 72, 1718. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Parker, C.; Castro, E.; Fizazi, K.; Heidenreich, A.; Ost, P.; Procopio, G.; Tombal, B.; Gillessen, S.; ESMO Guidelines Committee. Prostate cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1119–1134. [Google Scholar] [CrossRef]
- Gillessen, S.; Omlin, A.; Attard, G.; de Bono, J.S.; Efstathiou, E.; Fizazi, K.; Halabi, S.; Nelson, P.S.; Sartor, O.; Smith, M.R.; et al. Management of patients with advanced prostate cancer: Recommendations of the St Gallen Advanced Prostate Cancer Consensus Conference (APCCC) 2015. Ann. Oncol. 2015, 26, 1589–1604. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Agarwal, N.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez Soto, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide for Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2019, 381, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Chowdhury, S.; Bjartell, A.; Chung, B.H.; Pereira de Santana Gomes, A.J.; Given, R.; Juarez, A.; Merseburger, A.S.; Ozguroglu, M.; Uemura, H.; et al. Apalutamide in Patients With Metastatic Castration-Sensitive Prostate Cancer: Final Survival Analysis of the Randomized, Double-Blind, Phase III TITAN Study. J. Clin. Oncol. 2021, 39, 2294–2303. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Azad, A.A.; Iguchi, T.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Alcaraz, A.; Alekseev, B.; Shore, N.D.; et al. Improved Survival With Enzalutamide in Patients With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2022, 40, 1616–1622. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Fizazi, K.; Foulon, S.; Carles, J.; Roubaud, G.; McDermott, R.; Flechon, A.; Tombal, B.; Supiot, S.; Berthold, D.; Ronchin, P.; et al. Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castration-sensitive prostate cancer (PEACE-1): A multicentre, open-label, randomised, phase 3 study with a 2 x 2 factorial design. Lancet 2022, 399, 1695–1707. [Google Scholar] [CrossRef]
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, A.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2022, 386, 1132–1142. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Shore, N.D.; Chowdhury, S.; Villers, A.; Klotz, L.; Siemens, D.R.; Phung, D.; van Os, S.; Hasabou, N.; Wang, F.; Bhattacharya, S.; et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): A randomised, double-blind, phase 2 study. Lancet Oncol. 2016, 17, 153–163. [Google Scholar] [CrossRef]
- de Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- de Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wulfing, C.; Kramer, G.; Eymard, J.C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Saad, F.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Loredo, E.; Procopio, G.; Menezes, J.J.d.; Girotto, G.C.; Arslan, C.; Mehra, N.; et al. PROpel: Phase III trial of olaparib (ola) and abiraterone (abi) versus placebo (pbo) and abi as first-line (1L) therapy for patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 11. [Google Scholar] [CrossRef]
- Popovic, L.S.; Matovina-Brko, G.; Popovic, M. Checkpoint inhibitors in the treatment of urological malignancies. ESMO Open 2017, 2, e000165. [Google Scholar] [CrossRef]
- Popovic, M.; Matovina-Brko, G.; Jovic, M.; Popovic, L.S. Immunotherapy: A new standard in the treatment of metastatic clear cell renal cell carcinoma. World J. Clin. Oncol. 2022, 13, 28–38. [Google Scholar] [CrossRef]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E., 3rd; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: A randomized phase 3 trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; et al. NCCN Guidelines Insights: Prostate Cancer, Version 1.2021. J. Natl. Compr. Canc. Netw. 2021, 19, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.S.; Schweizer, M.T. Mismatch repair deficiency and clinical implications in prostate cancer. Prostate 2022, 82 (Suppl. 1), S37–S44. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Chen, Y.; Feng, Z.; Cullen, J.; Trock, B.J.; Suzman, D.; Antonarakis, E.S.; Paller, C.J.; Rosner, I.; Han, M.; et al. PSA Doubling Time and Absolute PSA Predict Metastasis-free Survival in Men With Biochemically Recurrent Prostate Cancer After Radical Prostatectomy. Clin. Genitourin. Cancer 2019, 17, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Fendler, W.P.; Weber, M.; Iravani, A.; Hofman, M.S.; Calais, J.; Czernin, J.; Ilhan, H.; Saad, F.; Small, E.J.; Smith, M.R.; et al. Prostate-Specific Membrane Antigen Ligand Positron Emission Tomography in Men with Nonmetastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 7448–7454. [Google Scholar] [CrossRef] [PubMed]
- Whelan, H.T. Neuromuscular syndromes associated with cancer. Compr. Ther. 1985, 11, 50–57. [Google Scholar]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Yang, J.C.; Kung, H.J.; Shi, X.B.; Tilki, D.; Lara, P.N., Jr.; DeVere White, R.W.; Gao, A.C.; Evans, C.P. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Zhang, W.; Zhang, D.; Li, Y.; Zhang, J.; Zhang, Y.; Diao, T.; Cui, L.; Li, W.; et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- Mortezavi, A.; Salemi, S.; Kranzbuhler, B.; Gross, O.; Sulser, T.; Simon, H.U.; Eberli, D. Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J. Urol. 2019, 37, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Eberli, D.; Kranzbuhler, B.; Mortezavi, A.; Sulser, T.; Salemi, S. Apalutamide in combination with autophagy inhibitors improves treatment effects in prostate cancer cells. Urol. Oncol. 2020, 38, 683.e19–683.e26. [Google Scholar] [CrossRef] [PubMed]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Farrow, J.M.; Yang, J.C.; Evans, C.P. Autophagy as a modulator and target in prostate cancer. Nat. Rev. Urol. 2014, 11, 508–516. [Google Scholar] [CrossRef]
- Veldhoen, R.A.; Banman, S.L.; Hemmerling, D.R.; Odsen, R.; Simmen, T.; Simmonds, A.J.; Underhill, D.A.; Goping, I.S. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene 2013, 32, 736–746. [Google Scholar] [CrossRef]
- Lin, J.Z.; Wang, W.W.; Hu, T.T.; Zhu, G.Y.; Li, L.N.; Zhang, C.Y.; Xu, Z.; Yu, H.B.; Wu, H.F.; Zhu, J.G. FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 2020, 469, 481–489. [Google Scholar] [CrossRef]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [(177)Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted alpha-Therapy of Metastatic Castration-Resistant Prostate Cancer with (225)Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef]
- Mukha, A.; Kahya, U.; Linge, A.; Chen, O.; Lock, S.; Lukiyanchuk, V.; Richter, S.; Alves, T.C.; Peitzsch, M.; Telychko, V.; et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics 2021, 11, 7844–7868. [Google Scholar] [CrossRef]
- Chan, T.G.; O’Neill, E.; Habjan, C.; Cornelissen, B. Combination Strategies to Improve Targeted Radionuclide Therapy. J. Nucl. Med. 2020, 61, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, P.A. Intermittent androgen deprivation therapy in patients with prostate cancer: Connecting the dots. Asian J Urol 2017, 4, 208–222. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Daniil, C.; Kalleda, R.; Pham, T.; Taplin, M.E.; Collaborators, P. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer with Rising Prostate-Specific Antigen During Enzalutamide Treatment. J. Clin. Oncol. 2018, 36, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Erkisa, M.; Aydinlik, S.; Cevatemre, B.; Aztopal, N.; Akar, R.O.; Celikler, S.; Yilmaz, V.T.; Ari, F.; Ulukaya, E. A promising therapeutic combination for metastatic prostate cancer: Chloroquine as autophagy inhibitor and palladium(II) barbiturate complex. Biochimie 2020, 175, 159–172. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting cancer cell metabolism: The combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Chen, C.; Wang, H.; Geng, X.; Zhang, D.; Zhu, Z.; Zhang, G.; Hou, J. Metformin exerts anti-AR-negative prostate cancer activity via AMPK/autophagy signaling pathway. Cancer Cell Int. 2021, 21, 404. [Google Scholar] [CrossRef]
- Luty, M.; Piwowarczyk, K.; Labedz-Maslowska, A.; Wrobel, T.; Szczygiel, M.; Catapano, J.; Drabik, G.; Ryszawy, D.; Kedracka-Krok, S.; Madeja, Z.; et al. Fenofibrate Augments the Sensitivity of Drug-Resistant Prostate Cancer Cells to Docetaxel. Cancers 2019, 11, 77. [Google Scholar] [CrossRef]
- Tan, Q.; Joshua, A.M.; Wang, M.; Bristow, R.G.; Wouters, B.G.; Allen, C.J.; Tannock, I.F. Up-regulation of autophagy is a mechanism of resistance to chemotherapy and can be inhibited by pantoprazole to increase drug sensitivity. Cancer Chemother. Pharmacol. 2017, 79, 959–969. [Google Scholar] [CrossRef]
- Hansen, A.R.; Tannock, I.F.; Templeton, A.; Chen, E.; Evans, A.; Knox, J.; Prawira, A.; Sridhar, S.S.; Tan, S.; Vera-Badillo, F.; et al. Pantoprazole Affecting Docetaxel Resistance Pathways via Autophagy (PANDORA): Phase II Trial of High Dose Pantoprazole (Autophagy Inhibitor) with Docetaxel in Metastatic Castration-Resistant Prostate Cancer (mCRPC). Oncologist 2019, 24, 1188–1194. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| The Name of the Antagonist | Affinity for Androgens Receptors (IC50) * |
|---|---|
| Hydroxyflutamide | 700 nM |
| Bicalutamide | 160 nM |
| Apalutamide | 93 nM |
| Enzalutamide | 86 nM |
| Darolutamide ** | 11 nM |
| A Drugs Class | A Molecular Target | Drug Example | Mechanism of Resistance |
|---|---|---|---|
| Androgen synthesis, serum testosterone levels | Gonadotropin synthesis in the adenohypophysis. GnRH agonists | Goserelin | Intratumoral androgen synthesis, AR gene amplification |
| Androgen synthesis | Intratumorally CYP17 | Abiraterone | Use of glucocorticoid receptors and glucocorticoids |
| Activation of testosterone to dihydrotestosterone | Steroid 5α-reductase | Dutasteride, finasteride | Changing the 5α-reductase isoform or using testosterone or adrenal androgens |
| AR inhibitors | Dihydrotestosterone binding to monomeric AR | Steroids: estrogens, cyproterone | AR inhibitors |
| AR dimerization | Androgen receptor | Stilbene, resveratrol | AR gene mutation or expression of ligand-independent splice variants |
| AR phosphorylation | Phosphorylation site AR kinase: | AR phosphorylation | |
| Translocation of AR in the nucleus | AR | Enzalutamide | Expression of AR splice variants that translocate to the nucleus in the absence of androgens |
| Binding of nuclear AR binding sites to DNA or to coactivators | AR and coactivators | DNA binding site: 4-(4-phenylthiazol-2-yl) | Changes in coactivator and corepressor balance and relative receptor affinity due to AR gene mutation |
| Effects of proteins that are influenced by AR (signaling pathways) | Androgen-responsive signaling molecules | morpholine | Activation of alternative salvage pathways that stimulate AR signaling pathways |
| Stability and degradation of AR | HSP90, serine proteases, calpain and caspases | Different | HS chaperone system becomes redundant, inhibition of AR proteolysis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pejčić, T.; Todorović, Z.; Đurašević, S.; Popović, L. Mechanisms of Prostate Cancer Cells Survival and Their Therapeutic Targeting. Int. J. Mol. Sci. 2023, 24, 2939. https://doi.org/10.3390/ijms24032939
Pejčić T, Todorović Z, Đurašević S, Popović L. Mechanisms of Prostate Cancer Cells Survival and Their Therapeutic Targeting. International Journal of Molecular Sciences. 2023; 24(3):2939. https://doi.org/10.3390/ijms24032939
Chicago/Turabian StylePejčić, Tomislav, Zoran Todorović, Siniša Đurašević, and Lazar Popović. 2023. "Mechanisms of Prostate Cancer Cells Survival and Their Therapeutic Targeting" International Journal of Molecular Sciences 24, no. 3: 2939. https://doi.org/10.3390/ijms24032939