
HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
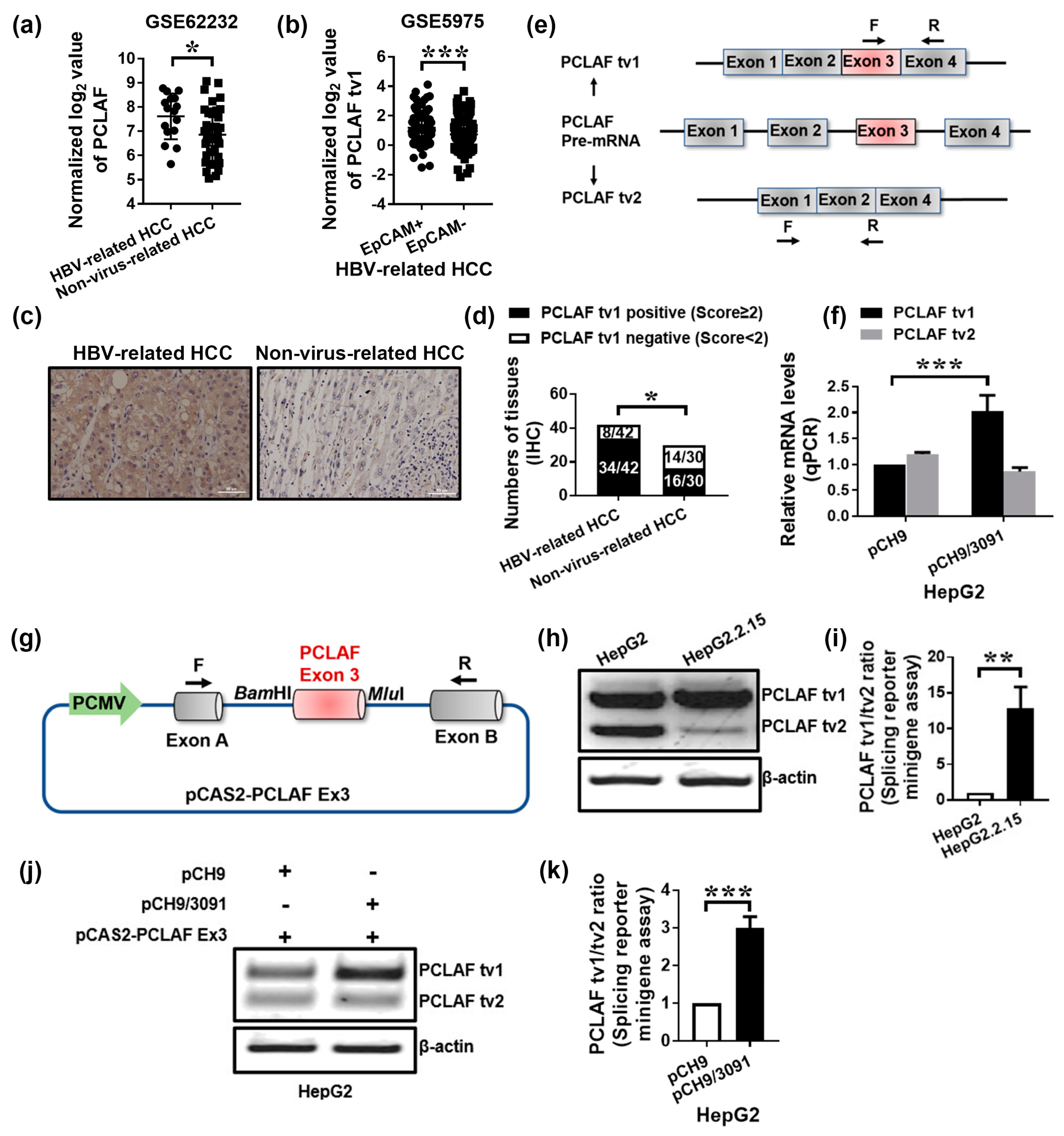
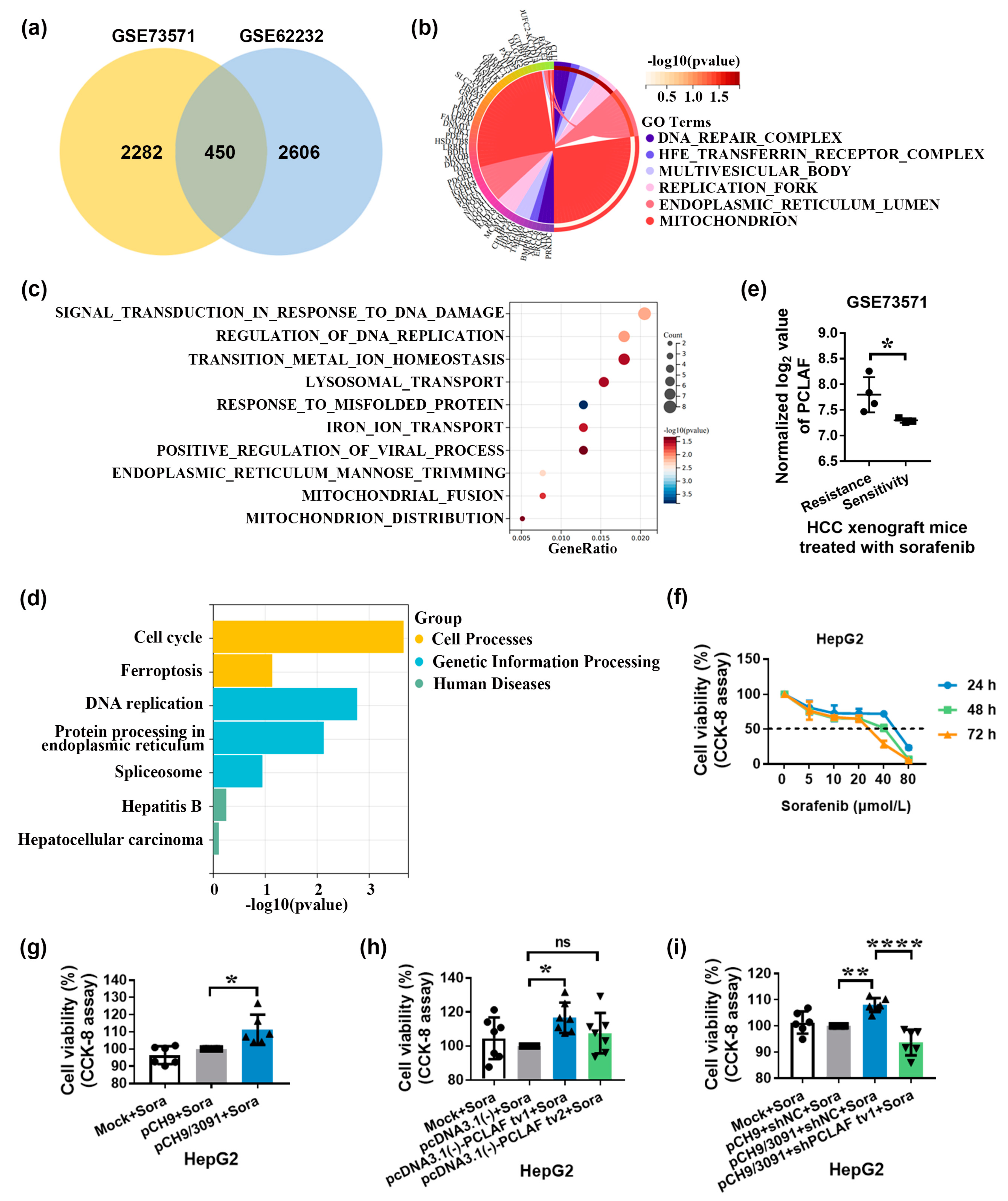
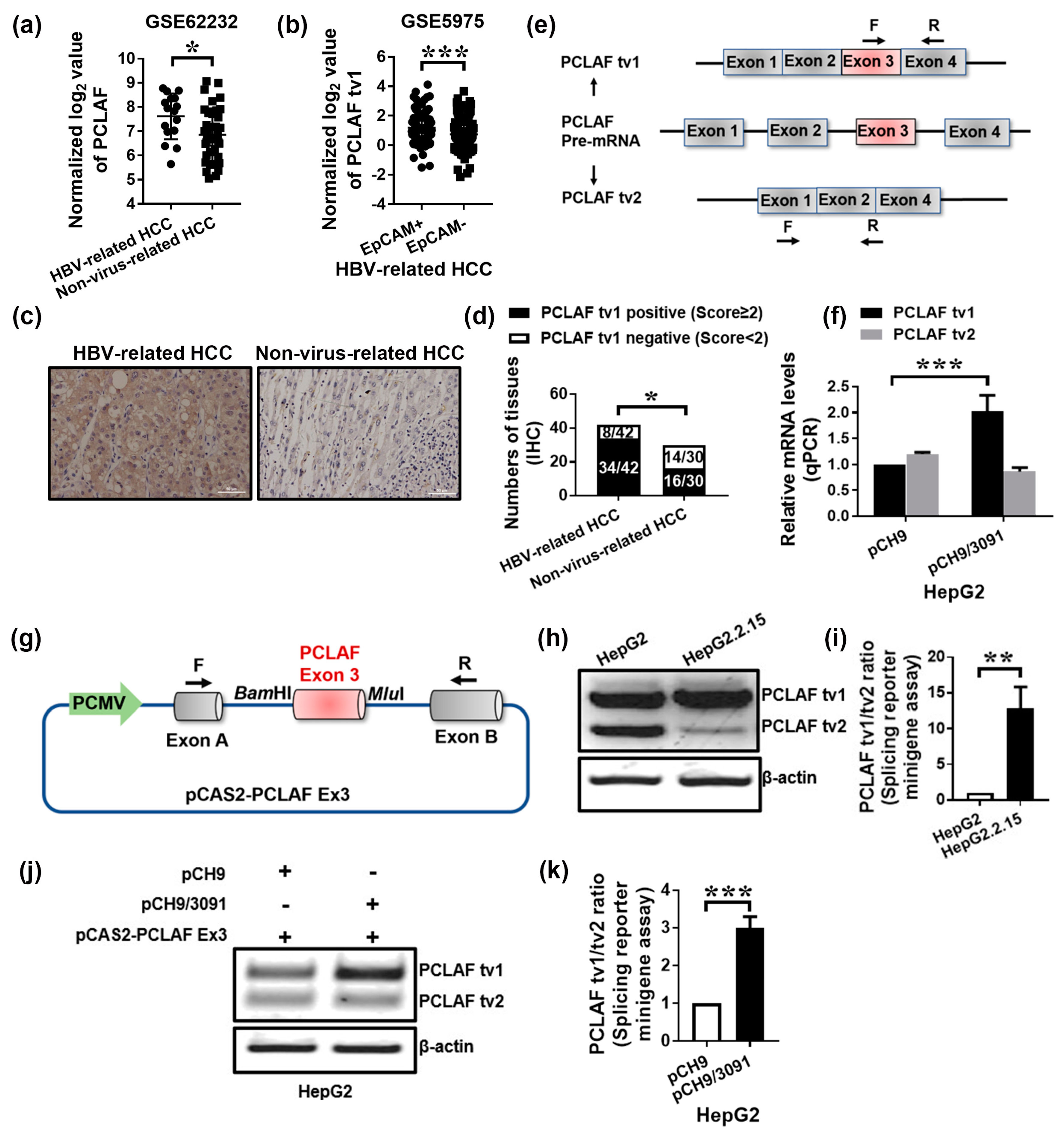
2.1. HBV Promotes Sorafenib Resistance in HCC by Increasing PCLAF tv1 Splicing
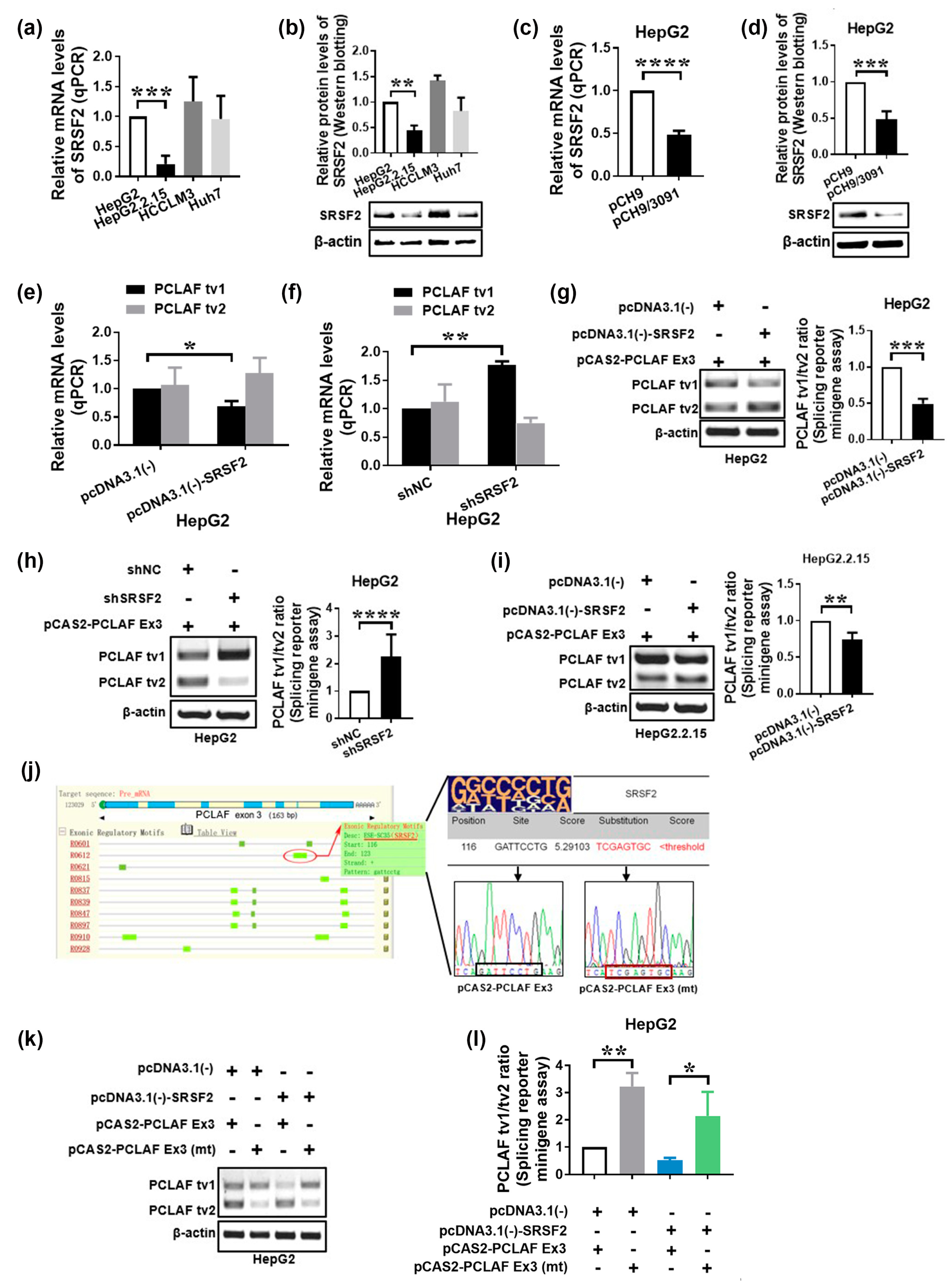
2.2. HBV Increases PCLAF tv1 Splicing by Inhibiting Trans-Acting Factor SRSF2
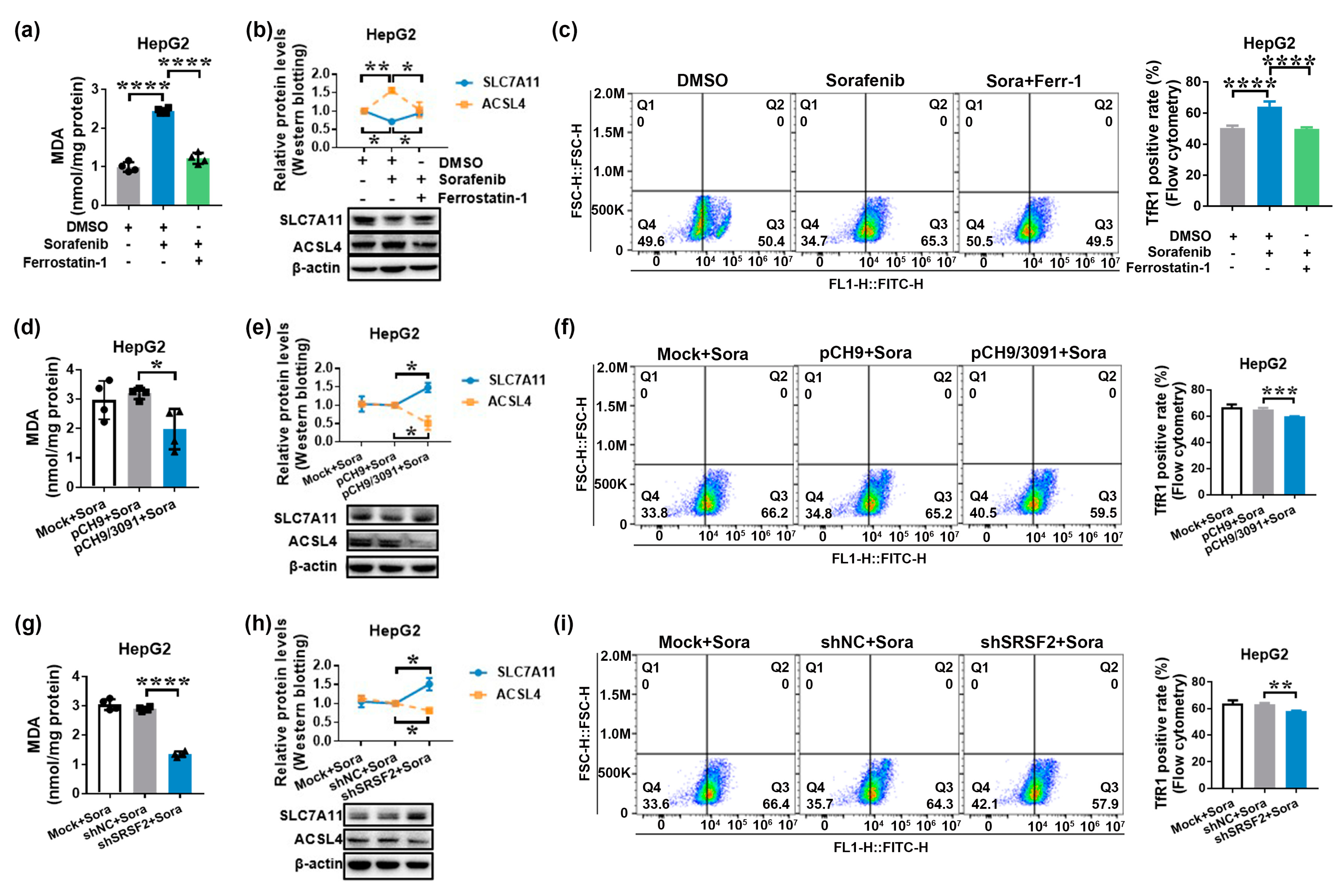
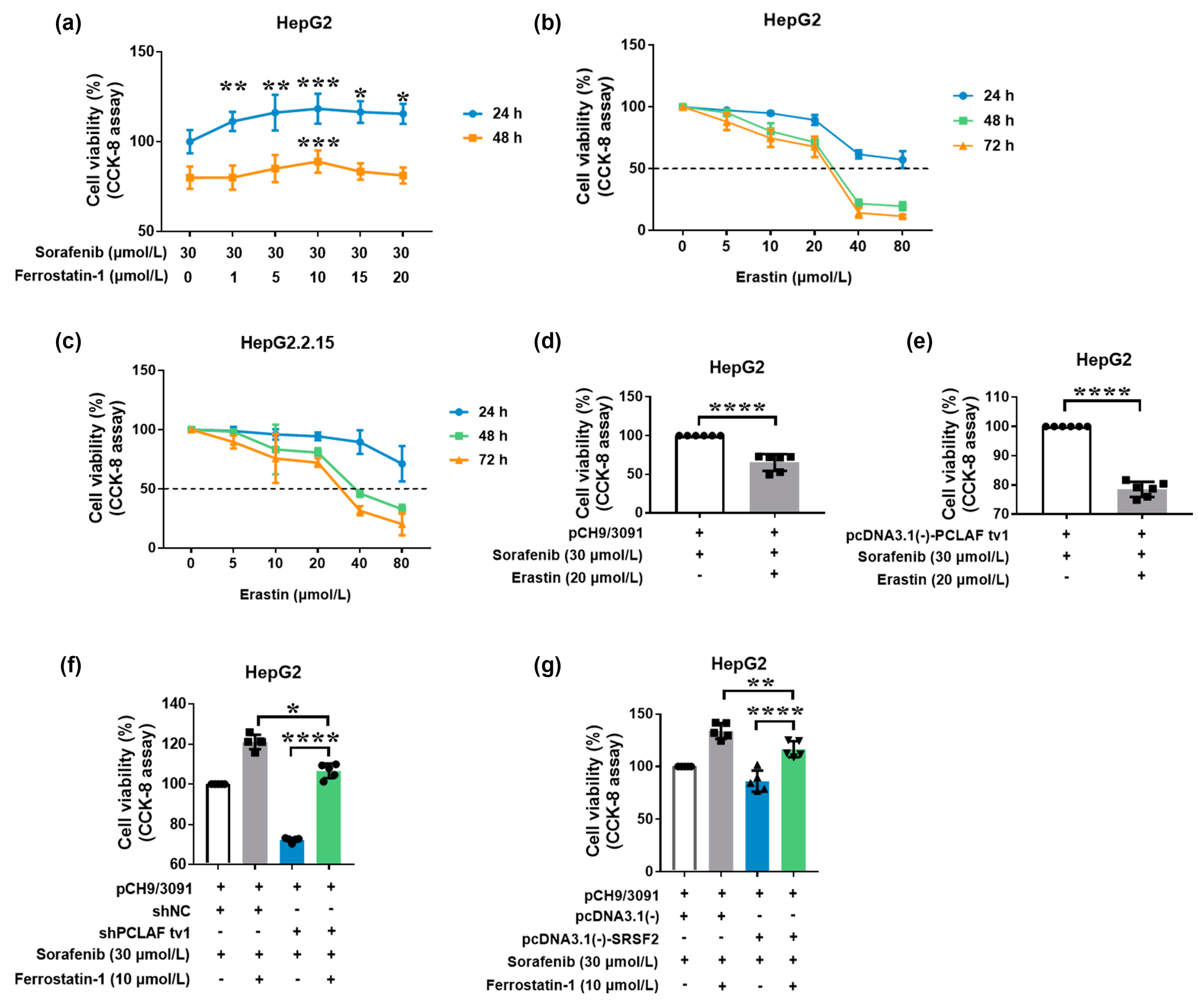
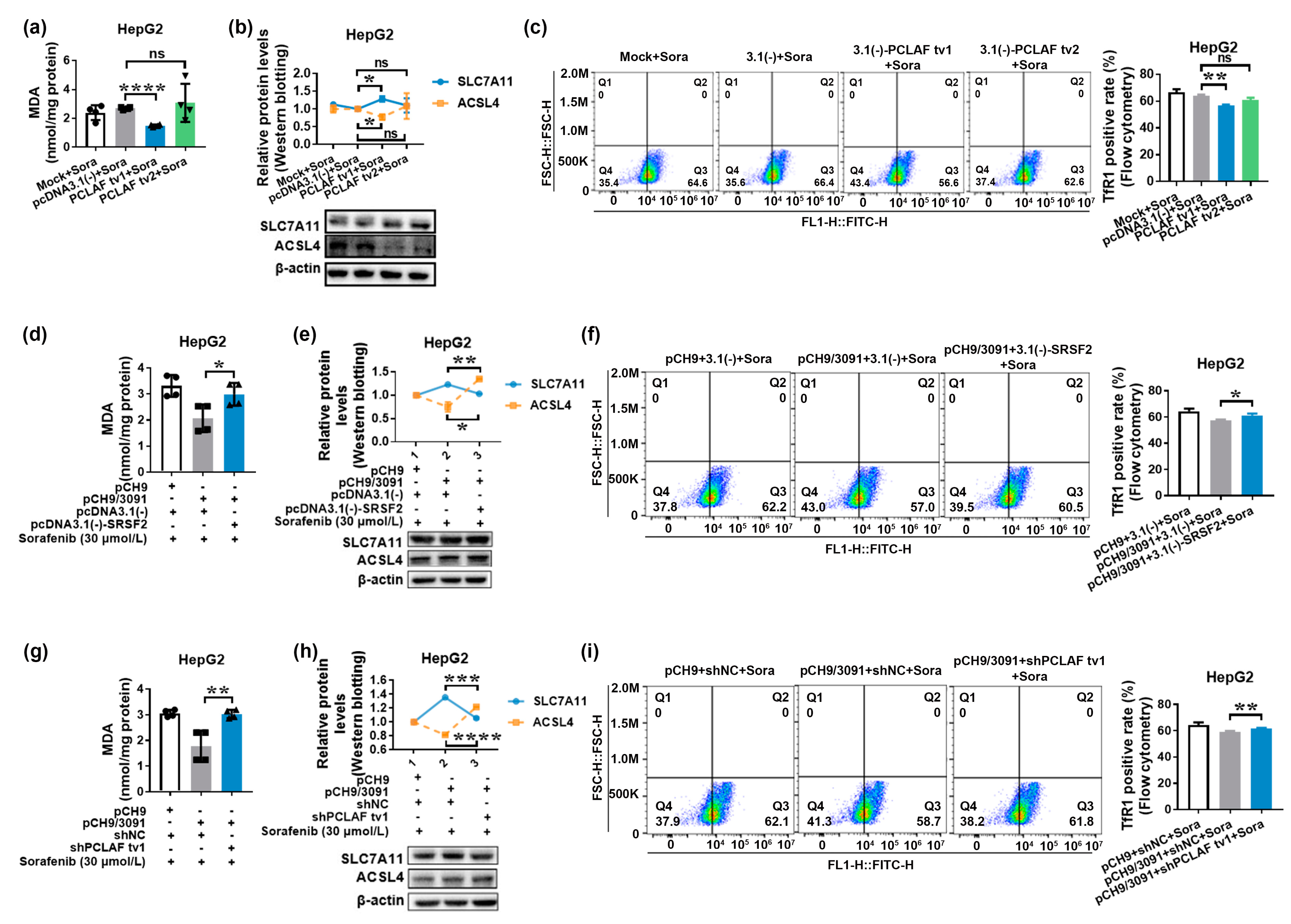
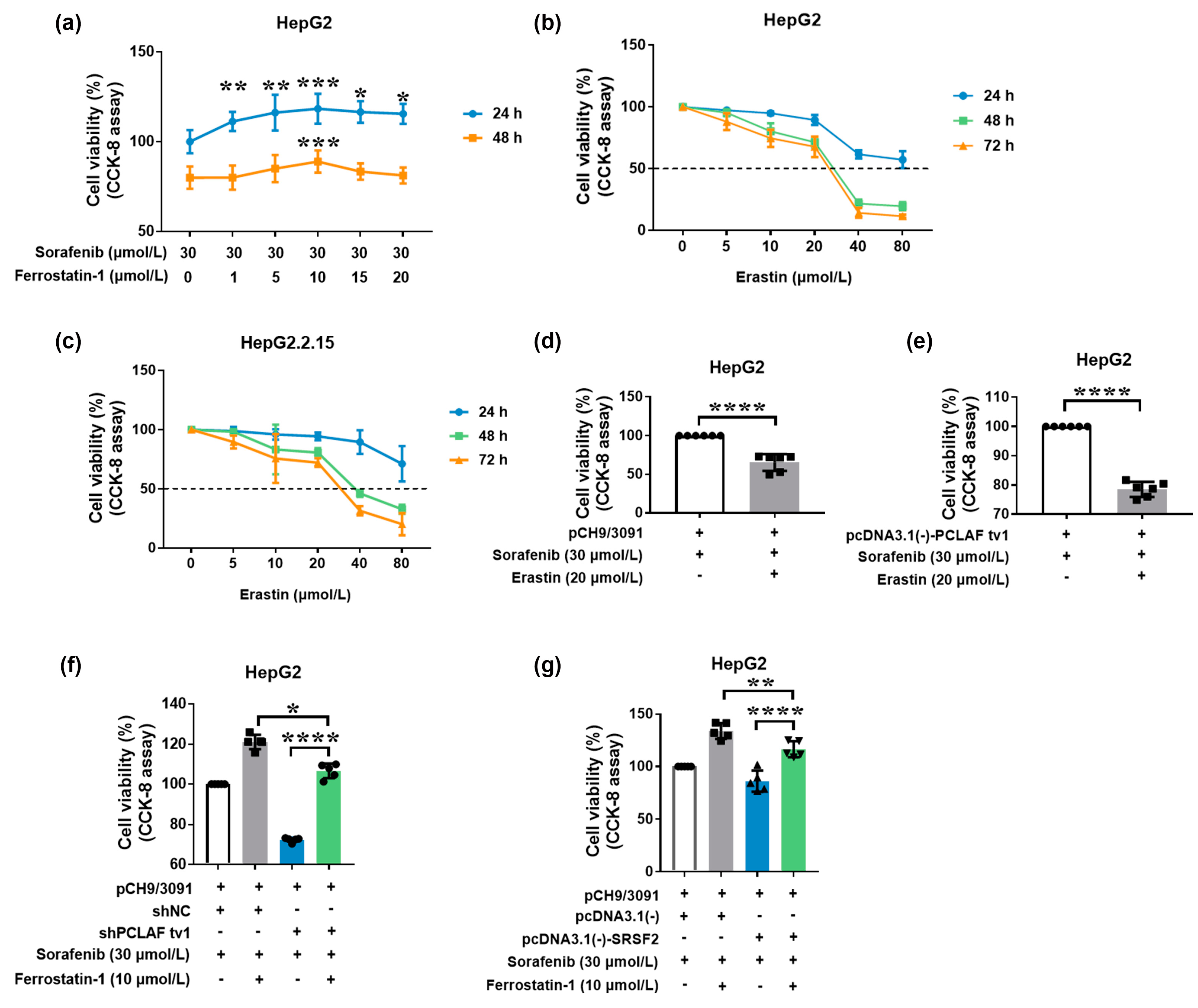
2.3. HBV Causes Sorafenib Resistance by Suppressing Ferroptosis via the SRSF2/PCLAF tv1 Pathway
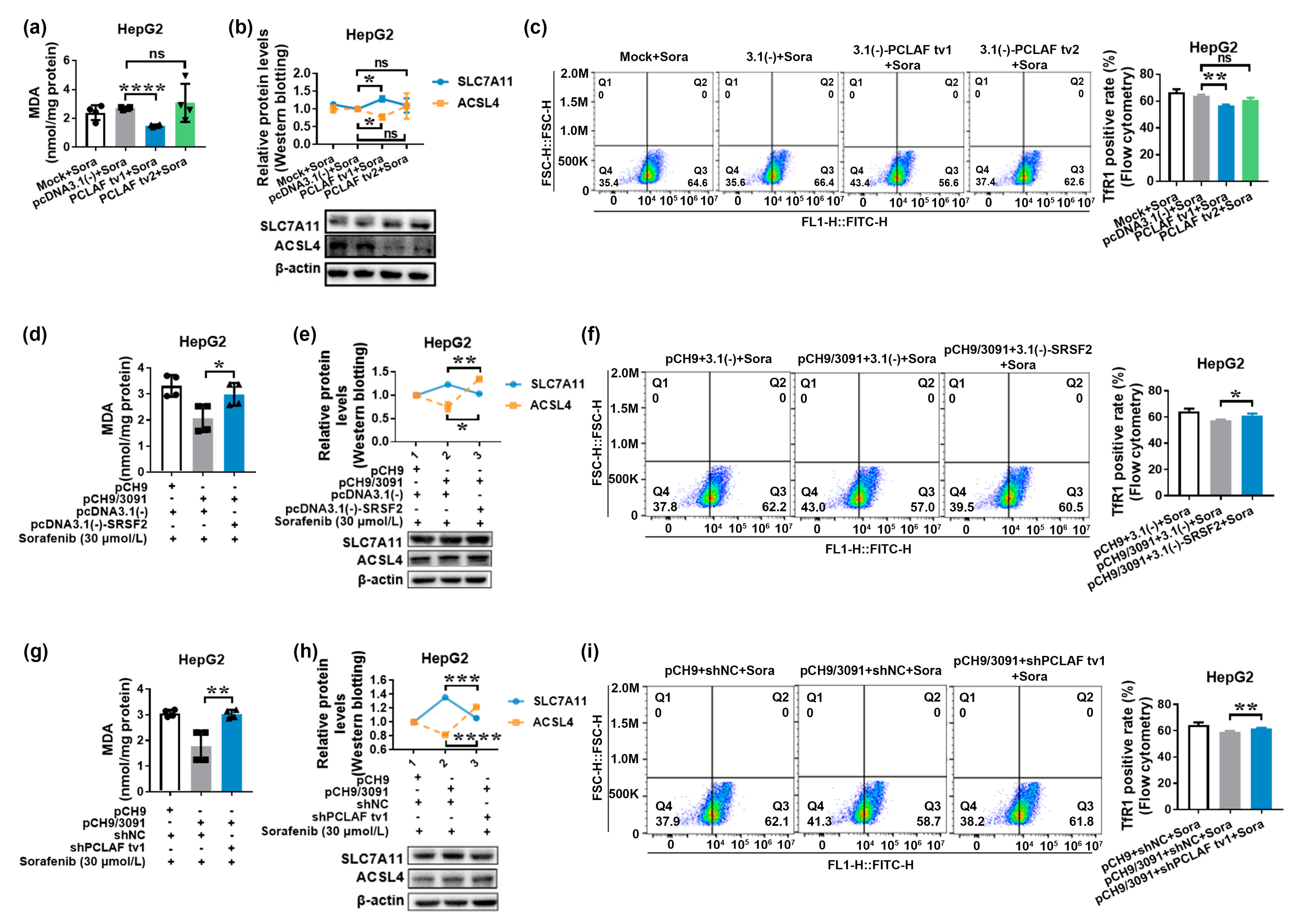
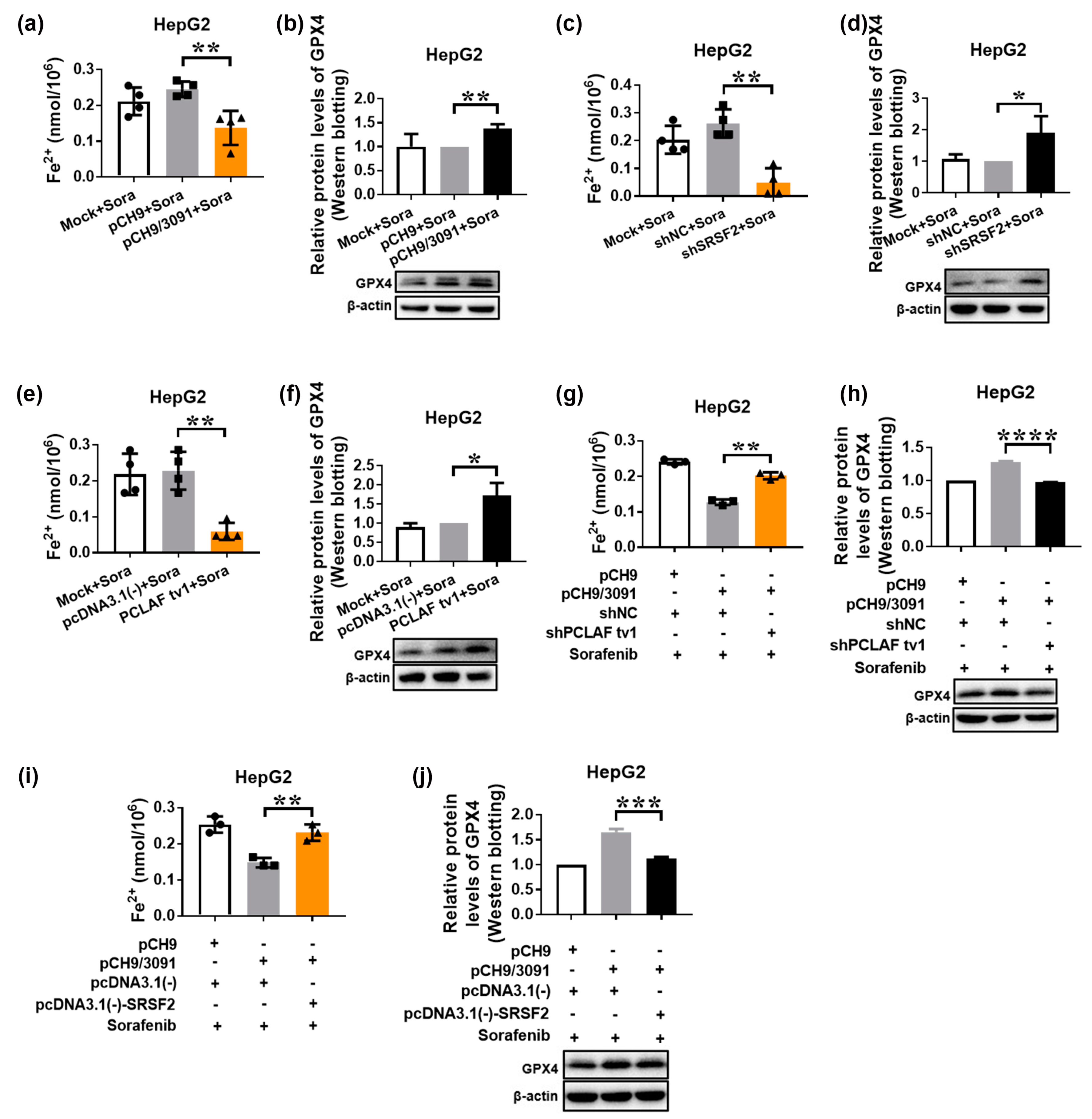
2.4. HBV Suppresses Sorafenib-Induced Ferroptosis through SRSF2/PCLAF tv1 via Both the Fe2+ and Glutathione Peroxidase 4 (GPX4) Pathways
2.5. The SRSF2/PCLAF tv1 Axis Is Engaged in HBV-Mediated Sorafenib Resistance by Suppressing Ferroptosis
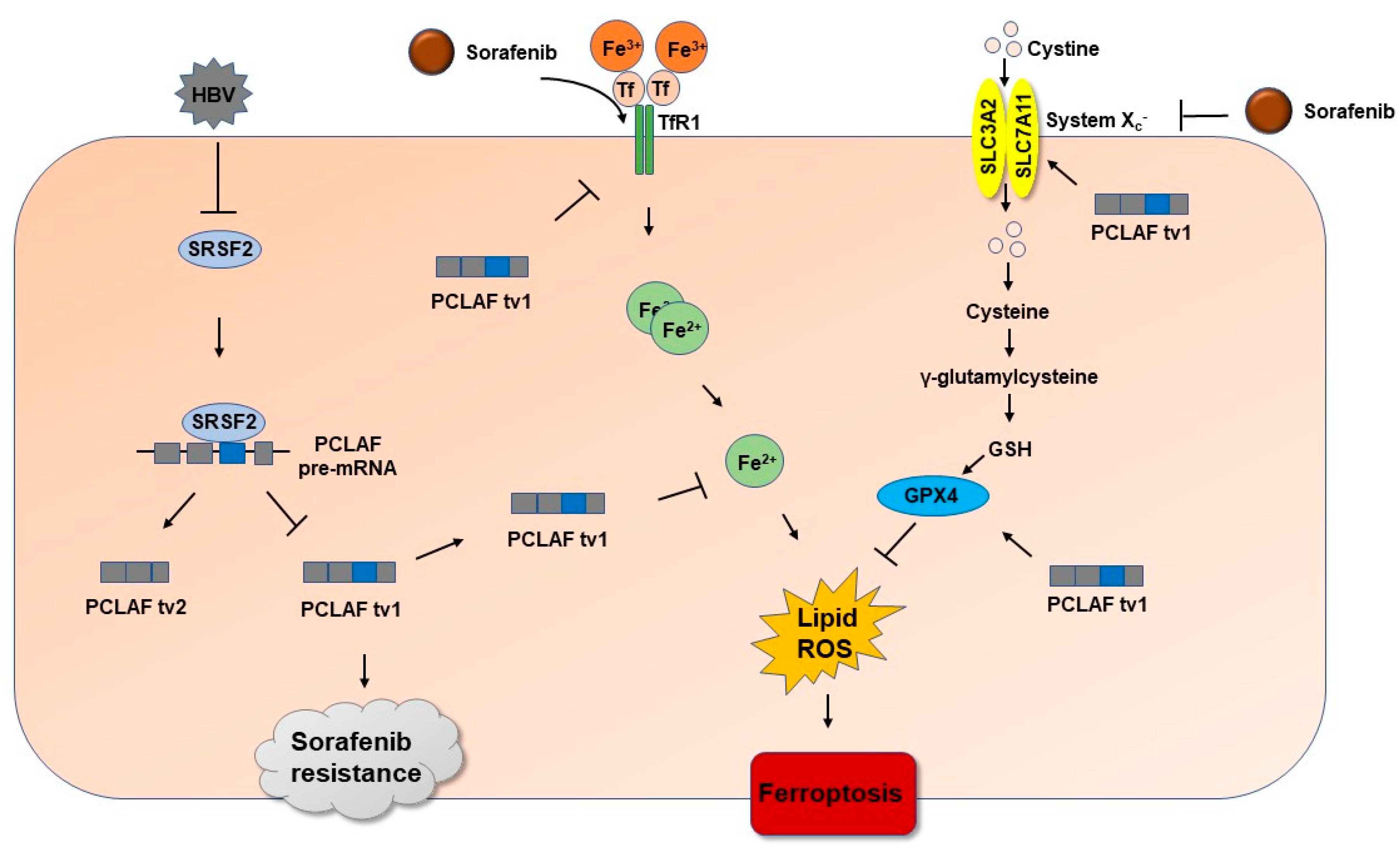
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. Clinical Tissues
4.3. IHC
4.4. Plasmids’ Construction
4.5. Cell Culture and Transient Transfection
4.6. RNA Extraction and Real-Time PCR
4.7. Splicing Reporter Minigene Assay
4.8. Drug Treatment
4.9. CCK-8 Assay
4.10. Protein Isolation and Western Blotting Analysis
4.11. Lipid Peroxidation Assessment
4.12. Flow Cytometry
4.13. Cell Ferrous Iron Colorimetric Assay
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Maucort-Boulch, D.; de Martel, C.; Franceschi, S.; Plummer, M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int. J. Cancer 2018, 142, 2471–2477. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Wei, X.C.; Xia, Y.R.; Zhou, P.; Xue, X.; Ding, S.; Liu, L.J.; Zhu, F. Hepatitis B core antigen modulates exosomal miR-135a to target vesicle-associated membrane protein 2 promoting chemoresistance in hepatocellular carcinoma. World J. Gastroenterol. 2021, 27, 8302–8322. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zheng, C.; Liu, M.; Xu, Y.; Qian, Y.; Zhang, Z.; Su, H.; Li, X.; Wu, H.; Gong, P.; et al. TRERNA1 upregulation mediated by HBx promotes sorafenib resistance and cell proliferation in HCC via targeting NRAS by sponging miR-22-3p. Mol. Ther. 2021, 29, 2601–2616. [Google Scholar] [CrossRef]
- Liu, L.J.; Lv, Z.; Xue, X.; Xing, Z.Y.; Zhu, F. Canonical WNT Signaling Activated by WNT7B Contributes to L-HBs-Mediated Sorafenib Resistance in Hepatocellular Carcinoma by Inhibiting Mitophagy. Cancers 2022, 14, 5781. [Google Scholar] [CrossRef] [PubMed]
- Verta, J.P.; Jacobs, A. The role of alternative splicing in adaptation and evolution. Trends Ecol. Evol. 2022, 37, 299–308. [Google Scholar] [CrossRef]
- Liu, L.; Xie, S.; Zhang, C.; Zhu, F. Aberrant regulation of alternative pre-mRNA splicing in hepatocellular carcinoma. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Assaraf, Y.G.; Maas, M.J.; Kaspers, G.J.; Jansen, G.; Cloos, J. Pre-mRNA splicing in cancer: The relevance in oncogenesis, treatment and drug resistance. Expert Opin. Drug Metab. Toxicol. 2015, 11, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Kokic, G.; Wagner, F.R.; Chernev, A.; Urlaub, H.; Cramer, P. Structural basis of human transcription-DNA repair coupling. Nature 2021, 598, 368–372. [Google Scholar] [CrossRef]
- Liu, X.; Cai, Y.; Cheng, C.; Gu, Y.; Hu, X.; Chen, K.; Wu, Y.; Wu, Z. PCLAF promotes neuroblastoma G1/S cell cycle progression via the E2F1/PTTG1 axis. Cell Death Dis. 2022, 13, 178. [Google Scholar] [CrossRef]
- Liu, L.J.; Liao, J.M.; Zhu, F. Proliferating cell nuclear antigen clamp associated factor, a potential proto-oncogene with increased expression in malignant gastrointestinal tumors. World J. Gastrointest. Oncol. 2021, 13, 1425–1439. [Google Scholar] [CrossRef]
- Magallanes-Garza, G.I.; Santuario-Facio, S.K.; Lira-Albarrán, S.; Varela-Varela, A.F.; Cardona-Huerta, S.; Ruiz-Flores, P.; Haro-Santa-Cruz, J.; Perez-Paramo, Y.X.; Gomez-Macias, G.S.; Davila-Gonzalez, D.; et al. NUSAP1 and PCLAF (KIA0101) Downregulation by Neoadjuvant Therapy is Associated with Better Therapeutic Outcomes and Survival in Breast Cancer. J. Oncol. 2022, 2022, 6001947. [Google Scholar] [CrossRef]
- Wang, K.; Li, J.; Zhou, B. KIAA0101 knockdown inhibits glioma progression and glycolysis by inactivating the PI3K/AKT/mTOR pathway. Metab. Brain Dis. 2022, 37, 489–499. [Google Scholar] [CrossRef]
- Pan, L.; Sha, J.; Lin, W.; Wang, Y.; Bian, T.; Guo, J. Curcumin inhibits prostate cancer progression by regulating the miR-30a-5p/PCLAF axis. Exp. Ther. Med. 2021, 22, 969. [Google Scholar] [CrossRef]
- Liu, L.; Chen, X.; Xie, S.; Zhang, C.; Qiu, Z.; Zhu, F. Variant 1 of KIAA0101, overexpressed in hepatocellular carcinoma, prevents doxorubicin-induced apoptosis by inhibiting p53 activation. Hepatology 2012, 56, 1760–1769. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Y.; Chen, X.; Wang, M.; Zhou, Y.; Zhou, P.; Li, W.; Zhu, F. Variant 2 of KIAA0101, antagonizing its oncogenic variant 1, might be a potential therapeutic strategy in hepatocellular carcinoma. Oncotarget 2017, 8, 43990–44003. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lin, Y.T.; Yan, X.L.; Ding, Y.L.; Wu, Y.L.; Chen, W.N.; Lin, X. Hepatitis B virus core protein inhibits Fas-mediated apoptosis of hepatoma cells via regulation of mFas/FasL and sFas expression. FASEB J. 2015, 29, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lv, G.; Chu, Z.; Piao, L.; Liu, X.; Wang, T.; Jiang, Y.; Zhang, P. Identification of natural splice variants of SAMHD1 in virus-infected HCC. Oncol. Rep. 2014, 31, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative splicing in Alzheimer’s disease. Aging Clin. Exp. Res. 2021, 33, 747–758. [Google Scholar] [CrossRef]
- Moon, H.; Cho, S.; Loh, T.J.; Jang, H.N.; Liu, Y.; Choi, N.; Oh, J.; Ha, J.; Zhou, J.; Cho, S.; et al. SRSF2 directly inhibits intron splicing to suppresses cassette exon inclusion. BMB Rep. 2017, 50, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef]
- Yuan, S.; Liao, G.; Zhang, M.; Zhu, Y.; Wang, K.; Xiao, W.; Jia, C.; Dong, M.; Sun, N.; Walch, A.; et al. Translatomic profiling reveals novel self-restricting virus-host interactions during HBV infection. J. Hepatol. 2021, 75, 74–85. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Mazière, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer Res. 2014, 34, 6417–6422. [Google Scholar] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [Green Version]
- Gaildrat, P.; Killian, A.; Martins, A.; Tournier, I.; Frébourg, T.; Tosi, M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol. Biol. 2010, 653, 249–257. [Google Scholar] [CrossRef]
- Cheng, A.L.; Guan, Z.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Yang, T.S.; Tak, W.Y.; Pan, H.; Yu, S.; et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: Subset analyses of the phase III Sorafenib Asia-Pacific trial. Eur. J. Cancer 2012, 48, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Chien, C.H.; Jen, K.H.; Huang, H.D. RegRNA: An integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 2006, 34, W429–W434. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto, F.F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kitano, M.; Sakurai, T.; Kudo, M. Molecular Mechanism and Prediction of Sorafenib Chemoresistance in Human Hepatocellular Carcinoma. Dig. Dis. 2015, 33, 771–779. [Google Scholar] [CrossRef]
- Liu, S.; Lai, J.; Lyu, N.; Xie, Q.; Cao, H.; Chen, D.; He, M.; Zhang, B.; Zhao, M. Effects of Antiviral Therapy on HBV Reactivation and Survival in Hepatocellular Carcinoma Patients Undergoing Hepatic Artery Infusion Chemotherapy. Front. Oncol. 2020, 10, 582504. [Google Scholar] [CrossRef]
- Kashyap, A.; Tripathi, G.; Tripathi, A.; Rao, R.; Kashyap, M.; Bhat, A.; Kumar, D.; Rajhans, A.; Kumar, P.; Chandrashekar, D.S.; et al. RNA splicing: A dual-edged sword for hepatocellular carcinoma. Med. Oncol. 2022, 39, 173. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L.; Zhou, Y.; Zhou, P.; Yan, Q.; Chen, X.; Ding, S.; Zhu, F. CKLF1 Enhances Inflammation-Mediated Carcinogenesis and Prevents Doxorubicin-Induced Apoptosis via IL6/STAT3 Signaling in HCC. Clin. Cancer Res. 2019, 25, 4141–4154. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yao, M.; Dong, Q. Proliferating cell unclear antigen-associated factor (PAF15): A novel oncogene. Int. J. Biochem. Cell Biol. 2014, 50, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.H.; Jeng, Y.M.; Pan, H.W.; Hu, F.C.; Lai, P.L.; Lee, P.H.; Hsu, H.C. Overexpression of KIAA0101 predicts high stage, early tumor recurrence, and poor prognosis of hepatocellular carcinoma. Clin. Cancer Res. 2007, 13, 5368–5376. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.; Wu, X.; Yu, S.; Xiong, J.; Sang, X.; Zheng, Y.; Zhang, Z. Prognostic value and underlying mechanism of KIAA0101 in hepatocellular carcinoma: Database mining and co-expression analysis. Aging 2020, 12, 16420–16436. [Google Scholar] [CrossRef]
- Jin, C.; Liu, Z.; Li, Y.; Bu, H.; Wang, Y.; Xu, Y.; Qiu, C.; Yan, S.; Yuan, C.; Li, R.; et al. PCNA-associated factor P15(PAF), targeted by FOXM1, predicts poor prognosis in high-grade serous ovarian cancer patients. Int. J. Cancer 2018, 143, 2973–2984. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, K.; Diao, D.; Zhu, K.; Shi, L.; Zhang, H.; Yuan, D.; Guo, Q.; Wu, X.; Liu, D.; et al. Expression of KIAA0101 protein is associated with poor survival of esophageal cancer patients and resistance to cisplatin treatment in vitro. Lab. Invest. 2013, 93, 1276–1287. [Google Scholar] [CrossRef]
- Gamazon, E.R.; Stranger, B.E. Genomics of alternative splicing: Evolution, development and pathophysiology. Hum. Genet. 2014, 133, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Lane, W.S.; Stolk, J.A.; Roth, M.B. SR proteins: A conserved family of pre-mRNA splicing factors. Genes Dev. 1992, 6, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Moon, H.; Jang, H.N.; Liu, Y.; Choi, N.; Oh, J.; Ha, J.; Zheng, X.; Shen, H. Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping. Cells 2019, 8, 696. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M.J.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef]
- Wang, P.; Guo, L.; Li, K.; Ning, S.; Shi, W.; Liu, Z.; Chen, Y. Serine/arginine rich splicing factor 2 expression and clinic pathological features indicating a prognostic factor in human hepatocellular carcinoma patients. Cancer Biomark. 2018, 21, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, L.; Yuan, W.; Liu, Y.; Guo, R.; Luo, Y.; Zhan, Z.; Xie, Z.; Wu, G.; Wu, W.; et al. Loss of SRSF2 triggers hepatic progenitor cell activation and tumor development in mice. Commun. Biol. 2020, 3, 210. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Zhang, Q.; Qu, Y.; Zhang, Q.; Li, F.; Li, B.; Li, Z.; Dong, Y.; Lu, L.; Cai, X. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol. Toxicol. 2022, 1–15. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Chiu, V.; Hsieh, P.C.; Huang, C.Y.; Huang, S.J.; Tzeng, I.S.; Tsai, F.M.; Chen, M.L.; Liu, C.T.; Chen, Y.R. Chrysophanol attenuates hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J. Pharmacol. Sci. 2020, 144, 172–182. [Google Scholar] [CrossRef]
- Haga, Y.; Kanda, T.; Nakamura, M.; Nakamoto, S.; Sasaki, R.; Takahashi, K.; Wu, S.; Yokosuka, O. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS ONE 2017, 12, e0174153. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhu, G.; Liu, Y.; Wu, Q.; Zhang, X.; Bian, Z.; Zhang, Y.; Pan, Q.; Sun, F. O-GlcNAcylated c-Jun antagonizes ferroptosis via inhibiting GSH synthesis in liver cancer. Cell. Signal. 2019, 63, 109384. [Google Scholar] [CrossRef]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kalathur, R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Lv, Z.; Wang, M.; Zhang, D.; Liu, D.; Zhu, F. HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing. Int. J. Mol. Sci. 2023, 24, 3263. https://doi.org/10.3390/ijms24043263
Liu L, Lv Z, Wang M, Zhang D, Liu D, Zhu F. HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing. International Journal of Molecular Sciences. 2023; 24(4):3263. https://doi.org/10.3390/ijms24043263
Chicago/Turabian StyleLiu, Lijuan, Zhao Lv, Miao Wang, Dongyan Zhang, Dongying Liu, and Fan Zhu. 2023. "HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing" International Journal of Molecular Sciences 24, no. 4: 3263. https://doi.org/10.3390/ijms24043263
APA StyleLiu, L., Lv, Z., Wang, M., Zhang, D., Liu, D., & Zhu, F. (2023). HBV Enhances Sorafenib Resistance in Hepatocellular Carcinoma by Reducing Ferroptosis via SRSF2-Mediated Abnormal PCLAF Splicing. International Journal of Molecular Sciences, 24(4), 3263. https://doi.org/10.3390/ijms24043263