


Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Most Common Mitochondria-Related Alterations Accompanying Heart Diseases
3. Mitochondrial Dynamics in Physiology
4. Impairments of Mitochondrial Dynamics in Coronary Artery Diseases
4.1. Ischemic Diseases
4.2. Heart Failure
5. Impairments of Mitochondrial Dynamics in Heart Rhythm Disorders
Atrial Fibrillation
6. Impairments of Mitochondrial Dynamics in Structural Heart Diseases
6.1. Valvular Heart Disease
6.2. Cardiomyopathies
7. Insights on the Role of Mitochondrial Dynamics in Atherosclerosis
8. Still Little Data from Clinical Studies
9. Mitophagy and Mitochondrial Unfolded Protein Response
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Giezen, M. Mitochondria and the Rise of Eukaryotes. BioScience 2011, 61, 594–601. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Joubert, F.; Puff, N. Mitochondrial Cristae Architecture and Functions: Lessons from Minimal Model Systems. Membranes 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and Structural Differences in Spatially Distinct Subpopulations of Cardiac Mitochondria: Influence of Cardiac Pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef]
- Dorn, G.W.; Vega, R.B.; Kelly, D.P. Mitochondrial Biogenesis and Dynamics in the Developing and Diseased Heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.M.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; Wieckowski, M.R.; Giorgi, C.; et al. Calcium Dysregulation in Heart Diseases: Targeting Calcium Channels to Achieve a Correct Calcium Homeostasis. Pharmacol. Res. 2022, 177, 106119. [Google Scholar] [CrossRef]
- Morciano, G.; Vitto, V.A.M.; Bouhamida, E.; Giorgi, C.; Pinton, P. Mitochondrial Bioenergetics and Dynamism in the Failing Heart. Life 2021, 11, 436. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, H.Z.E.; Zhao, G.; Shah, A.M.; Zhang, M. Role of Oxidative Stress in Calcific Aortic Valve Disease and Its Therapeutic Implications. Cardiovasc. Res. 2022, 118, 1433–1451. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Yamamoto, E.; Tokitsu, T.; Kusaka, H.; Fujisue, K.; Kurokawa, H.; Sugamura, K.; Maeda, H.; Tsujita, K.; Kaikita, K.; et al. Reactive Oxygen Metabolites Are Closely Associated with the Diagnosis and Prognosis of Coronary Artery Disease. J. Am. Heart Assoc. 2015, 4, e001451. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. TNF-Alpha and IL-1 Alpha Inhibit Both Pyruvate Dehydrogenase Activity and Mitochondrial Function in Cardiomyocytes: Evidence for Primary Impairment of Mitochondrial Function. Mol. Cell. Biochem. 1997, 177, 61–67. [Google Scholar] [CrossRef]
- Daiber, A. Redox Signaling (Cross-Talk) from and to Mitochondria Involves Mitochondrial Pores and Reactive Oxygen Species. Biochim. Biophys. Acta 2010, 1797, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular Identity of the Mitochondrial Permeability Transition Pore and Its Role in Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The Mitochondrial Permeability Transition Pore: An Evolving Concept Critical for Cell Life and Death. Biol. Rev. 2021, 96, 2489–2521. [Google Scholar] [CrossRef]
- Campo, G.; Morciano, G.; Pavasini, R.; Bonora, M.; Sbano, L.; Biscaglia, S.; Bovolenta, M.; Pinotti, M.; Punzetti, S.; Rizzo, P.; et al. Fo ATP Synthase C Subunit Serum Levels in Patients with ST-Segment Elevation Myocardial Infarction: Preliminary Findings. Int. J. Cardiol. 2016, 221, 993–997. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular Mechanisms and Consequences of Mitochondrial Permeability Transition. Nat. Rev. Mol. Cell Biol. 2021, 4, 266–285. [Google Scholar] [CrossRef]
- Gertz, E.W.; Wisneski, J.A.; Stanley, W.C.; Neese, R.A. Myocardial Substrate Utilization during Exercise in Humans. Dual Carbon-Labeled Carbohydrate Isotope Experiments. J. Clin. Investig. 1988, 82, 2017–2025. [Google Scholar] [CrossRef]
- Wisneski, J.A.; Gertz, E.W.; Neese, R.A.; Gruenke, L.D.; Craig, J.C. Dual Carbon-Labeled Isotope Experiments Using D-[6-14C] Glucose and L-[1,2,3-13C3] Lactate: A New Approach for Investigating Human Myocardial Metabolism during Ischemia. J. Am. Coll. Cardiol. 1985, 5, 1138–1146. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Koves, T.R.; Jaswal, J.S.; Zhang, L.; Ilkayeva, O.; Dyck, J.R.B.; Muoio, D.M.; Lopaschuk, G.D. Insulin-Stimulated Cardiac Glucose Oxidation Is Increased in High-Fat Diet-Induced Obese Mice Lacking Malonyl CoA Decarboxylase. Diabetes 2009, 58, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Leone, T.C.; Keller, M.P.; Martin, O.J.; Broman, A.T.; Nigro, J.; Kapoor, K.; Koves, T.R.; Stevens, R.; Ilkayeva, O.R.; et al. Energy Metabolic Reprogramming in the Hypertrophied and Early Stage Failing Heart: A Multisystems Approach. Circ. Heart Fail. 2014, 7, 1022–1031. [Google Scholar] [CrossRef]
- Bouhamida, E.; Morciano, G.; Perrone, M.; Kahsay, A.E.; Della Sala, M.; Wieckowski, M.R.; Fiorica, F.; Pinton, P.; Giorgi, C.; Patergnani, S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology 2022, 11, 300. [Google Scholar] [CrossRef] [PubMed]
- Zhabyeyev, P.; Gandhi, M.; Mori, J.; Basu, R.; Kassiri, Z.; Clanachan, A.; Lopaschuk, G.D.; Oudit, G.Y. Pressure-Overload-Induced Heart Failure Induces a Selective Reduction in Glucose Oxidation at Physiological Afterload. Cardiovasc. Res. 2013, 97, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired Myocardial Fatty Acid Oxidation and Reduced Protein Expression of Retinoid X Receptor-Alpha in Pacing-Induced Heart Failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased Rates of Substrate Oxidation Ex Vivo Predict the Onset of Heart Failure and Contractile Dysfunction in Rats with Pressure Overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and Nuclear DNA Oxidative Damage in Physiological and Pathological Aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the Mitochondrial DNA Replication Machinery in Mitochondrial DNA Mutagenesis, Aging and Age-Related Diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Strauss, K.A.; DuBiner, L.; Simon, M.; Zaragoza, M.; Sengupta, P.P.; Li, P.; Narula, N.; Dreike, S.; Platt, J.; Procaccio, V.; et al. Severity of Cardiomyopathy Associated with Adenine Nucleotide Translocator-1 Deficiency Correlates with MtDNA Haplogroup. Proc. Natl. Acad. Sci. USA 2013, 110, 3453–3458. [Google Scholar] [CrossRef] [Green Version]
- Narula, N.; Zaragoza, M.V.; Sengupta, P.P.; Li, P.; Haider, N.; Verjans, J.; Waymire, K.; Vannan, M.; Wallace, D.C. Adenine Nucleotide Translocase 1 Deficiency Results in Dilated Cardiomyopathy with Defects in Myocardial Mechanics, Histopathological Alterations, and Activation of Apoptosis. JACC Cardiovasc. Imaging 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Santorelli, F.M.; Tanji, K.; Manta, P.; Casali, C.; Krishna, S.; Hays, A.P.; Mancini, D.M.; DiMauro, S.; Hirano, M. Maternally Inherited Cardiomyopathy: An Atypical Presentation of the MtDNA 12S RRNA Gene A1555G Mutation. Am. J. Hum. Genet. 1999, 64, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Pyle, A.; Griffin, H.; Coxhead, J.; Hussain, R.; Braund, P.S.; Li, L.; Burgess, A.; Munroe, P.B.; Little, L.; et al. Heteroplasmic Mitochondrial DNA Variants in Cardiovascular Diseases. PLoS Genet. 2022, 18, e1010068. [Google Scholar] [CrossRef]
- Andreassi, M.G.; Botto, N.; Colombo, M.G.; Biagini, A.; Clerico, A. Genetic Instability and Atherosclerosis: Can Somatic Mutations Account for the Development of Cardiovascular Diseases? Environ. Mol. Mutagen. 2000, 35, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and Dysfunctions of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Knowlton, A.A. Mitochondrial Dynamics in Heart Failure. Congest. Heart Fail. 2011, 17, 257–261. [Google Scholar] [CrossRef]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics in Mammalian Health and Disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial Fusion Is Essential for Organelle Function and Cardiac Homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Zhao, F.; Zhang, Z.; Kobayashi, T.; Huang, Y.; Shi, B.; Wu, W.; Liang, Q. Mitochondrial Fission and Mitophagy Coordinately Restrict High Glucose Toxicity in Cardiomyocytes. Front. Physiol. 2020, 11, 604069. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Bilyy, R. Mitochondrial Dynamics during Cell Cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Silva Ramos, E.; Larsson, N.-G.; Mourier, A. Bioenergetic Roles of Mitochondrial Fusion. Biochim. Biophys. Acta 2016, 1857, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-Mitochondria Contacts Couple MtDNA Synthesis with Mitochondrial Division in Human Cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef]
- Kraus, F.; Ryan, M.T. The Constriction and Scission Machineries Involved in Mitochondrial Fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational Changes in Dnm1 Support a Contractile Mechanism for Mitochondrial Fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff Is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, New Components of the Mitochondrial Fission Machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Ji, W.; Hatch, A.L.; Merrill, R.A.; Strack, S.; Higgs, H.N. Actin Filaments Target the Oligomeric Maturation of the Dynamin GTPase Drp1 to Mitochondrial Fission Sites. eLife 2015, 4, e11553. [Google Scholar] [CrossRef]
- Zhu, P.-P.; Patterson, A.; Stadler, J.; Seeburg, D.P.; Sheng, M.; Blackstone, C. Intra- and Intermolecular Domain Interactions of the C-Terminal GTPase Effector Domain of the Multimeric Dynamin-like GTPase Drp1. J. Biol. Chem. 2004, 279, 35967–35974. [Google Scholar] [CrossRef]
- Fröhlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural Insights into Oligomerization and Mitochondrial Remodelling of Dynamin 1-like Protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 Mediate Drp1 Recruitment in Mitochondrial Fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.-B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 Regulates Mitochondrial Dynamics through Inhibition of the Fusion Machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.I. Human Cells Are Protected from Mitochondrial Dysfunction by Complementation of DNA Products in Fused Mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 Is Required for Stress-Induced Mitochondrial Hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of Human Mitofusin 1 Provide Insight into Mitochondrial Tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural Basis for GTP Hydrolysis and Conformational Change of MFN1 in Mediating Membrane Fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Mattie, S.; Riemer, J.; Wideman, J.G.; McBride, H.M. A New Mitofusin Topology Places the Redox-Regulated C Terminus in the Mitochondrial Intermembrane Space. J. Cell Biol. 2018, 217, 507–515. [Google Scholar] [CrossRef]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The Human Dynamin-Related Protein OPA1 Is Anchored to the Mitochondrial Inner Membrane Facing the Inter-Membrane Space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of Mitochondrial Morphology through Proteolytic Cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular Basis of Selective Mitochondrial Fusion by Heterotypic Action between OPA1 and Cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 Links Human Mitochondrial Genome Maintenance to MtDNA Replication and Distribution. Genome Res. 2011, 21, 12–20. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of Mitochondrial Fission and Fusion Machinery with Apoptotic Pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the Banquet: Mitochondrial Biogenesis, Mitophagy, and Cell Survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial Homeostasis: The Interplay between Mitophagy and Mitochondrial Biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- López-Lluch, G.; Irusta, P.M.; Navas, P.; de Cabo, R. Mitochondrial Biogenesis and Healthy Aging. Exp. Gerontol. 2008, 43, 813–819. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional Integration of Mitochondrial Biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human Mitochondrial Transcription Revisited: Only TFAM and TFB2M Are Required for Transcription of the Mitochondrial Genes In Vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, X.; Feng, Y.; Handschin, C.; Feng, Y.; Gullicksen, P.S.; Bare, O.; Labow, M.; Spiegelman, B.; Stevenson, S.C. Transducer of Regulated CREB-Binding Proteins (TORCs) Induce PGC-1alpha Transcription and Mitochondrial Biogenesis in Muscle Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14379–14384. [Google Scholar] [CrossRef] [Green Version]
- De Lange, P.; Farina, P.; Moreno, M.; Ragni, M.; Lombardi, A.; Silvestri, E.; Burrone, L.; Lanni, A.; Goglia, F. Sequential Changes in the Signal Transduction Responses of Skeletal Muscle Following Food Deprivation. FASEB J. 2006, 20, 2579–2581. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-Activated Protein Kinase (AMPK) Action in Skeletal Muscle via Direct Phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic Regulation of PGC-1alpha Localization and Turnover Implicates Mitochondrial Adaptation in Calorie Restriction and the Stress Response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef]
- Vachharajani, V.T.; Liu, T.; Wang, X.; Hoth, J.J.; Yoza, B.K.; McCall, C.E. Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016, 2016, 8167273. [Google Scholar] [CrossRef]
- Oka, S.-I.; Sabry, A.D.; Cawley, K.M.; Warren, J.S. Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Biscaglia, S.; Erriquez, A.; Serenelli, M.; D’Ascenzo, F.; De Ferrari, G.; Ariza Sole, A.; Sanchis, J.; Giannini, F.; Gallo, F.; Scala, A.; et al. Complete versus Culprit-Only Strategy in Older MI Patients with Multivessel Disease. Catheter. Cardiovasc. Interv. 2022, 99, 970–978. [Google Scholar] [CrossRef]
- Wong, N.D. Epidemiological Studies of CHD and the Evolution of Preventive Cardiology. Nat. Rev. Cardiol. 2014, 11, 276–289. [Google Scholar] [CrossRef]
- Campo, G.; Pavasini, R.; Morciano, G.; Lincoff, A.M.; Gibson, C.M.; Kitakaze, M.; Lonborg, J.; Ahluwalia, A.; Ishii, H.; Frenneaux, M.; et al. Clinical Benefit of Drugs Targeting Mitochondrial Function as an Adjunct to Reperfusion in ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of Randomized Clinical Trials. Int. J. Cardiol. 2017, 244, 59–66. [Google Scholar] [CrossRef]
- Campo, G.; Pavasini, R.; Morciano, G.; Lincoff, M.A.; Gibson, M.C.; Kitakaze, M.; Lonborg, J.; Ahluwalia, A.; Ishii, H.; Frenneaux, M.; et al. Data on Administration of Cyclosporine, Nicorandil, Metoprolol on Reperfusion Related Outcomes in ST-Segment Elevation Myocardial Infarction Treated with Percutaneous Coronary Intervention. Data Brief 2017, 14, 197–205. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-Related Protein 1 (Drp1)-Mediated Diastolic Dysfunction in Myocardial Ischemia-Reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Guan, P.; Ye, X.; Lu, Y.; Hang, Y.; Su, Y.; Hu, W. SOCS6 Promotes Mitochondrial Fission and Cardiomyocyte Apoptosis and Is Negatively Regulated by Quaking-Mediated MiR-19b. Oxid. Med. Cell. Longev. 2022, 2022, 1121323. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Disatnik, M.-H.; Ferreira, J.C.B.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.M.; Qi, X.; Mochly-Rosen, D. Acute Inhibition of Excessive Mitochondrial Fission after Myocardial Infarction Prevents Long-Term Cardiac Dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, S.; Jin, Q.; Shi, C.; Zhang, Y.; Zhu, P.; Ma, Q.; Tian, F.; Chen, Y. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of MROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved MPTP Opening. J. Am. Heart Assoc. 2017, 6, e005328. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Kerdphoo, S.; Jaiwongkam, T.; Chattipakorn, S.C.; Chattipakorn, N. Balancing Mitochondrial Dynamics via Increasing Mitochondrial Fusion Attenuates Infarct Size and Left Ventricular Dysfunction in Rats with Cardiac Ischemia/Reperfusion Injury. Clin. Sci. 2019, 133, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Che, Z.; Meng, X.; Yu, Y.; Li, M.; Yu, Z.; Shi, H.; Yang, D.; Yu, M. MCU Up-Regulation Contributes to Myocardial Ischemia-Reperfusion Injury through Calpain/OPA-1-Mediated Mitochondrial Fusion/Mitophagy Inhibition. J. Cell. Mol. Med. 2019, 23, 7830–7843. [Google Scholar] [CrossRef]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 Processing and Mitochondrial Fragmentation Cause Heart Failure in Mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, Apoptosis, and Heart Failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of Mitochondrial Fusion-Fission Proteins during Post-Infarction Remodeling: The Effect of NHE-1 Inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Z.; Qin, X.; Xu, J.; Hou, Z.; Yang, H.; Mao, X.; Xing, W.; Li, X.; Zhang, X.; et al. Enhancing Fatty Acid Utilization Ameliorates Mitochondrial Fragmentation and Cardiac Dysfunction via Rebalancing Optic Atrophy 1 Processing in the Failing Heart. Cardiovasc. Res. 2018, 114, 979–991. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Z.; Yin, Q.; Fu, C.; Barszczyk, A.; Zhang, X.; Wang, J.; Yang, D. Cardiac-Specific Overexpression of MiR-122 Induces Mitochondria-Dependent Cardiomyocyte Apoptosis and Promotes Heart Failure by Inhibiting Hand2. J. Cell. Mol. Med. 2021, 25, 5326–5334. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a Sodium Glucose Co-Transporter-2 Inhibitor, Alleviates Atrial Remodeling and Improves Mitochondrial Function in High-Fat Diet/Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.-M.; Mouton, S.; Sebti, Y.; et al. Myocardial Contractile Dysfunction Is Associated with Impaired Mitochondrial Function and Dynamics in Type 2 Diabetic but Not in Obese Patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef]
- Rogers, M.A.; Maldonado, N.; Hutcheson, J.D.; Goettsch, C.; Goto, S.; Yamada, I.; Faits, T.; Sesaki, H.; Aikawa, M.; Aikawa, E. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ. Res. 2017, 121, 220–233. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Pedriali, G.; Cimaglia, P.; Mikus, E.; Calvi, S.; Albertini, A.; Giorgi, C.; Campo, G.; Ferrari, R.; et al. Impairment of Mitophagy and Autophagy Accompanies Calcific Aortic Valve Stenosis Favoring Cell Death and the Severity of Disease. Cardiovasc. Res. 2021, 118, 2548–2559. [Google Scholar] [CrossRef]
- Abudupataer, M.; Zhu, S.; Yan, S.; Xu, K.; Zhang, J.; Luo, S.; Ma, W.; Alam, M.F.; Tang, Y.; Huang, H.; et al. Aorta Smooth Muscle-on-a-Chip Reveals Impaired Mitochondrial Dynamics as a Therapeutic Target for Aortic Aneurysm in Bicuspid Aortic Valve Disease. eLife 2021, 10, e69310. [Google Scholar] [CrossRef]
- Cahill, T.J.; Leo, V.; Kelly, M.; Stockenhuber, A.; Kennedy, N.W.; Bao, L.; Cereghetti, G.M.; Harper, A.R.; Czibik, G.; Liao, C.; et al. Resistance of Dynamin-Related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure. J. Biol. Chem. 2015, 290, 25907–25919. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M.; et al. Dynamics of Mitochondrial DNA Nucleoids Regulated by Mitochondrial Fission Is Essential for Maintenance of Homogeneously Active Mitochondria during Neonatal Heart Development. Mol. Cell. Biol. 2015, 35, 211–223. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Kikuchi, R.; Ngoh, G.A.; Coughlan, K.A.; Dominguez, I.; Stanley, W.C.; Walsh, K. Mitofusins 1 and 2 Are Essential for Postnatal Metabolic Remodeling in Heart. Circ. Res. 2012, 111, 1012–1026. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, M.; Torres, G.; Wu, S.; Ouyang, C.; Xie, Z.; Zou, M.-H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef]
- Chehaitly, A.; Guihot, A.-L.; Proux, C.; Grimaud, L.; Aurrière, J.; Legouriellec, B.; Rivron, J.; Vessieres, E.; Tétaud, C.; Zorzano, A.; et al. Altered Mitochondrial Opa1-Related Fusion in Mouse Promotes Endothelial Cell Dysfunction and Atherosclerosis. Antioxidants 2022, 11, 1078. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1-Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Salabei, J.K.; Hill, B.G. Mitochondrial Fission Induced by Platelet-Derived Growth Factor Regulates Vascular Smooth Muscle Cell Bioenergetics and Cell Proliferation. Redox Biol. 2013, 1, 542–551. [Google Scholar] [CrossRef]
- Guo, X.; Chen, K.-H.; Guo, Y.; Liao, H.; Tang, J.; Xiao, R.-P. Mitofusin 2 Triggers Vascular Smooth Muscle Cell Apoptosis via Mitochondrial Death Pathway. Circ. Res. 2007, 101, 1113–1122. [Google Scholar] [CrossRef]
- Guo, Y.-H.; Chen, K.; Gao, W.; Li, Q.; Chen, L.; Wang, G.-S.; Tang, J. Overexpression of Mitofusin 2 Inhibited Oxidized Low-Density Lipoprotein Induced Vascular Smooth Muscle Cell Proliferation and Reduced Atherosclerotic Lesion Formation in Rabbit. Biochem. Biophys. Res. Commun. 2007, 363, 411–417. [Google Scholar] [CrossRef]
- He, L.; Zhou, Q.; Huang, Z.; Xu, J.; Zhou, H.; Lv, D.; Lu, L.; Huang, S.; Tang, M.; Zhong, J.; et al. PINK1/Parkin-Mediated Mitophagy Promotes Apelin-13-Induced Vascular Smooth Muscle Cell Proliferation by AMPKα and Exacerbates Atherosclerotic Lesions. J. Cell. Physiol. 2019, 234, 8668–8682. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, J.; Liu, X.; Zhang, Q. MicroRNA-138 Attenuates Myocardial Ischemia Reperfusion Injury through Inhibiting Mitochondria-Mediated Apoptosis by Targeting HIF1-α. Exp. Ther. Med. 2019, 18, 3325–3332. [Google Scholar] [CrossRef]
- Aishwarya, R.; Alam, S.; Abdullah, C.S.; Morshed, M.; Nitu, S.S.; Panchatcharam, M.; Miriyala, S.; Kevil, C.G.; Bhuiyan, M.S. Pleiotropic Effects of Mdivi-1 in Altering Mitochondrial Dynamics, Respiration, and Autophagy in Cardiomyocytes. Redox Biol. 2020, 36, 101660. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Liu, F.; Fang, B.-B.; Tian, T.; Li, Y.-H.; Zhang, T.; Li, X.-M.; Yang, Y.-N. NFKB1 Gene Mutant Was Associated with Prognosis of Coronary Artery Disease and Exacerbated Endothelial Mitochondrial Fission and Dysfunction. Oxid. Med. Cell. Longev. 2022, 2022, 9494926. [Google Scholar] [CrossRef]
- Jin, Q.; Li, R.; Hu, N.; Xin, T.; Zhu, P.; Hu, S.; Ma, S.; Zhu, H.; Ren, J.; Zhou, H. DUSP1 Alleviates Cardiac Ischemia/Reperfusion Injury by Suppressing the Mff-Required Mitochondrial Fission and Bnip3-Related Mitophagy via the JNK Pathways. Redox Biol. 2018, 14, 576–587. [Google Scholar] [CrossRef]
- Rodríguez-Graciani, K.M.; Chapa-Dubocq, X.R.; MacMillan-Crow, L.A.; Javadov, S. Association Between L-OPA1 Cleavage and Cardiac Dysfunction During Ischemia-Reperfusion Injury in Rats. Cell. Physiol. Biochem. 2020, 54, 1101–1114. [Google Scholar] [CrossRef]
- Kulek, A.R.; Undyala, V.V.R.; Anzell, A.R.; Raghunayakula, S.; MacMillan-Crow, L.A.; Sanderson, T.H.; Przyklenk, K. Does Disruption of Optic Atrophy-1 (OPA1) Contribute to Cell Death in HL-1 Cardiomyocytes Subjected to Lethal Ischemia-Reperfusion Injury? Cells 2022, 11, 3083. [Google Scholar] [CrossRef]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.D.; Lucena, J.R.; et al. Impaired Mitochondrial Biogenesis Is a Common Feature to Myocardial Hypertrophy and End-Stage Ischemic Heart Failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha Target Gene Downregulation Is a Signature of the Failing Human Heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA Replication Impairs Mitochondrial Biogenesis in Human Failing Hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Garnier, A.; Fortin, D.; Deloménie, C.; Momken, I.; Veksler, V.; Ventura-Clapier, R. Depressed Mitochondrial Transcription Factors and Oxidative Capacity in Rat Failing Cardiac and Skeletal Muscles. J. Physiol. 2003, 551, 491–501. [Google Scholar] [CrossRef]
- Fabregat-Andrés, Ó.; Ridocci-Soriano, F.; Estornell-Erill, J.; Corbí-Pascual, M.; Valle-Muñoz, A.; Berenguer-Jofresa, A.; Barrabés, J.A.; Mata, M.; Monsalve, M. Blood PGC-1α Concentration Predicts Myocardial Salvage and Ventricular Remodeling After ST-Segment Elevation Acute Myocardial Infarction. Rev. Esp. Cardiol. 2015, 68, 408–416. [Google Scholar] [CrossRef]
- Fabregat-Andrés, Ó.; Tierrez, A.; Mata, M.; Estornell-Erill, J.; Ridocci-Soriano, F.; Monsalve, M. Induction of PGC-1α Expression Can Be Detected in Blood Samples of Patients with ST-Segment Elevation Acute Myocardial Infarction. PLoS ONE 2011, 6, e26913. [Google Scholar] [CrossRef]
- Fabregat-Andres, O.; Paredes, F.; Monsalve, M.; Milara, J.; Ridocci-Soriano, F.; Gonzalez-Hervas, S.; Mena, A.; Facila, L.; Hornero, F.; Morell, S.; et al. MRNA PGC-1α Levels in Blood Samples Reliably Correlates with Its Myocardial Expression: Study in Patients Undergoing Cardiac Surgery. Anatol. J. Cardiol. 2016, 16, 622–629. [Google Scholar] [CrossRef]
- Ramaccini, D.; Pedriali, G.; Perrone, M.; Bouhamida, E.; Modesti, L.; Wieckowski, M.R.; Giorgi, C.; Pinton, P.; Morciano, G. Some Insights into the Regulation of Cardiac Physiology and Pathology by the Hippo Pathway. Biomedicines 2022, 10, 726. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Li, J.; Zhou, P.; Zhao, X.; Chen, R.; Song, L.; Zhao, H.; Yan, H. LATS2 Deletion Attenuates Myocardial Ischemia-Reperfusion Injury by Promoting Mitochondrial Biogenesis. Oxid. Med. Cell. Longev. 2021, 2021, 1058872. [Google Scholar] [CrossRef]
- Guariento, A.; Blitzer, D.; Doulamis, I.; Shin, B.; Moskowitzova, K.; Orfany, A.; Ramirez-Barbieri, G.; Staffa, S.J.; Zurakowski, D.; Del Nido, P.J.; et al. Preischemic Autologous Mitochondrial Transplantation by Intracoronary Injection for Myocardial Protection. J. Thorac. Cardiovasc. Surg. 2020, 160, e15–e29. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The Emerging Role of Nrf2 in Mitochondrial Function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Jia, L.; Yang, L.; Tian, Y.; Yang, L.; Wu, D.; Zhang, H.; Li, M.; Wu, N. Nrf2 Participates in the Protective Effect of Exogenous Mitochondria against Mitochondrial Dysfunction in Myocardial Ischaemic and Hypoxic Injury. Cell. Signal. 2022, 92, 110266. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Costantino, S.; Paneni, F. MicroRNA-122 in Heart Failure with Reduced Ejection Fraction: Epiphenomenon or Causal? Int. J. Cardiol. 2020, 303, 66–67. [Google Scholar] [CrossRef]
- Sabbah, H.N.; Gupta, R.C.; Singh-Gupta, V.; Zhang, K.; Lanfear, D.E. Abnormalities of Mitochondrial Dynamics in the Failing Heart: Normalization Following Long-Term Therapy with Elamipretide. Cardiovasc. Drugs Ther. 2018, 32, 319–328. [Google Scholar] [CrossRef]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of Mitochondrial Fusion Rescues Mff-Deficient Cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse Aortic Constriction Leads to Accelerated Heart Failure in Mice Lacking PPAR-Gamma Coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1β Deficiency Accelerates the Transition to Heart Failure in Pressure Overload Hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar] [CrossRef]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and Arrhythmias. Free Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef]
- Westerman, S.; Wenger, N. Gender Differences in Atrial Fibrillation: A Review of Epidemiology, Management, and Outcomes. Curr. Cardiol. Rev. 2019, 15, 136–144. [Google Scholar] [CrossRef]
- Thiedemann, K.U.; Ferrans, V.J. Left Atrial Ultrastructure in Mitral Valvular Disease. Am. J. Pathol. 1977, 89, 575–604. [Google Scholar]
- Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; de Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, J.; Zhang, M.; Liu, G.; Wang, X.; Liu, Y.; Yang, N.; Liu, Y.; Zhao, G.; Sun, J.; et al. Β3-Adrenoceptor Impairs Mitochondrial Biogenesis and Energy Metabolism During Rapid Atrial Pacing-Induced Atrial Fibrillation. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 114–126. [Google Scholar] [CrossRef]
- Bell, D.S.H.; Goncalves, E. Atrial Fibrillation and Type 2 Diabetes: Prevalence, Etiology, Pathophysiology and Effect of Anti-Diabetic Therapies. Diabetes Obes. Metab. 2019, 21, 210–217. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmacol. 2021, 12, 658362. [Google Scholar] [CrossRef]
- Jeganathan, J.; Saraf, R.; Mahmood, F.; Pal, A.; Bhasin, M.K.; Huang, T.; Mittel, A.; Knio, Z.; Simons, R.; Khabbaz, K.; et al. Mitochondrial Dysfunction in Atrial Tissue of Patients Developing Postoperative Atrial Fibrillation. Ann. Thorac. Surg. 2017, 104, 1547–1555. [Google Scholar] [CrossRef]
- Kumar, A.; Avishay, D.M.; Jones, C.R.; Shaikh, J.D.; Kaur, R.; Aljadah, M.; Kichloo, A.; Shiwalkar, N.; Keshavamurthy, S. Sudden Cardiac Death: Epidemiology, Pathogenesis and Management. Rev. Cardiovasc. Med. 2021, 22, 147–158. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Devalla, H.D.; Gélinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.-A.; Tadros, R.; Jiang, H.; Le Béchec, A.; et al. TECRL, a New Life-Threatening Inherited Arrhythmia Gene Associated with Overlapping Clinical Features of Both LQTS and CPVT. EMBO Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef]
- Hou, C.; Jiang, X.; Zhang, H.; Zheng, J.; Qiu, Q.; Zhang, Y.; Sun, X.; Xu, M.; Chang, A.C.Y.; Xie, L.; et al. TECRL Deficiency Results in Aberrant Mitochondrial Function in Cardiomyocytes. Commun. Biol. 2022, 5, 470. [Google Scholar] [CrossRef]
- Valli, H.; Ahmad, S.; Chadda, K.R.; Al-Hadithi, A.B.A.K.; Grace, A.A.; Jeevaratnam, K.; Huang, C.L.-H. Age-Dependent Atrial Arrhythmic Phenotype Secondary to Mitochondrial Dysfunction in Pgc-1β Deficient Murine Hearts. Mech. Ageing Dev. 2017, 167, 30–45. [Google Scholar] [CrossRef]
- Valli, H.; Ahmad, S.; Fraser, J.A.; Jeevaratnam, K.; Huang, C.L.-H. Pro-Arrhythmic Atrial Phenotypes in Incrementally Paced Murine Pgc1β-/- Hearts: Effects of Age. Exp. Physiol. 2017, 102, 1619–1634. [Google Scholar] [CrossRef]
- Ahmad, S.; Valli, H.; Chadda, K.R.; Cranley, J.; Jeevaratnam, K.; Huang, C.L.-H. Ventricular Pro-Arrhythmic Phenotype, Arrhythmic Substrate, Ageing and Mitochondrial Dysfunction in Peroxisome Proliferator Activated Receptor-γ Coactivator-1β Deficient (Pgc-1β-/-) Murine Hearts. Mech. Ageing Dev. 2018, 173, 92–103. [Google Scholar] [CrossRef]
- Iung, B.; Vahanian, A. Epidemiology of Acquired Valvular Heart Disease. Can. J. Cardiol. 2014, 30, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Iung, B.; Vahanian, A. Epidemiology of Valvular Heart Disease in the Adult. Nat. Rev. Cardiol. 2011, 8, 162–172. [Google Scholar] [CrossRef]
- Lincoln, J.; Garg, V. Etiology of Valvular Heart Disease-Genetic and Developmental Origins. Circ. J. 2014, 78, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Brieler, J.; Breeden, M.A.; Tucker, J. Cardiomyopathy: An Overview. Am. Fam. Physician 2017, 96, 640–646. [Google Scholar]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Merlo, M.; Cannatà, A.; Gobbo, M.; Stolfo, D.; Elliott, P.M.; Sinagra, G. Evolving Concepts in Dilated Cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 228–239. [Google Scholar] [CrossRef]
- Pedriali, G.; Morciano, G.; Patergnani, S.; Cimaglia, P.; Morelli, C.; Mikus, E.; Ferrari, R.; Gasbarro, V.; Giorgi, C.; Wieckowski, M.R.; et al. Aortic Valve Stenosis and Mitochondrial Dysfunctions: Clinical and Molecular Perspectives. Int. J. Mol. Sci. 2020, 21, 4899. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Hu, W.; Gao, C.; Zeng, Z.; Cheng, S.; Yu, K.; Qian, Y.; Xu, D.; Zhu, G.; et al. PTP1B Inhibition Improves Mitochondrial Dynamics to Alleviate Calcific Aortic Valve Disease via Regulating OPA1 Homeostasis. JACC Basic Transl. Sci. 2022, 7, 697–712. [Google Scholar] [CrossRef]
- Wang, L.; Ming Wang, L.; Chen, W.; Chen, X. Bicuspid Aortic Valve: A Review of Its Genetics and Clinical Significance. J. Heart Valve Dis. 2016, 25, 568–573. [Google Scholar]
- Ashrafian, H.; Docherty, L.; Leo, V.; Towlson, C.; Neilan, M.; Steeples, V.; Lygate, C.A.; Hough, T.; Townsend, S.; Williams, D.; et al. A Mutation in the Mitochondrial Fission Gene Dnm1l Leads to Cardiomyopathy. PLoS Genet. 2010, 6, e1001000. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 Mutation and Late-Onset Cardiomyopathy: Mitochondrial Dysfunction and MtDNA Instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef]
- Hsiao, Y.T.; Shimizu, I.; Wakasugi, T.; Yoshida, Y.; Ikegami, R.; Hayashi, Y.; Suda, M.; Katsuumi, G.; Nakao, M.; Ozawa, T.; et al. Cardiac Mitofusin-1 Is Reduced in Non-Responding Patients with Idiopathic Dilated Cardiomyopathy. Sci. Rep. 2021, 11, 6722. [Google Scholar] [CrossRef]
- Ahuja, P.; Wanagat, J.; Wang, Z.; Wang, Y.; Liem, D.A.; Ping, P.; Antoshechkin, I.A.; Margulies, K.B.; Maclellan, W.R. Divergent Mitochondrial Biogenesis Responses in Human Cardiomyopathy. Circulation 2013, 127, 1957–1967. [Google Scholar] [CrossRef]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of Mitochondrial Biogenesis Is a Maladaptive Mechanism in Mitochondrial Cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Liu, Y.; Zhou, W.; Sun, R.; Xia, M. Retinol Binding Protein 4 Induces Mitochondrial Dysfunction and Vascular Oxidative Damage. Atherosclerosis 2015, 240, 335–344. [Google Scholar] [CrossRef]
- Wei, Z.; Chong, H.; Jiang, Q.; Tang, Y.; Xu, J.; Wang, H.; Shi, Y.; Cui, L.; Li, J.; Zhang, Y.; et al. Smooth Muscle Overexpression of PGC1α Attenuates Atherosclerosis in Rabbits. Circ. Res. 2021, 129, e72–e86. [Google Scholar] [CrossRef]
- Stein, S.; Lohmann, C.; Handschin, C.; Stenfeldt, E.; Borén, J.; Lüscher, T.F.; Matter, C.M. ApoE-/- PGC-1α-/- Mice Display Reduced IL-18 Levels and Do Not Develop Enhanced Atherosclerosis. PLoS ONE 2010, 5, e13539. [Google Scholar] [CrossRef]
- Vandenbeek, R.; Khan, N.P.; Estall, J.L. Linking Metabolic Disease With the PGC-1α Gly482Ser Polymorphism. Endocrinology 2018, 159, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, W.; Li, X.; Tang, Y.; Xie, P.; Ji, Y.; Fan, L.; Chen, Q. Association between PPARGC1A Gene Polymorphisms and Coronary Artery Disease in a Chinese Population. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Guo, D.; Li, Y.; Liang, S.; Wu, Y. The Impact of Severity of Hypertension on Association of PGC-1alpha Gene with Blood Pressure and Risk of Hypertension. BMC Cardiovasc. Disord. 2007, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Fitchett, D.; Inzucchi, S.E.; Cannon, C.P.; McGuire, D.K.; Scirica, B.M.; Johansen, O.E.; Sambevski, S.; Kaspers, S.; Pfarr, E.; George, J.T.; et al. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019, 139, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Cardioprotective Effects of Sirtuin-1 and Its Downstream Effectors: Potential Role in Mediating the Heart Failure Benefits of SGLT2 (Sodium-Glucose Cotransporter 2) Inhibitors. Circ. Heart Fail. 2020, 13, e007197. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Zhuravlev, A.D.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. Mitochondria-Mediated Cardiovascular Benefits of Sodium-Glucose Co-Transporter 2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin Attenuates Vascular Calcification by Inhibiting Mitochondria Fission via an AMPK/Drp1 Signalling Pathway. J. Cell. Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Zhou, Y.J.; Yang, J.Q.; Liu, F.; Wu, X.P.; Sha, Y. Melatonin Attenuates Calcium Deposition from Vascular Smooth Muscle Cells by Activating Mitochondrial Fusion and Mitophagy via an AMPK/OPA1 Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5298483. [Google Scholar] [CrossRef]
- Domínguez-Rodríguez, A.; Abreu-González, P.; Báez-Ferrer, N.; Reiter, R.J.; Avanzas, P.; Hernández-Vaquero, D. Melatonin and Cardioprotection in Humans: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Cardiovasc. Med. 2021, 8, 635083. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef]
- Vögtle, F.-N. Open Questions on the Mitochondrial Unfolded Protein Response. FEBS J. 2021, 288, 2856–2869. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell. Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy Limits Acute Myocardial Infarction Induced by Permanent Coronary Artery Occlusion. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2261–H2271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Siraj, S.; Zhang, R.; Chen, Q. Mitophagy Receptor FUNDC1 Regulates Mitochondrial Homeostasis and Protects the Heart from I/R Injury. Autophagy 2017, 13, 1080–1081. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-Inducible Kinase 1 (PINK1)/Park6 Is Indispensable for Normal Heart Function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Liu, L.; McKeehan, W.L.; Wang, F. The Fibroblast Growth Factor Signaling Axis Controls Cardiac Stem Cell Differentiation through Regulating Autophagy. Autophagy 2012, 8, 690–691. [Google Scholar] [CrossRef]
- Cadete, V.J.J.; Vasam, G.; Menzies, K.J.; Burelle, Y. Mitochondrial Quality Control in the Cardiac System: An Integrative View. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 782–796. [Google Scholar] [CrossRef]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Reche-López, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; Suárez-Carrillo, A.; Romero-González, A.; et al. MtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]

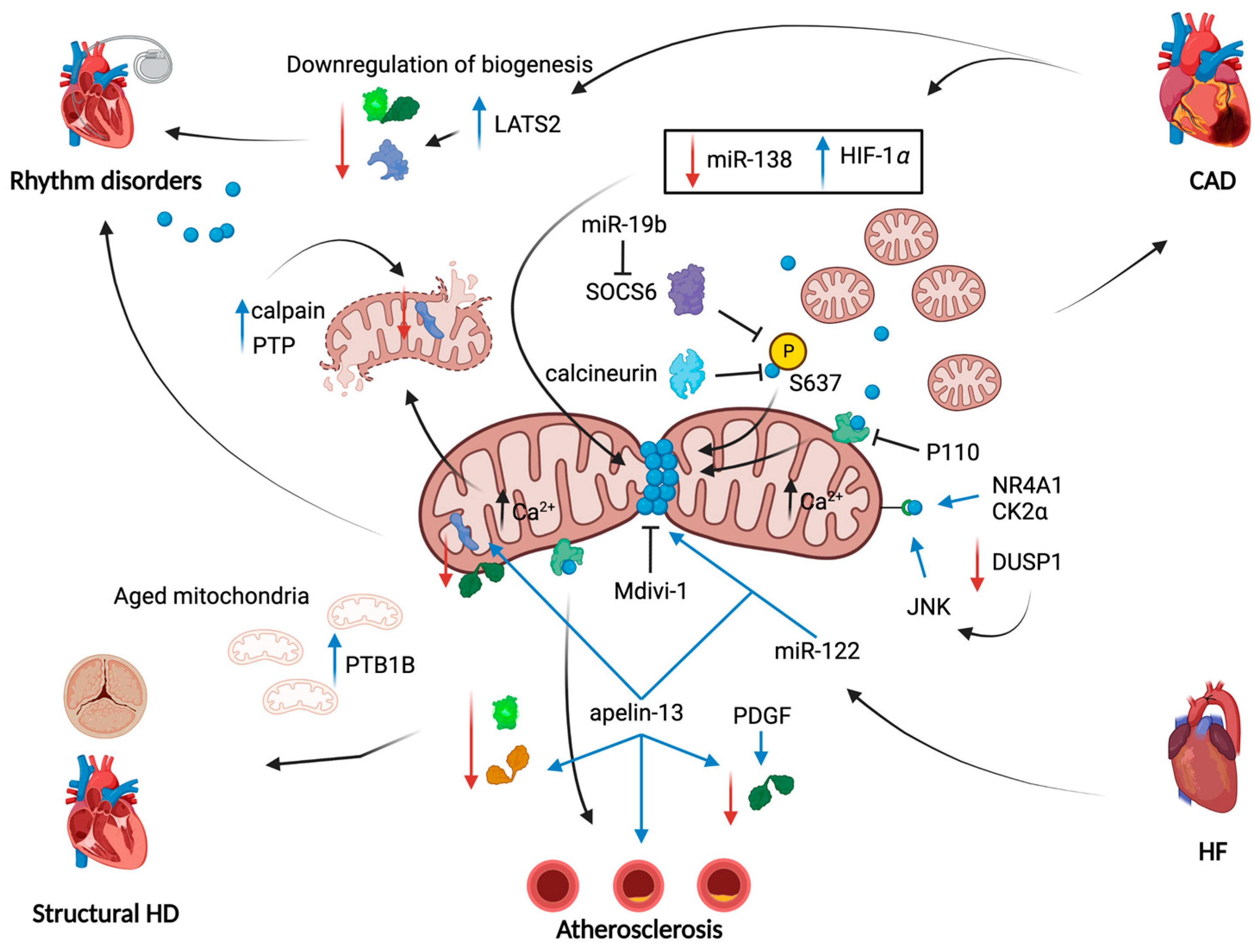

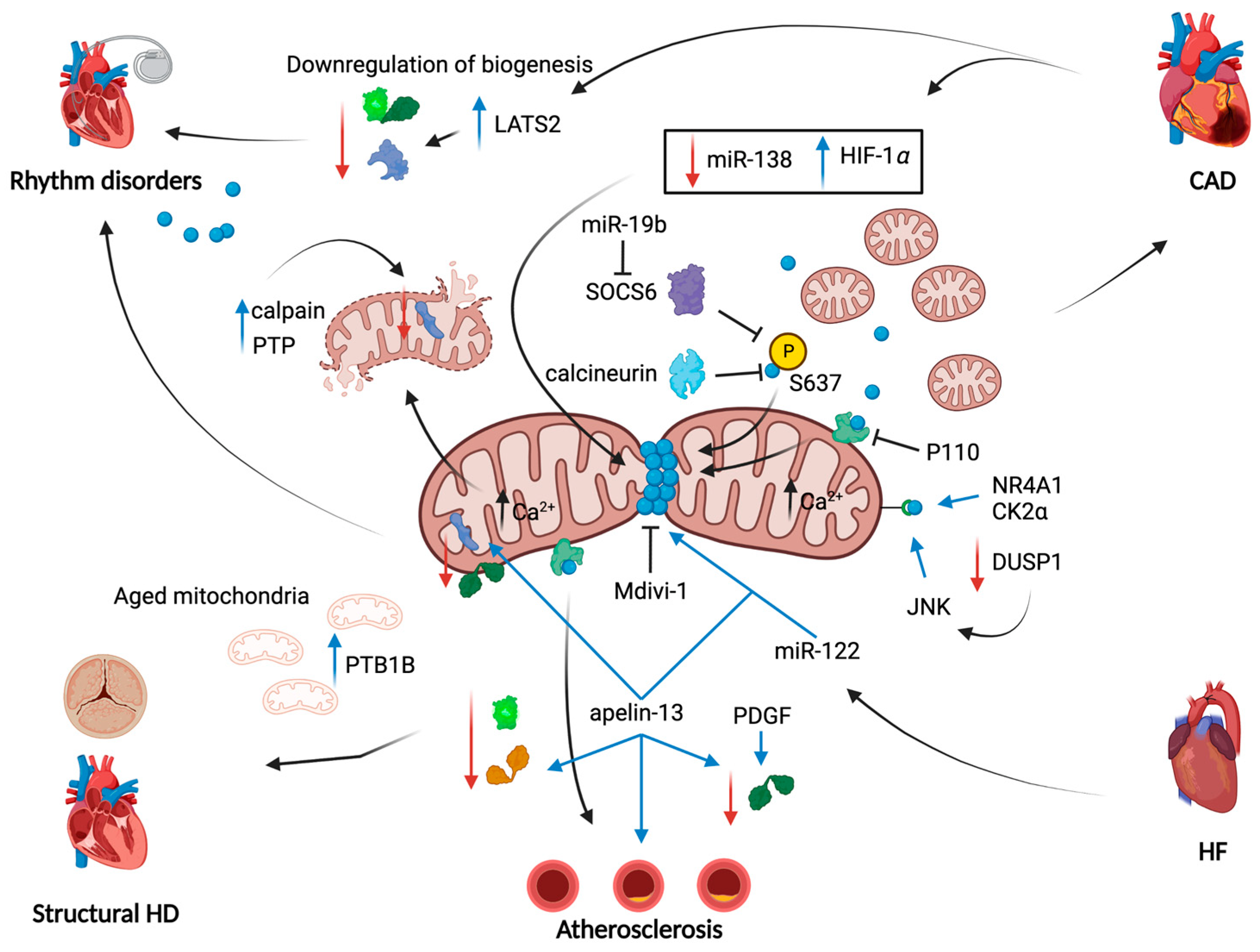
{kind=link}
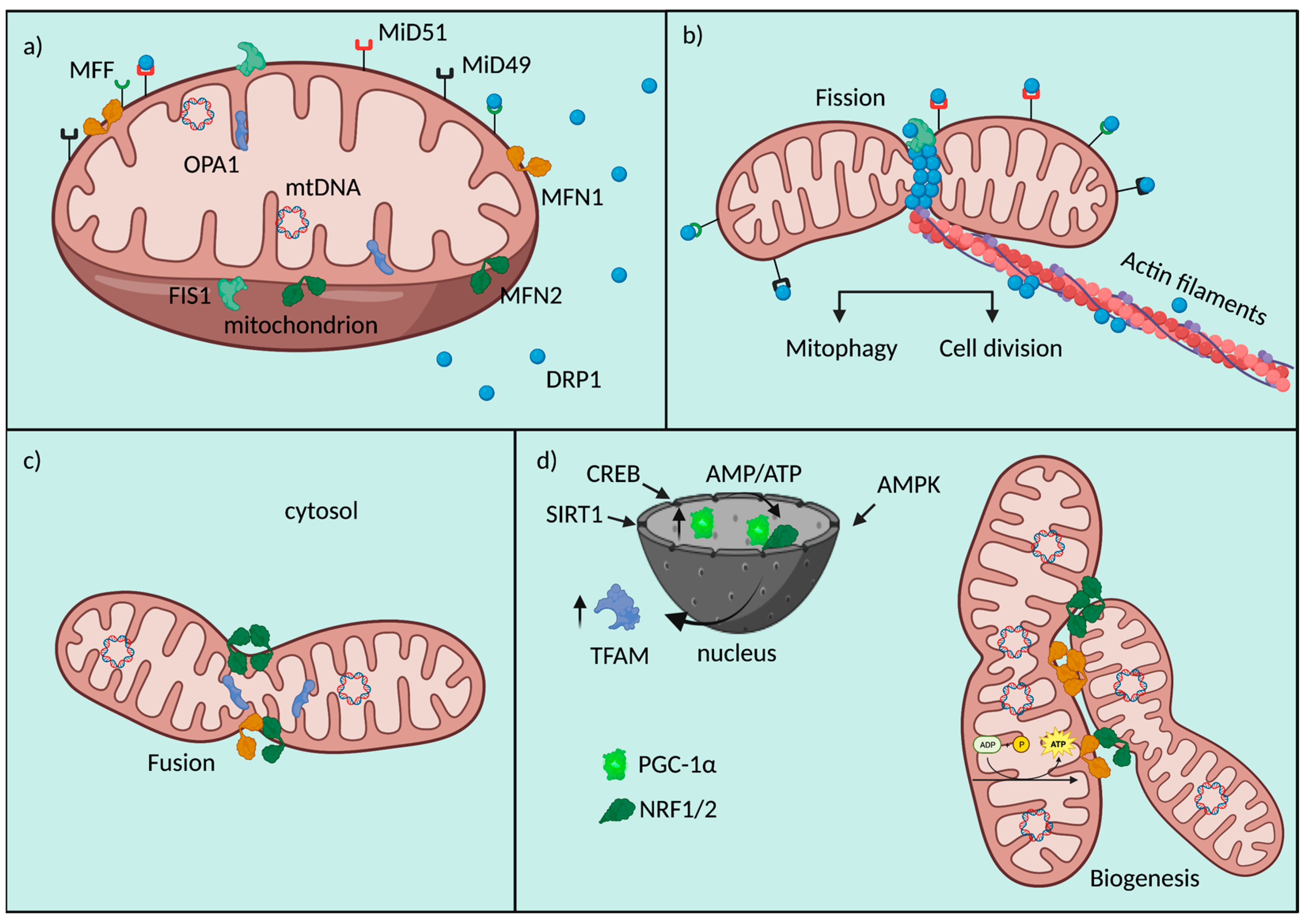
{kind=link}
| Cardiovascular Diseases | Protein Expression Alterations | Phenotype Manifestations | Ref. |
|---|---|---|---|
| Ischemia-reperfusion injury | DRP1 excessive activations | Increase of mitochondrial fragmentation ROS production, Ca2+ overload, cell death and LV impairment | [80,81,82,83] |
| Persistent DRP1 downregulation | Excessively elongated mitochondria, significant downregulation of the mitophagic process, LV dysfunction and cell death | ||
| MFF loss | Reduction of mitochondrial fission, microvascular mitochondrial structure amelioration | [84] | |
| MFF activation | Opening of PTP, apoptosis and infarct size expansion | [85] | |
| MFN2 and OPA1 reduction | Mitochondrial membrane depolarization and swelling | [86] | |
| OPA1 downregulation | Overactivation of mitochondrial fission and fusion, inhibition of mitophagy and apoptosis | [87] | |
| Heart Failure | OPA1 proteolysis upregulation | Enhanced mitochondrial fragmentation, increased cell death and metabolism alteration | [88] |
| OPA1, MFN2 downregulation and FIS1 upregulation | Mitochondrial impairment and apoptosis | [89,90] | |
| OPA1 upregulation | Improved cardiac function, reduced mitochondrial fragmentation | [91] | |
| DRP1 upregulation | Increase of mitochondrial fragmentation Increase mitochondrial fission and increase apoptosis Higher mitochondrial structural morphological modifications | [92,93] | |
| Coronary artery disease | OPA1 overexpression | Reduction of MI size | [91] |
| DRP1 inhibition | Preservation of mitochondrial network and morphology, reduction of MI size | [80,81,82] | |
| DRP1 inhibition/OPA1 proteolytic cleavage | Uncontrolled mitochondrial fission and mitochondrial alterations | [82] | |
| DRP1 upregulation | Reduction mitochondrial fission, mitochondrial dysfunction and apoptosis | [94] | |
| Atrial Fibrillation | Reduction of DRP1, MFN1 and OPA1 | Alteration in mitochondrial fission-fusion and mitochondrial biogenesis | [95] |
| MFN2 downregulation | Mitochondrial fragmentation | [96] | |
| Calcific aortic valve | DRP1 upregulation | High mitochondrial fragmentation | [97,98] |
| DRP1 inhibition | Reduction of the calcification process | ||
| Bicuspid aortic valve/thoracic aortic aneurysm | DRP1 and MFF reduction/MFN1 and MFN2 downregulation | Unbalanced mitochondrial dynamics | [99] |
| Cardiomyopathy | DRP1 Disruption | Elongated mitochondrial morphology and disruption in mitochondrial fission process, mitophagy alteration | [100,101] |
| MFN1/2 deletion | Severe mitochondrial fragmentation and reduced mtDNA | [102,103] | |
| Atherosclerosis | Upregulation of FIS1 expression | High mitochondrial fragmentation | [104,105] |
| Induction of DRP1 and FIS1 expression | Loss mitochondrial network | [97] | |
| DRP1 inhibition | Lower VSM calcification, matrix mineralization Reduction of mitochondrial degradation and prevent atherosclerosis lesions Restoration of mitochondria network and slow pulmonary artery smooth muscle proliferation cells | [106] | |
| OPA1 downregulation | Reduction of mitochondrial length | [107] | |
| MFN2 downregulation | Loss of mitochondrial network | [108] | |
| MFN2 overexpression | Reduction of VSMCs proliferation | [109,110] | |
| MFN1/2, OPA1 downregulation and DRP1 upregulation | Mitophagy impairment Accelerate atherosclerosis | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morciano, G.; Boncompagni, C.; Ramaccini, D.; Pedriali, G.; Bouhamida, E.; Tremoli, E.; Giorgi, C.; Pinton, P. Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases. Int. J. Mol. Sci. 2023, 24, 3414. https://doi.org/10.3390/ijms24043414
Morciano G, Boncompagni C, Ramaccini D, Pedriali G, Bouhamida E, Tremoli E, Giorgi C, Pinton P. Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases. International Journal of Molecular Sciences. 2023; 24(4):3414. https://doi.org/10.3390/ijms24043414
Chicago/Turabian StyleMorciano, Giampaolo, Caterina Boncompagni, Daniela Ramaccini, Gaia Pedriali, Esmaa Bouhamida, Elena Tremoli, Carlotta Giorgi, and Paolo Pinton. 2023. "Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases" International Journal of Molecular Sciences 24, no. 4: 3414. https://doi.org/10.3390/ijms24043414
APA StyleMorciano, G., Boncompagni, C., Ramaccini, D., Pedriali, G., Bouhamida, E., Tremoli, E., Giorgi, C., & Pinton, P. (2023). Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart Diseases. International Journal of Molecular Sciences, 24(4), 3414. https://doi.org/10.3390/ijms24043414