
bioTCIs: Middle-to-Macro Biomolecular Targeted Covalent Inhibitors Possessing Both Semi-Permanent Drug Action and Stringent Target Specificity as Potential Antibody Replacements

Abstract
:1. Introduction
Targeted Covalent Inhibitors (TCI) as Potential Antibody Replacements
2. History and General Principle of bioTCI
2.1. From Small Molecular TCI to bioTCI
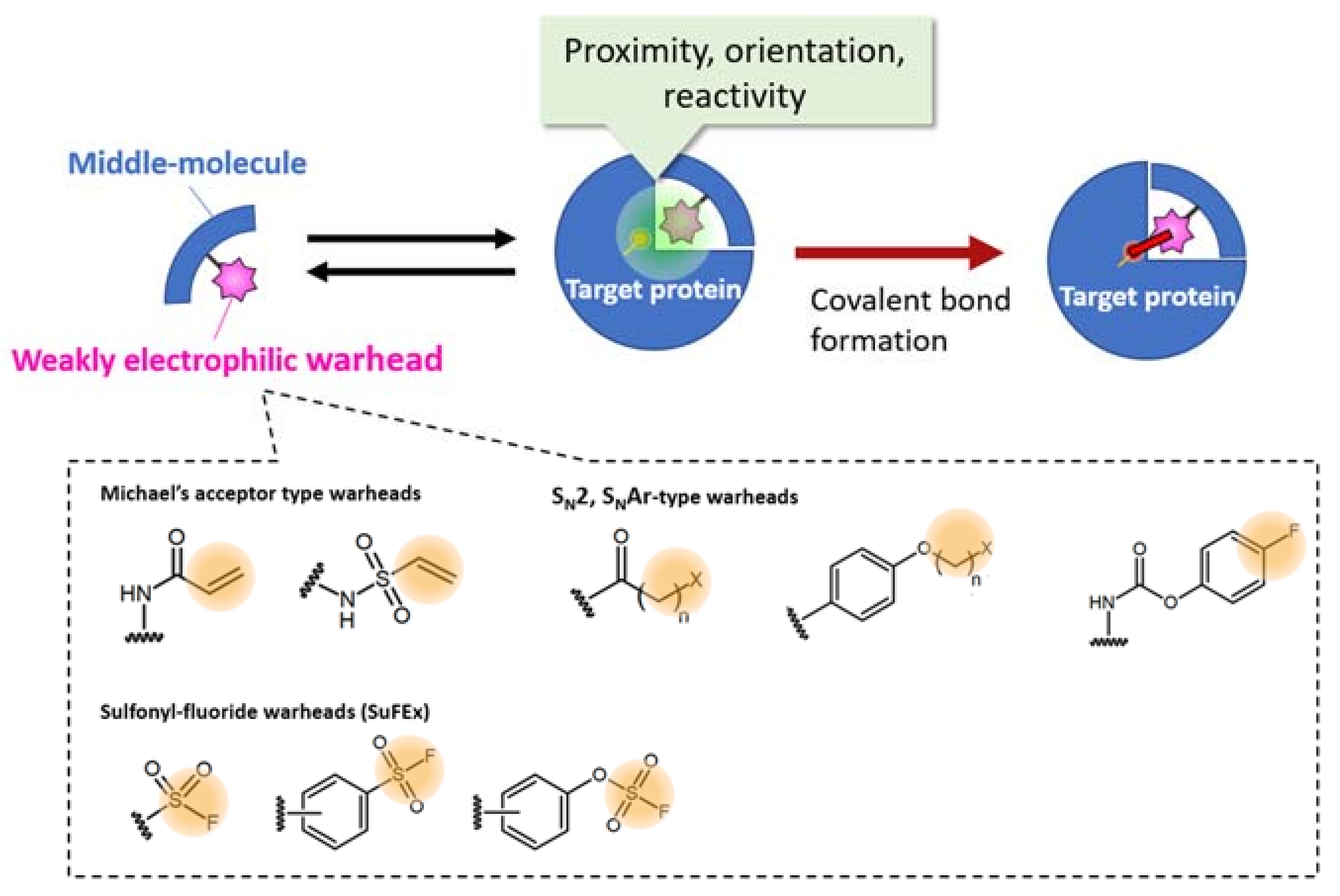
2.2. Warhead Design and Introduction into Middle/Macro-Biomolecules
2.3. Pros and Cons of bioTCI over Non-Covalent Biomolecular Targeted Inhibitors
3. Recent Hot Topics of bioTci
3.1. Combinatorial Screening of Peptidic TCI: A Well-Developed Modality
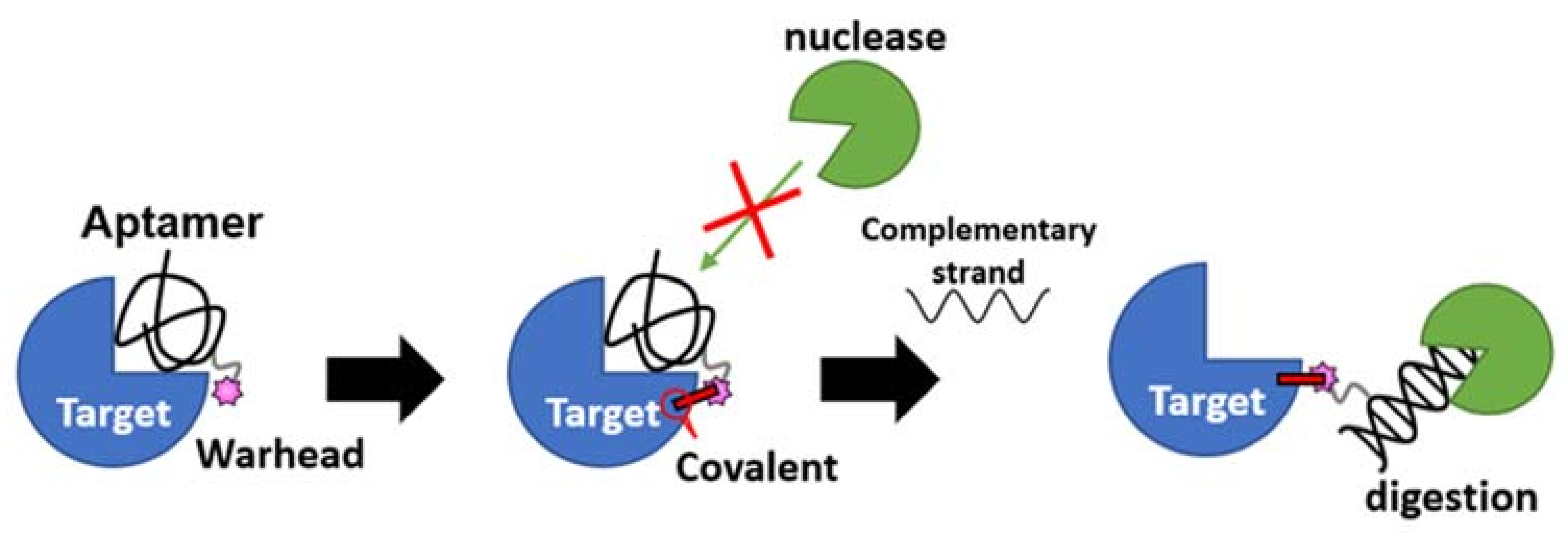
3.2. Nucleotidic TCI: A Developing Modality
4. Future Perspectives of bioTCI Research: Technical Challenges and Critical Questions That Need to Be Answered
4.1. Design and Selection Methodology of bioTCI
4.2. Beyond the Target Affinity/Specificity: Additional Functionalization of bioTCI
4.3. Other Possibilities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADE: | adverse drug effect |
| AFS: | aryl-fluorosulfate (aryl-OSO2F) |
| bioTCI: | biomolecular targeted covalent inhibitor |
| CD: | cluster of differentiation |
| CHO: | Chinese hamster ovary |
| CS: | complimentary strand |
| CuAAC: | copper(I)-catalyzed azide-alkyne cycloaddition |
| Da: | Dalton |
| DS: | double strand |
| EdUTP: | 5-ethynyl-dUTP |
| EGFR: | epidermal growth factor receptor |
| ELISA: | enzyme-linked immunosorbent assay |
| Fab: | fragment antigen binding (variable) |
| FasL: | Fas ligand (CD95L, CD178) |
| Fc: | fragment crystallizable (constant) |
| FcR: | Fc receptor |
| FDA: | Food and Drug Administration (USA |
| GPCR: | G-protein-coupled receptor |
| HER2: | human epidermal growth factor 2 |
| mAb: | monoclonal antibody |
| NK: | natural killer |
| NGS: | next-generation sequencing |
| OctdU: | octadiynyl-dU |
| PCR: | polymerase chain reaction |
| PD1: | programmed cell death protein 1 (CD279) |
| PD-L1: | programmed death-ligand 1 (CD274) |
| PK: | pharmacokinetic |
| PPI: | protein-protein interaction |
| PS: | phosphorothioate |
| RBD: | receptor binding domain |
| SARS-CoV-2: | severe acute respiratory syndrome-associated coronavirus-2 |
| SELEX: | Systematic Evolution of Ligands by EXponential enrichment |
| SN2: | bimolecular nucleophilic substitution |
| SNAr: | nucleophilic aromatic substitution |
| SuFEx: | sulfur (VI) fluoride exchange |
| SDS: | sodium dodecyl sulfate |
| TBA: | thrombin binding aptamer |
| TCI: | targeted covalent inhibitor |
| TeTCI: | tethered targeted covalent inhibitor |
| TRAIL: | TNF-related apoptosis-inducing ligand |
| Uaa: | unnatural amino acid |
References
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2019, 109, 74–103. [Google Scholar] [CrossRef] [PubMed]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Zhao, Q.; Hui, J.; Wang, T.; Lin, M.; Wang, K.; Zhang, J.; Shentu, J.; Dalby, P.A.; Zhang, H.; et al. The Global Landscape of Approved Antibody Therapies. Antib. Ther. 2022, 5, 233–257. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C.; Oshiro, M.Y.; Albericio, F.; de la Torre, B.G.; Pereira, G.J.V.; Gonzaga, R.V. Trends and Perspectives of Biological Drug Approvals by the FDA: A Review from 2015 to 2021. Biomedicines 2022, 10, 2325. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Ma, H.; Ó’Fágáin, C.; O’Kennedy, R. Antibody stability: A key to performance—Analysis, influences and improvement. Biochimie 2020, 177, 213–225. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.Z.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Fu, Z.; Xiang, J. Aptamers, the Nucleic Acid Antibodies, in Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 2793. [Google Scholar] [CrossRef]
- Lundin, K.E.; Gissberg, O.; Smith, C.I.E. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef] [PubMed]
- DeFrees, S.A.; Phillips, L.; Guo, L.; Zalipsky, S. Sialyl Lewis x Liposomes as a Multivalent Ligand and Inhibitor of E-Selectin Mediated Cellular Adhesion. J. Am. Chem. Soc. 1996, 118, 6101–6104. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- De Vita, E. 10 years into the resurgence of covalent drugs. Futur. Med. Chem. 2021, 13, 193–210. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2018, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Potashman, M.H.; Duggan, M.E. Covalent Modifiers: An Orthogonal Approach to Drug Design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Halford, B. Covalent drugs go from fringe field to fashionable endeavor. Chem. Eng. News 2020, 98, 28–33. [Google Scholar] [CrossRef]
- Roth, G.J.; Majerus, P.W. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J. Clin. Investig. 1975, 56, 624–632. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef]
- Singh, J.; Dobrusin, E.M.; Fry, D.W.; Haske, T.; Whitty, A.; McNamara, D.J. Structure-Based Design of a Potent, Selective, and Irreversible Inhibitor of the Catalytic Domain of the erbB Receptor Subfamily of Protein Tyrosine Kinases. J. Med. Chem. 1997, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Bridges, A.J.; Denny, W.A.; Doherty, A.; Greis, K.D.; Hicks, J.L.; Hook, K.E.; Keller, P.R.; Leopold, W.R.; Loo, J.A.; et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 12022–12027. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.S.; Gupta, R.; Liguori, M.J.; Hu, M.; Huang, X.; Mantena, S.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Van Vleet, T.R. Novel Computational Approach to Predict Off-Target Interactions for Small Molecules. Front. Big Data 2019, 2, 25. [Google Scholar] [CrossRef]
- Lanning, B.R.; Whitby, L.R.; Dix, M.M.; Douhan, J.; Gilbert, A.M.; Hett, E.C.; Johnson, T.O.; Joslyn, C.M.; Kath, J.C.; Niessen, S.; et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. [Google Scholar] [CrossRef]
- Paulussen, F.M.; Grossmann, T.N. Peptide-based covalent inhibitors of protein–protein interactions. J. Pept. Sci. 2022, 29, e3457. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Yang, J.; Taki, M. Inhibition of thrombin activity by a covalent-binding aptamer and reversal by the complementary strand antidote. Chem. Commun. 2021, 57, 2483–2486. [Google Scholar] [CrossRef]
- Berdan, V.Y.; Klauser, P.C.; Wang, L. Covalent peptides and proteins for therapeutics. Bioorganic Med. Chem. 2020, 29, 115896. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Q.; Klauser, P.C.; Li, M.; Zheng, F.; Wang, N.; Li, X.; Zhang, Q.; Fu, X.; Wang, Q.; et al. Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 2020, 182, 85–97.e16. [Google Scholar] [CrossRef]
- Gambini, L.; Baggio, C.; Udompholkul, P.; Jossart, J.; Salem, A.F.; Perry, J.J.P.; Pellecchia, M. Covalent Inhibitors of Protein–Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J. Med. Chem. 2019, 62, 5616–5627. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, Y.; Katsuki, R.; Ito, Y.; Taki, M. Direct Combinatorial Screening of a Target-Specific Covalent Binding Peptide: Ac-tivation of the Warhead Reactivity in a Pseudo-Catalytic Microenvironment. Pept. Sci. 2021, 2022, 15–16. [Google Scholar]
- Tabuchi, Y.; Yang, J.; Taki, M. Relative Nuclease Resistance of a DNA Aptamer Covalently Conjugated to a Target Protein. Int. J. Mol. Sci. 2022, 23, 7778. [Google Scholar] [CrossRef] [PubMed]
- Tivon, Y.; Falcone, G.; Deiters, A. Protein Labeling and Crosslinking by Covalent Aptamers. Angew. Chem. Int. Ed. 2021, 60, 15899–15904. [Google Scholar] [CrossRef] [PubMed]
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorganic Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265. [Google Scholar]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 345–382. [Google Scholar] [CrossRef]
- McAulay, K.; Bilsland, A.; Bon, M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals 2022, 15, 1366. [Google Scholar] [CrossRef]
- Carneiro, S.N.; Khasnavis, S.R.; Lee, J.; Butler, T.W.; Majmudar, J.D.; Ende, C.W.A.; Ball, N.D. Sulfur(vi) fluorides as tools in biomolecular and medicinal chemistry. Org. Biomol. Chem. 2023. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, J.; Huang, Z.; Yang, Y.; Fu, T.; Yang, Y.; Lyu, Y.; Jiang, J.; Qiu, L.; Cao, Z.; et al. Robust Covalent Aptamer Strategy Enables Sensitive Detection and Enhanced Inhibition of SARS-CoV-2 Proteins. ACS Central Sci. 2023, 9, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.R. Tissue-Specific Irreversible Inhibitors of Dihydrofolic Reductase1. Acc. Chem. Res. 1969, 2, 129–136. [Google Scholar] [CrossRef]
- Dong, J.; Krasnova, L.; Finn, M.G.; Sharpless, K.B. Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.S.; Smedley, C.J.; Zheng, Q.; Li, S.; Dong, J.; Moses, J.E. The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 2019, 48, 4731–4758. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Woehl, J.L.; Kitamura, S.; Santos-Martins, D.; Smedley, C.J.; Li, G.; Forli, S.; Moses, J.E.; Wolan, D.W.; Sharpless, K.B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814. [Google Scholar] [CrossRef]
- Narayanan, A.; Jones, L.H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659. [Google Scholar] [CrossRef]
- Mukherjee, H.; Debreczeni, J.; Breed, J.; Tentarelli, S.; Aquila, B.; Dowling, J.E.; Whitty, A.; Grimster, N.P. A study of the reactivity of S(VI)–F containing warheads with nucleophilic amino-acid side chains under physiological conditions. Org. Biomol. Chem. 2017, 15, 9685–9695. [Google Scholar] [CrossRef]
- Hoppmann, C.; Wang, L. Proximity-enabled bioreactivity to generate covalent peptide inhibitors of p53–Mdm4. Chem. Commun. 2016, 52, 5140–5143. [Google Scholar] [CrossRef]
- Xiang, Z.; Ren, H.; Hu, Y.S.; Coin, I.; Wei, J.; Cang, H.; Wang, L. Adding an unnatural covalent bond to proteins through proximity-enhanced bioreactivity. Nat. Methods 2013, 10, 885–888. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Watanabe, T.; Katsuki, R.; Ito, Y.; Taki, M. Direct screening of a target-specific covalent binder: Stringent regulation of warhead reactivity in a matchmaking environment. Chem. Commun. 2021, 57, 5378–5381. [Google Scholar] [CrossRef]
- Guldalian, J., Jr.; Lawson, W.B.; Brown, R.K. Formation of a Covalent Linkage between Bromoacetylated Antigen and Its Antibody. J. Biol. Chem. 1965, 240, PC2757–PC2759. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Hubbell, G.E.; Tepe, J.J. Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors. RSC Chem. Biol. 2020, 1, 305–332. [Google Scholar] [CrossRef]
- Hanada, M.; Sugawara, K.; Kaneta, K.; Toda, S.; Nishiyama, Y.; Tomita, K.; Yamamoto, H.; Konishi, M.; Oki, T. Epoxomicin, a new antitumor agent of microbial origin. J. Antibiot. 1992, 45, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McClure, R.A.; Zheng, Y.; Thomson, R.J.; Kelleher, N.L. Proteomics Guided Discovery of Flavopeptins: Anti-proliferative Aldehydes Synthesized by a Reductase Domain-Containing Non-ribosomal Peptide Synthetase. J. Am. Chem. Soc. 2013, 135, 10449–10456. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898. [Google Scholar] [CrossRef]
- Wang, L. Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Accounts Chem. Res. 2017, 50, 2767–2775. [Google Scholar] [CrossRef]
- Yu, B.; Li, S.; Tabata, T.; Wang, N.; Cao, L.; Kumar, G.R.; Sun, W.; Liu, J.; Ott, M.; Wang, L. Accelerating PERx reaction enables covalent nanobodies for potent neutralization of SARS-CoV-2 and variants. Chem 2022, 8, 2766–2783. [Google Scholar] [CrossRef]
- Han, Y.; Yang, Z.; Hu, H.; Zhang, H.; Chen, L.; Li, K.; Kong, L.; Wang, Q.; Liu, B.; Wang, M.; et al. Covalently Engineered Protein Minibinders with Enhanced Neutralization Efficacy against Escaping SARS-CoV-2 Variants. J. Am. Chem. Soc. 2022, 144, 5702–5707. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, L. New covalent bonding ability for proteins. Protein Sci. 2021, 31, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. Adv. Sci. Drug Discov. 2017, 22, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Ward, R.A.; Doig, P.; Argyrou, A. Insight into the Therapeutic Selectivity of the Irreversible EGFR Tyrosine Kinase Inhibitor Osimertinib through Enzyme Kinetic Studies. Biochemistry 2020, 59, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Mons, E.; Roet, S.; Kim, R.Q.; Mulder, M.P.C. A Comprehensive Guide for Assessing Covalent Inhibition in Enzymatic Assays Illustrated with Kinetic Simulations. Curr. Protoc. 2022, 2, e419. [Google Scholar] [CrossRef]
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. USA 2013, 111, 173–178. [Google Scholar] [CrossRef]
- Qin, Z.; Zhu, Y.; Xiang, Y. Covalent inhibition of SARS-CoV-2 RBD-ACE2 interaction by aptamers with multiple sulfur(VI) fluoride exchange modifications. ChemRxiv 2021, 1–22. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Uematsu, S.; Tabuchi, Y.; Ito, Y.; Taki, M. Combinatorially Screened Peptide as Targeted Covalent Binder: Alteration of Bait-Conjugated Peptide to Reactive Modifier. Bioconjugate Chem. 2018, 29, 1866–1871. [Google Scholar] [CrossRef]
- Chen, S.; Lovell, S.; Lee, S.; Fellner, M.; Mace, P.D.; Bogyo, M. Identification of highly selective covalent inhibitors by phage display. Nat. Biotechnol. 2020, 39, 490–498. [Google Scholar] [CrossRef]
- Choi, E.J.; Jung, D.; Kim, J.-S.; Lee, Y.; Kim, B.M. Chemoselective Tyrosine Bioconjugation through Sulfate Click Reaction. Chem. A Eur. J. 2018, 24, 10948–10952. [Google Scholar] [CrossRef] [PubMed]
- Bolding, J.E.; Martín-Gago, P.; Rajabi, N.; Gamon, L.F.; Hansen, T.N.; Bartling, C.R.O.; Strømgaard, K.; Davies, M.J.; Olsen, C.A. Aryl Fluorosulfate Based Inhibitors That Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem. Int. Ed. 2022, 61, e202204565. [Google Scholar] [CrossRef] [PubMed]
- van der Zouwen, A.J.; Witte, M.D. Modular Approaches to Synthesize Activity- and Affinity-Based Chemical Probes. Front. Chem. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Taki, M. Practical Tips for Construction of Custom Peptide Libraries and Affinity Selection by Using Commercially Available Phage Display Cloning Systems. J. Nucl. Acids 2012, 2012, 295719. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Chen, F.-J.; Li, K.; Reja, R.M.; Haeffner, F.; Gao, J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893. [Google Scholar] [CrossRef]
- Serafimova, I.M.; Pufall, M.; Krishnan, S.; Duda, K.; Cohen, M.S.; Maglathlin, R.L.; McFarland, J.M.; Miller, R.M.; Frödin, M.; Taunton, J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Gao, J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016, 34, 110–116. [Google Scholar] [CrossRef]
- Abe, T.; Tabuchi, Y.; Taki, M. Reversal of covalent conjugation of benzoxaborole-modified targeted peptide by reduction. Pept. Sci. 2021, 2022, 17–18. [Google Scholar]
- Graham, B.J.; Windsor, I.W.; Gold, B.; Raines, R.T. Boronic acid with high oxidative stability and utility in biological contexts. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Byun, J. Recent Progress and Opportunities for Nucleic Acid Aptamers. Life 2021, 11, 193. [Google Scholar] [CrossRef]
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Nakamura, Y. Aptamers: A Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef]
- Xiao, X.; Li, H.; Zhao, L.; Zhang, Y.; Liu, Z. Oligonucleotide aptamers: Recent advances in their screening, molecular conformation and therapeutic applications. Biomed. Pharmacother. 2021, 143, 112232. [Google Scholar] [CrossRef]
- Smith, D.; Kirschenheuter, G.P.; Charlton, J.; Guidot, D.M.; Repine, J.E. In vitro selection of RNA-based irreversible inhibitors of human neutrophil elastase. Chem. Biol. 1995, 2, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.; Kirschenheuter, G.P.; Smith, D. Highly Potent Irreversible Inhibitors of Neutrophil Elastase Generated by Selection from a Randomized DNA−Valine Phosphonate Library. Biochemistry 1997, 36, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Tamura, T.; Kawano, M.; Shiono, K.; Hobor, F.; Wilson, A.J.; Hamachi, I. Enhanced Suppression of a Protein–Protein Interaction in Cells Using Small-Molecule Covalent Inhibitors Based on an N-Acyl-N-alkyl Sulfonamide Warhead. J. Am. Chem. Soc. 2021, 143, 4766–4774. [Google Scholar] [CrossRef] [PubMed]
- Corso, A.D.; Catalano, M.; Schmid, A.; Scheuermann, J.; Neri, D. Affinity Enhancement of Protein Ligands by Reversible Covalent Modification of Neighboring Lysine Residues. Angew. Chem. Int. Ed. 2018, 57, 17178–17182. [Google Scholar] [CrossRef]
- Krauss, I.R.; Merlino, A.; Randazzo, A.; Novellino, E.; Mazzarella, L.; Sica, F. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012, 40, 8119–8128. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef]
- Wakui, K.; Yoshitomi, T.; Yamaguchi, A.; Tsuchida, M.; Saito, S.; Shibukawa, M.; Furusho, H.; Yoshimoto, K. Rapidly Neutralizable and Highly Anticoagulant Thrombin-Binding DNA Aptamer Discovered by MACE SELEX. Mol. Ther. - Nucleic Acids 2019, 16, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Liu, S.; Wei, X.; Wan, S.; Huang, M.; Song, T.; Lu, Y.; Weng, X.; Lin, Z.; Chen, H.; et al. Aptamer Blocking Strategy Inhibits SARS-CoV-2 Virus Infection. Angew. Chem. Int. Ed. 2021, 60, 10266–10272. [Google Scholar] [CrossRef] [PubMed]
- Chrominski, M.; Ziemkiewicz, K.; Kowalska, J.; Jemielity, J. Introducing SuFNucs: Sulfamoyl-Fluoride-Functionalized Nucleosides That Undergo Sulfur Fluoride Exchange Reaction. Org. Lett. 2022, 24, 4977–4981. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, A.; Ferenczy, G.G.; Keserű, G.M. Covalent Docking in Drug Discovery: Scope and Limitations. Curr. Pharm. Des. 2020, 26, 5684–5699. [Google Scholar] [CrossRef]
- Oyedele, A.-Q.K.; Ogunlana, A.T.; Boyenle, I.D.; Adeyemi, A.O.; Rita, T.O.; Adelusi, T.I.; Abdul-Hammed, M.; Elegbeleye, O.E.; Odunitan, T.T. Docking covalent targets for drug discovery: Stimulating the computer-aided drug design community of possible pitfalls and erroneous practices. Mol. Divers. 2022, 1–25. [Google Scholar] [CrossRef]
- Pfeiffer, F.; Tolle, F.; Rosenthal, M.; Brändle, G.M.; Ewers, J.; Mayer, G. Identification and characterization of nucleobase-modified aptamers by click-SELEX. Nat. Protoc. 2018, 13, 1153–1180. [Google Scholar] [CrossRef]
- Schmitz, A.; Weber, A.; Bayin, M.; Breuers, S.; Fieberg, V.; Famulok, M.; Mayer, G. A SARS-CoV-2 Spike Binding DNA Aptamer that Inhibits Pseudovirus Infection by an RBD-Independent Mechanism. Angew. Chem. Int. Ed. 2021, 60, 10279–10285. [Google Scholar] [CrossRef]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Gao, F.; Yin, J.; Chen, Y.; Guo, C.; Hu, H.; Su, J. Recent advances in aptamer-based targeted drug delivery systems for cancer therapy. Front. Bioeng. Biotechnol. 2022, 10, 972933. [Google Scholar] [CrossRef]
- Allemailem, K.S.; Almatroudi, A.; Alsahli, M.A.; Basfar, G.T.; Alrumaihi, F.; Rahmani, A.H.; Khan, A.A. Recent advances in understanding oligonucleotide aptamers and their applications as therapeutic agents. 3 Biotech 2020, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Haßel, S.K.; Mayer, G. Aptamers as Therapeutic Agents: Has the Initial Euphoria Subsided? Mol. Diagn. Ther. 2019, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Boshtam, M.; Asgary, S.; Kouhpayeh, S.; Shariati, L.; Khanahmad, H. Aptamers Against Pro- and Anti-Inflammatory Cytokines: A Review. Inflammation 2016, 40, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Ha, S.-J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320. [Google Scholar] [CrossRef]
- Kraehenbuehl, L.; Weng, C.-H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2021, 19, 37–50. [Google Scholar] [CrossRef]
- McNamara, J.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig. 2008, 118, 376–386. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- Huang, B.-T.; Lai, W.-Y.; Chang, Y.-C.; Wang, J.-W.; Yeh, S.-D.; Lin, E.P.-Y.; Yang, P.-C. A CTLA-4 Antagonizing DNA Aptamer with Antitumor Effect. Mol. Ther. - Nucleic Acids 2017, 8, 520–528. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Meraviglia-Crivelli, D.H.; Menon, A.P.; Ruiz, M.; Cebollero, J.; Villalba, M.; Moreno, B.; Lozano, T.; Llopiz, D.; et al. ICOS Costimulation at the Tumor Site in Combination with CTLA-4 Blockade Therapy Elicits Strong Tumor Immunity. Mol. Ther. 2019, 27, 1878–1891. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Hervas, S.; Villanueva, H.; Lozano, T.; Rabal, O.; Oyarzabal, J.; Lasarte, J.J.; Bendandi, M.; Inogès, S.; De Cerio, A.L.-D.; et al. Identification of LAG3 high affinity aptamers by HT-SELEX and Conserved Motif Accumulation (CMA). PLOS ONE 2017, 12, e0185169. [Google Scholar] [CrossRef]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.-B.; Cydzik, M.; Gariépy, J. Targeting the PD-1/PD-L1 Immune Evasion Axis With DNA Aptamers as a Novel Therapeutic Strategy for the Treatment of Disseminated Cancers. Mol. Ther. - Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-F.; Chen, C.; Zeng, J.-Y.; Wang, S.; Han, S.-Q.; Zhang, Y.-Q.; Qiu, D.-D.; Guo, H.-X. Screening and characterization of aptamers for recombinant human programmed death-1 and recombinant extracellular domain of human programmed death ligand-1. Eur. Rev. Med Pharmacol. Sci. 2021, 25, 3997–4004. [Google Scholar] [CrossRef] [PubMed]
- Khedri, M.; Abnous, K.; Rafatpanah, H.; Nabavinia, M.S.; Taghdisi, S.M.; Ramezani, M. Development and Evaluation of Novel Aptamers Specific for Human PD1 Using Hybrid Systematic Evolution of Ligands by Exponential Enrichment Approach. Immunol. Investig. 2020, 49, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Pei, R. Isolation of DNA Aptamer Targeting PD-1 with an Antitumor Immunotherapy Effect. ACS Appl. Bio Mater. 2020, 3, 7080–7086. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Li, L.; Yang, G.; Qu, F. Investigating the Influences of Random-Region Length on Aptamer Selection Efficiency Based on Capillary Electrophoresis–SELEX and High-Throughput Sequencing. Anal. Chem. 2021, 93, 17030–17035. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, F.; Li, Y.; Yu, Y.; Liang, C.; Zhang, B.; Zhao, C.; Lu, A.; Zhang, G. A PD-L1 Aptamer Selected by Loss-Gain Cell-SELEX Conjugated with Paclitaxel for Treating Triple-Negative Breast Cancer. Med. Sci. Monit. 2020, 26, e925583. [Google Scholar] [CrossRef]
- Wang, H.; Lam, C.H.; Li, X.; West, D.L.; Yang, X. Selection of PD1/PD-L1 X-Aptamers. Biochimie 2017, 145, 125–130. [Google Scholar] [CrossRef]
- Ren, X.; Li, J.; Wu, X.; Zhao, J.; Yang, Q.; Lou, X. A highly specific aptamer probe targeting PD-L1 in tumor tissue sections: Mutation favors specificity. Anal. Chim. Acta 2021, 1185, 339066. [Google Scholar] [CrossRef]
- Malicki, S.; Pucelik, B.; Żyła, E.; Benedyk-Machaczka, M.; Gałan, W.; Golda, A.; Sochaj-Gregorczyk, A.; Kamińska, M.; Encarnação, J.C.; Chruścicka, B.; et al. Imaging of Clear Cell Renal Carcinoma with Immune Checkpoint Targeting Aptamer-Based Probe. Pharmaceuticals 2022, 15, 697. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Yu, H. Selection of threose nucleic acid aptamers to block PD-1/PD-L1 interaction for cancer immunotherapy. Chem. Commun. 2020, 56, 14653–14656. [Google Scholar] [CrossRef]
- Li, J.; Ren, X.; Zhao, J.; Lou, X. PD-L1 aptamer isolation via Modular-SELEX and its applications in cancer cell detection and tumor tissue section imaging. Anal. 2021, 146, 2910–2918. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.-Y.; Huang, B.-T.; Wang, J.-W.; Lin, P.-Y.; Yang, P.-C. A Novel PD-L1-targeting Antagonistic DNA Aptamer With Antitumor Effects. Mol. Ther. - Nucleic Acids 2016, 5, e397. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Mao, Z.; Li, W.; Pei, R. Anti-PD-L1 DNA aptamer antagonizes the interaction of PD-1/PD-L1 with antitumor effect. J. Mater. Chem. B 2020, 9, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Soldevilla, M.M.; Villanueva, H.; Mancheño, U.; Bendandi, M.; Pastor, F. Identification of TIM3 2′-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget 2015, 7, 4522–4530. [Google Scholar] [CrossRef]
- Gefen, T.; Castro, I.; Muharemagic, D.; Puplampu-Dove, Y.; Patel, S.; Gilboa, E. A TIM-3 Oligonucleotide Aptamer Enhances T Cell Functions and Potentiates Tumor Immunity in Mice. Mol. Ther. 2017, 25, 2280–2288. [Google Scholar] [CrossRef]
- Zhou, J.; Tiemann, K.; Chomchan, P.; Alluin, J.; Swiderski, P.; Burnett, J.; Zhang, X.; Forman, S.; Chen, R.; Rossi, J. Dual functional BAFF receptor aptamers inhibit ligand-induced proliferation and deliver siRNAs to NHL cells. Nucleic Acids Res. 2013, 41, 4266–4283. [Google Scholar] [CrossRef]
- Ho, D.-R.; Chang, P.-J.; Lin, W.-Y.; Huang, Y.-C.; Lin, J.-H.; Huang, K.-T.; Chan, W.-N.; Chen, C.-S. Beneficial Effects of Inflammatory Cytokine-Targeting Aptamers in an Animal Model of Chronic Prostatitis. Int. J. Mol. Sci. 2020, 21, 3953. [Google Scholar] [CrossRef]
- Momeni, M.; Mashayekhi, K.; Navashenaq, J.G.; Sankian, M. Identification of G-quadruplex anti-Interleukin-2 aptamer with high specificity through SELEX stringency. Heliyon 2022, 8. [Google Scholar] [CrossRef]
- Khanahmad, H.; Jamalvandi, M.; Irani, S.; Bastaminezhad, S. Selection and characterization of single-stranded DNA aptamers against interleukin-5. Res. Pharm. Sci. 2019, 14, 515–523. [Google Scholar] [CrossRef]
- Meyer, C.; Berg, K.; Eydeler-Haeder, K.; Lorenzen, I.; Grötzinger, J.; Rose-John, S.; Hahn, U. Stabilized Interleukin-6 receptor binding RNA aptamers. RNA Biol. 2013, 11, 57–65. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hirota, M.; Waugh, S.M.; Murakami, I.; Suzuki, T.; Muraguchi, M.; Shibamori, M.; Ishikawa, Y.; Jarvis, T.C.; Carter, J.D.; et al. Chemically Modified DNA Aptamers Bind Interleukin-6 with High Affinity and Inhibit Signaling by Blocking Its Interaction with Interleukin-6 Receptor. J. Biol. Chem. 2014, 289, 8706–8719. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, A.; Akiyama, T.; Adachi, H.; Inoue, J.-I.; Nakamura, Y. Therapeutic potential of anti-interleukin-17A aptamer: Suppression of interleukin-17A signaling and attenuation of autoimmunity in two mouse models. Arthritis Rheum. 2010, 63, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Takamori, Y.; Ando, T.; Sato, M.; Vedi, S.; Fuji, D.; Yokoyama, T.; Tsukamoto, K.; Yamamoto, M.; Kawakami, T. Artificial aptamer that inhibits interleukin-23/interleukin-23 receptor interaction discovered via SELEX. Biochem. Biophys. Res. Commun. 2022, 614, 17–21. [Google Scholar] [CrossRef]
- Sardou, H.S.; Jebali, A.; Iman, M. Dual function of interleukin-23 Aptamer to suppress brain inflammation via attachment to macrophage stimulating 1 kinase and interleukin-23. Colloids Surfaces B: Biointerfaces 2020, 185, 110619. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, P.E.; Wang, C.; Killough, J.R.; Lewis, S.D.; Horwitz, L.R.; Ferguson, A.; Thompson, K.M.; Pendergrast, P.S.; McCauley, T.G.; Kurz, M.; et al. 2´-Deoxy Purine, 2´-O-Methyl Pyrimidine (dRmY) Aptamers as Candidate Therapeutics. Oligonucleotides 2006, 16, 337–351. [Google Scholar] [CrossRef]
- Orava, E.W.; Jarvik, N.; Shek, Y.L.; Sidhu, S.S.; Gariépy, J. A Short DNA Aptamer that Recognizes TNFα and Blocks Its Activity In Vitro. ACS Chem. Biol. 2012, 8, 170–178. [Google Scholar] [CrossRef]
- Freage, L.; Jamal, D.; Williams, N.B.; Mallikaratchy, P.R. A Homodimeric Aptamer Variant Generated from Ligand-Guided Selection Activates the T Cell Receptor Cluster of Differentiation 3 Complex. Mol. Ther. - Nucleic Acids 2020, 22, 167–178. [Google Scholar] [CrossRef]
- Fellows, T.; Ho, L.; Flanagan, S.; Fogel, R.; Ojo, D.; Limson, J. Gold nanoparticle-streptavidin conjugates for rapid and efficient screening of aptamer function in lateral flow sensors using novel CD4-binding aptamers identified through Crossover-SELEX. Analyst 2020, 145, 5180–5193. [Google Scholar] [CrossRef]
- Esposito, C.L.; Van Roosbroeck, K.; Santamaria, G.; Rotoli, D.; Sandomenico, A.; Wierda, W.G.; Ferrajoli, A.; Ruvo, M.; Calin, G.A.; de Franciscis, V.; et al. Selection of a Nuclease-Resistant RNA Aptamer Targeting CD19. Cancers 2021, 13, 5220. [Google Scholar] [CrossRef]
- Haghighi, M.; Khanahmad, H.; Palizban, A. Selection and Characterization of Single-Stranded DNA Aptamers Binding Human B-Cell Surface Protein CD20 by Cell-SELEX. Molecules 2018, 23, 715. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.; Kamble, S.; Zhao, N.; Zeng, Z.; Portier, B.P.; Zu, Y. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials 2013, 34, 8909–8917. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Y.; Ge, M.H.; Fu, Y.J.; Hao, R.; Islam, K.; Huang, P.; Chen, F.; Sun, J.; Hong, D.F.; et al. Rapid identification of specific DNA aptamers precisely targeting CD33 positive leukemia cells through a paired cell-based approach. Biomater. Sci. 2018, 7, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Tao, W.; Hao, S.; Iyer, S.P.; Zu, Y. A unique aptamer-drug conjugate for targeted therapy of multiple myeloma. Leukemia 2015, 30, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, I.M.; Zhu, Z.; Ding, C.; Chen, S.; Wu, H.; Yang, Y.; Che, F.; Li, Q.; Li, H. A Novel pH-Sensitive Multifunctional DNA Nanomedicine: An Enhanced and Harmless GD2 Aptamer-Mediated Strategy for Guiding Neuroblastoma Antitumor Therapy. Int. J. Nanomed. 2021, 16, 3217–3240. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, P.; Li, S.; Geng, X.; Zou, L.; Jin, M.; Zou, Q.; Wang, Q.; Yang, X.; Wang, K. Development of DNA Aptamer as a β-Amyloid Aggregation Inhibitor. ACS Appl. Bio Mater. 2020, 3, 8611–8618. [Google Scholar] [CrossRef]
- Chakravarthy, M.; AlShamaileh, H.; Huang, H.; Tannenberg, R.K.; Chen, S.; Worrall, S.; Dodd, P.R.; Veedu, R.N. Development of DNA aptamers targeting low-molecular-weight amyloid-β peptide aggregates in vitro. Chem. Commun. 2018, 54, 4593–4596. [Google Scholar] [CrossRef]
- Juhl, L.; Edvinsson, L.; Olesen, J.; Jansen-Olesen, I. Effect of two novel CGRP-binding compounds in a closed cranial window rat model. Eur. J. Pharmacol. 2007, 567, 117–124. [Google Scholar] [CrossRef]
- Liu, Z.; Duan, J.-H.; Song, Y.-M.; Ma, J.; Wang, F.-D.; Lu, X.; Yang, X.-D. Novel HER2 Aptamer Selectively Delivers Cytotoxic Drug to HER2-positive Breast Cancer Cells In Vitro. J. Transl. Med. 2012, 10, 148. [Google Scholar] [CrossRef]
- Wang, K.; Yao, H.; Meng, Y.; Wang, Y.; Yan, X.; Huang, R. Specific aptamer-conjugated mesoporous silica–carbon nanoparticles for HER2-targeted chemo-photothermal combined therapy. Acta Biomater. 2015, 16, 196–205. [Google Scholar] [CrossRef]
- Liang, T.; Yao, Z.; Ding, J.; Min, Q.; Jiang, L.-P.; Zhu, J.-J. Cascaded Aptamers-Governed Multistage Drug-Delivery System Based on Biodegradable Envelope-Type Nanovehicle for Targeted Therapy of HER2-Overexpressing Breast Cancer. ACS Appl. Mater. Interfaces 2018, 10, 34050–34059. [Google Scholar] [CrossRef] [PubMed]
- Gijs, M.; Penner, G.; Blackler, G.B.; Impens, N.R.; Baatout, S.; Luxen, A.; Aerts, A.M. Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals 2016, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Wu, X.; Armstrong, B.; Habib, N.; Rossi, J.J. An RNA Aptamer Targeting the Receptor Tyrosine Kinase PDGFRα Induces Anti-tumor Effects through STAT3 and p53 in Glioblastoma. Mol. Ther. - Nucleic Acids 2018, 14, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Melone, M.A.B.; Montesarchio, D. Anti-VEGF DNA-Based Aptamers in Cancer Therapeutics and Diagnostics. Med. Res. Rev. 2020, 41, 464–506. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Yung, L.-Y.L. Probing High Affinity Sequences of DNA Aptamer against VEGF165. PLOS ONE 2012, 7, e31196. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Yoshitomi, T.; Hayashi, M.; Hayashi, M.; Oguro, T.; Oguro, T.; Kimura, K.; Kimura, K.; Wayama, F.; Wayama, F.; et al. Binding and Structural Properties of DNA Aptamers with VEGF-A-Mimic Activity. Mol. Ther. - Nucleic Acids 2020, 19, 1145–1152. [Google Scholar] [CrossRef]
- Kim, E.S.; Serur, A.; Huang, J.; Manley, C.A.; McCrudden, K.W.; Frischer, J.S.; Soffer, S.Z.; Ring, L.; New, T.; Zabski, S.; et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc. Natl. Acad. Sci. USA 2002, 99, 11399–11404. [Google Scholar] [CrossRef]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortel, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar] [CrossRef]
- Diener, J.L.; Daniel Lagasse, H.A.; Duerschmied, D.; Merhi, Y.; Tanguay, J.F.; Hutabarat, R.; Gilbert, J.; Wagner, D.D.; Schaub, R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J. Thromb. Haemost. 2009, 7, 1155–1162. [Google Scholar] [CrossRef]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Murin, C.D. Considerations of Antibody Geometric Constraints on NK Cell Antibody Dependent Cellular Cytotoxicity. Front. Immunol. 2020, 11, 1635. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W.; Spirig, R.; Morelli, A.B.; Rowe, T.; Käsermann, F. Next-generation Fc receptor–targeting biologics for autoimmune diseases. Autoimmun. Rev. 2019, 18, 102366. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, S.; Kitahara, Y.; Okamatsu, Y.; Fujii, T.; Nakayama, A.; Ueno, S.; Ijichi, C.; Futaki, F.; Nakata, K.; Taki, M. Facile and Efficient Chemoenzymatic Semisynthesis of Fc-Fusion Compounds for Half-Life Extension of Pharmaceutical Components. Bioconjugate Chem. 2019, 30, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Di, J.; Xie, F.; Xu, Y. When liposomes met antibodies: Drug delivery and beyond. Adv. Drug Deliv. Rev. 2020, 154-155, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.L.; Wagstaff, K.M. Internalized Functional DNA Aptamers as Alternative Cancer Therapies. Front. Pharmacol. 2020, 11, 1115. [Google Scholar] [CrossRef]
- Hickey, A.J.; Stewart, I.E. Inhaled antibodies: Quality and performance considerations. Hum. Vaccines Immunother. 2021, 18, 1940650. [Google Scholar] [CrossRef]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. Mabs 2018, 10, 693–711. [Google Scholar] [CrossRef]
- Carter, P.J.; Rajpal, A. Designing antibodies as therapeutics. Cell 2022, 185, 2789–2805. [Google Scholar] [CrossRef]
- Palluk, S.; Arlow, D.H.; de Rond, T.; Barthel, S.; Kang, J.S.; Bector, R.; Baghdassarian, H.M.; Truong, A.N.; Kim, P.W.; Singh, A.K.; et al. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018, 36, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Edmunds, G.; Gibbons, C.; Zhang, J.; Gadi, M.R.; Zhu, H.; Fang, J.; Liu, X.; Kong, Y.; Wang, P.G. Toward Automated Enzymatic Synthesis of Oligosaccharides. Chem. Rev. 2018, 118, 8151–8187. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Antibody | Peptide | Nucleotide |
|---|---|---|---|
| Molecular mass | ~150 kDa for IgG | variable (~110Da/residue) | variable (~340Da/residue) |
| Production | complex biologic | Synthetic | Synthetic |
| Mechanisms of target elimination | multiple | neutralization | neutralization |
| Combinatorial screening | limited | yes | limited |
| In vivo half-life | long | Short | extremely short |
| Reversibility | no | yes | yes (on-demand) |
| Immunogenicity | yes | depends | low |
| Generation | Lead Pharmacophore | Obtained Function by Lead Engineering | Warhead Incorporated | Breakthrough Point |
|---|---|---|---|---|
| 1st: Charlton, 1997 | Low molecule; TCI | Increasing affinity/specificity by DNA library conjugation | valyl phosphonate |
|
| 2nd: Tabuchi, 2021 | Middle molecule; non-covalent DNA aptamer | Nuclease resistance by targeted covalent binding via warhead introduction | 4-(acetyl)-benzene-1- sulfonyl fluoride |
|
| 2nd: Tivon (2021) | “ | “ | acyl-sulfonamide |
|
| 2nd: Qin (2021) | “ | “ | 4-(methyl)-benzene-1- sulfonyl fluoride |
|
| Molecular Target | Name | mAb | Aptamer | Reference | |
|---|---|---|---|---|---|
| Check point proteins | |||||
| 4-1BB | TNF ligand superfamily member 9 | Utomilumab * | M12-23 | [107] | |
| B7-H3 | B7 homolog 3 | Enoblituzumab * | none | ||
| BLTA | B and T lymphocyte attenuator | Icatolimab * | none | ||
| CTLA-4 | cytotoxic T lymphocyte-associate antigen 4 | Ipilimumab, Tremelimumab | aptCTLA-4 | [108,109] | |
| ICOS | inducible costimulatory | Vopratelimab * | MRP1-ICOS | [110] | |
| Lag-3 | lymphocyte-activation gene 3 | Relatimab | Apt1 | [111] | |
| PD1 | programmed cell death protein 1 | Cemiplimab, Dostarlimab, Nivolumab | MP7 | [112,113,114,115] | |
| PD-L1 | programmed death ligand 1 | Atezolizumab, Avelumab, Durvalumab | aptPD-L1 | [116,117,118,119,120,121,122,123,124] | |
| TIGIT | T cell immunoreceptor with IgG and ITIM domains | Tirabolumab * | none | ||
| TIM-3 | T cell immunoglobulin mucin 3 | Sabatolimab *, Cobolimab * | TIJM3Apt | [125,126] | |
| Cytokines/Chemokine | |||||
| BAFF | B-cell activating factor | Belimumab | R1-14 | [127] | |
| IL1b | interleukin 1 beta | Canakinumab | AptIL-1b, many | [128] | |
| IL2 | interleukin 2 | Basiliximab, Reslizumab | M20 (@mouse) | [129] | |
| IL5 | interleukin 5 | Mepolizumab, Reslizumab | 19 clones in 5 families | [130] | |
| IL6 | interleukin 6 | Tocilizumab | S1025, S1026, AIR-3 | [131,132,133] | |
| IL12 | interleukin 12 | Ustekinumab | none | ||
| IL-13 | interleukin 13 | Tralokinumab | none | ||
| IL17a | interleukin 17a | Ixekizumab, Secukinumab | Apt21-2 | [134] | |
| IL23 | interleukin 23 | Guselkumab, Tildrakizumab | clone 1, clone A5, C3 | [135,136,137] | |
| IL36 | interleukin | Spesolimab | none | ||
| TNFa | tumor necrosis factor alpha | Adalimumab, Certolizumab, Golimumab | AptTNF-a, VR11 | [128,138] | |
| Tumor markers | |||||
| CD3 | cluster of differentiation | 3 | Teplizumab * | J7, OSJ-T1-4 | [139] |
| CD4 | “ | 4 | Ibalizumab | U26 | [140] |
| CD19 | “ | 19 | Tafasitamab, Blinatumomab | B83.T2 | [141] |
| CD20 | “ | 20 | Rituximab, Ibritumomab, Obinutuzumab | AP1-3 | [142] |
| CD22 | “ | 22 | Inotuzumab, Moxetunomab, epcortamab * | none | |
| CD30 | “ | 30 | Brentuximab | C2, NGS6.0 | [143] |
| CD33 | “ | 33 | Gemtuzumab | S30 | [144] |
| CD38 | “ | 38 | Daratumumab, Isatuximab | aptamer#1 | [145] |
| CD52 | “ | 52 | Alzetuzumab | none | |
| CD79b | “ | 79b | Polatuzumab | none | |
| GD2 | disialogangloside 2 | Dinutuximab, Naxitamab, Margetuximab | DB67 | [146] | |
| Growth factors/receptor | |||||
| a-beta | amyloid protein beta | Donanemab * | Ab-Apt, RNV95 | [147,148] | |
| CGRP | calcitonin gene-related peptide | Eptinezumab, Frenenezumab, Galcanezumab | Star-F12 | [149] | |
| HER2 | human epidermal growth factor receptor 2 | Trastuzumab, Pertuzumab, Margetuximab | HB5, HeA2_3 | [150,151,152,153] | |
| PDGFR-a | platelet-derived growth factor receptor alpha | Olaratumab | PDR3 | [154] | |
| VEGF | vascular endothelial growth factor | Bevacozumab, Ranibizumab, Brolucizumab | SL2b, many more | [155,156] | |
| VEGFR2 | vascular endothelial growth factor receptor 2 | Ramucrumab | Apt01, 02, NX1838 | [157,158] | |
| Factor IXa/X | X coagulation factor IXa/X | 5Epicizamab | 9.3t, RB006 | [159] | |
| vWF | Von Willebrand factor | Caplacizumab | ARC1779 | [160] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Tabuchi, Y.; Katsuki, R.; Taki, M. bioTCIs: Middle-to-Macro Biomolecular Targeted Covalent Inhibitors Possessing Both Semi-Permanent Drug Action and Stringent Target Specificity as Potential Antibody Replacements. Int. J. Mol. Sci. 2023, 24, 3525. https://doi.org/10.3390/ijms24043525
Yang J, Tabuchi Y, Katsuki R, Taki M. bioTCIs: Middle-to-Macro Biomolecular Targeted Covalent Inhibitors Possessing Both Semi-Permanent Drug Action and Stringent Target Specificity as Potential Antibody Replacements. International Journal of Molecular Sciences. 2023; 24(4):3525. https://doi.org/10.3390/ijms24043525
Chicago/Turabian StyleYang, Jay, Yudai Tabuchi, Riku Katsuki, and Masumi Taki. 2023. "bioTCIs: Middle-to-Macro Biomolecular Targeted Covalent Inhibitors Possessing Both Semi-Permanent Drug Action and Stringent Target Specificity as Potential Antibody Replacements" International Journal of Molecular Sciences 24, no. 4: 3525. https://doi.org/10.3390/ijms24043525