
Novel Variants of SOX4 in Patients with Intellectual Disability
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
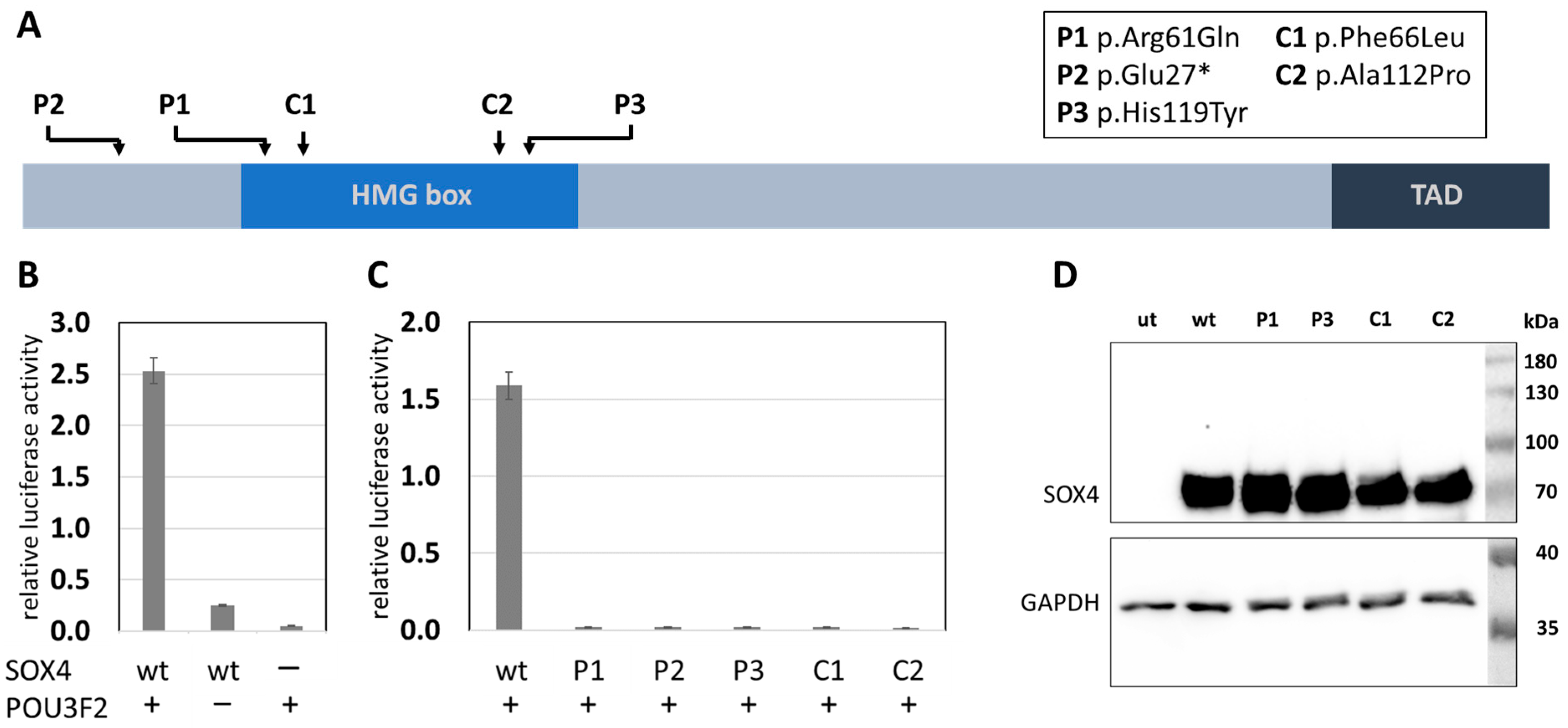
2. Results
3. Discussions
4. Material and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef]
- Geijsen, N.; Uings, I.J.; Pals, C.; Armstrong, J.; McKinnon, M.; Raaijmakers, J.A.; Lammers, J.-W.J.; Koenderman, L.; Coffer, P.J. Cytokine-specific transcriptional regulation through an IL-5Rα interacting protein. Science 2001, 293, 1136–1138. [Google Scholar] [CrossRef] [PubMed]
- Kuhlbrodt, K.; Herbarth, B.; Sock, E.; Enderich, J.; Hermans-Borgmeyer, I.; Wegner, M. Cooperative function of POU proteins and SOX proteins in glial cells. J. Biol. Chem. 1998, 273, 16050–16057. [Google Scholar] [CrossRef] [PubMed]
- Dy, P.; Penzo-Mendez, A.; Wang, H.; Pedraza, C.E.; Macklin, W.B.; Lefebvre, V. The three SoxC proteins—Sox4, Sox11 and Sox12—Exhibit overlapping expression patterns and molecular properties. Nucleic Acids Res. 2008, 36, 3101–3117. [Google Scholar] [CrossRef] [PubMed]
- Angelozzi, M.; Lefebvre, V. SOXopathies: Growing family of developmental disorders due to SOX mutations. Trends Genet. 2019, 35, 658–671. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Oosterwegel, M.; van Norren, K.; Clevers, H. Sox-4, an Sry-like HMG box protein, is a transcriptional activator in lymphocytes. EMBO J. 1993, 12, 3847–3854. [Google Scholar] [CrossRef]
- Zawerton, A.; Yao, B.; Yeager, J.P.; Pippucci, T.; Haseeb, A.; Smith, J.D.; Wischmann, L.; Kühl, S.J.; Dean, J.C.; Pilz, D.T. De novo SOX4 variants cause a neurodevelopmental disease associated with mild dysmorphism. Am. J. Hum. Genet. 2019, 104, 246–259. [Google Scholar] [CrossRef]
- Shim, S.; Kwan, K.Y.; Li, M.; Lefebvre, V.; Šestan, N. Cis-regulatory control of corticospinal system development and evolution. Nature 2012, 486, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Tsurusaki, Y.; Koshimizu, E.; Ohashi, H.; Phadke, S.; Kou, I.; Shiina, M.; Suzuki, T.; Okamoto, N.; Imamura, S.; Yamashita, M. De novo SOX11 mutations cause Coffin–Siris syndrome. Nat. Commun. 2014, 5, 4011. [Google Scholar] [CrossRef] [Green Version]
- Angelozzi, M.; Karvande, A.; Molin, A.N.; Ritter, A.L.; Leonard, J.M.M.; Savatt, J.M.; Douglass, K.; Myers, S.M.; Grippa, M.; Tolchin, D.; et al. Consolidation of the clinical and genetic definition of a SOX4-related neurodevelopmental syndrome. J. Med. Genet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ghaffar, A.; Rasheed, F.; Rashid, M.; van Bokhoven, H.; Ahmed, Z.M.; Riazuddin, S.; Riazuddin, S. Biallelic in-frame deletion of SOX4 is associated with developmental delay, hypotonia and intellectual disability. Eur. J. Hum. Genet. 2022, 30, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Jauch, R.; Ng, C.K.; Narasimhan, K.; Kolatkar, P.R. The crystal structure of the Sox4 HMG domain–DNA complex suggests a mechanism for positional interdependence in DNA recognition. Biochem. J. 2012, 443, 39–47. [Google Scholar] [CrossRef]
- Malki, S.; Boizet-Bonhoure, B.; Poulat, F. Shuttling of SOX proteins. Int. J. Biochem. Cell. Biol. 2010, 42, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Love, D.C.; Prinz, W.A. Calmodulin-driven nuclear entry: Trigger for sex determination and terminal differentiation. J. Biol. Chem. 2009, 284, 12593–12597. [Google Scholar] [CrossRef]
- Wilke, C.O. Transcriptional robustness complements nonsense-mediated decay in humans. PLoS Genet. 2011, 7, e1002296. [Google Scholar] [CrossRef] [PubMed]
- Reményi, A.; Lins, K.; Nissen, L.J.; Reinbold, R.; Schöler, H.R.; Wilmanns, M. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev. 2003, 17, 2048–2059. [Google Scholar] [CrossRef]
- Kuwahara, M.; Yamashita, M.; Shinoda, K.; Tofukuji, S.; Onodera, A.; Shinnakasu, R.; Motohashi, S.; Hosokawa, H.; Tumes, D.; Iwamura, C. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-β and suppresses TH 2 differentiation. Nat. Immunol. 2012, 13, 778–786. [Google Scholar] [CrossRef]
- Kamachi, Y.; Cheah, K.S.; Kondoh, H. Mechanism of regulatory target selection by the SOX high-mobility-group domain proteins as revealed by comparison of SOX1/2/3 and SOX9. Mol. Cell. Biol. 1999, 19, 107–120. [Google Scholar] [CrossRef]
- Cameron, F.J.; Hageman, R.M.; Cooke-Yarborough, C.; Kwok, C.; Goodwin, L.L.; Sillence, D.O.; Sinclair, A.H. A novel germ line mutation in SOX9 causes familial campomelic dysplasia and sex reversal. Hum. Mol. Genet. 1996, 5, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Benko, S.; Gordon, C.T.; Mallet, D.; Sreenivasan, R.; Thauvin-Robinet, C.; Brendehaug, A.; Thomas, S.; Bruland, O.; David, M.; Nicolino, M. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J. Med. Genet. 2011, 48, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.; Weller, P.A.; Guioli, S.; Foster, J.W.; Mansour, S.; Zuffardi, O.; Punnett, H.H.; Dominguez-Steglich, M.A.; Brook, J.D.; Young, I.D. Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am. J. Hum. Genet. 1995, 57, 1028. [Google Scholar] [PubMed]
- Pillai-Kastoori, L.; Wen, W.; Wilson, S.G.; Strachan, E.; Lo-Castro, A.; Fichera, M.; Musumeci, S.A.; Lehmann, O.J.; Morris, A.C. Sox11 is required to maintain proper levels of Hedgehog signaling during vertebrate ocular morphogenesis. PLoS Genet. 2014, 10, e1004491. [Google Scholar] [CrossRef] [PubMed]
- Zawerton, A.; Mignot, C.; Sigafoos, A.; Blackburn, P.R.; Haseeb, A.; McWalter, K.; Ichikawa, S.; Nava, C.; Keren, B.; Charles, P. Widening of the genetic and clinical spectrum of Lamb–Shaffer syndrome, a neurodevelopmental disorder due to SOX5 haploinsufficiency. Genet. Med. 2020, 22, 524–537. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grosse, M.; Kuechler, A.; Dabir, T.; Spranger, S.; Beck-Wödl, S.; Bertrand, M.; Haack, T.B.; Grasemann, C.; Manka, E.; Depienne, C.; et al. Novel Variants of SOX4 in Patients with Intellectual Disability. Int. J. Mol. Sci. 2023, 24, 3519. https://doi.org/10.3390/ijms24043519
Grosse M, Kuechler A, Dabir T, Spranger S, Beck-Wödl S, Bertrand M, Haack TB, Grasemann C, Manka E, Depienne C, et al. Novel Variants of SOX4 in Patients with Intellectual Disability. International Journal of Molecular Sciences. 2023; 24(4):3519. https://doi.org/10.3390/ijms24043519
Chicago/Turabian StyleGrosse, Martin, Alma Kuechler, Tabib Dabir, Stephanie Spranger, Stefanie Beck-Wödl, Miriam Bertrand, Tobias B. Haack, Corinna Grasemann, Eva Manka, Christel Depienne, and et al. 2023. "Novel Variants of SOX4 in Patients with Intellectual Disability" International Journal of Molecular Sciences 24, no. 4: 3519. https://doi.org/10.3390/ijms24043519