
Crucial Role of Lysine-Specific Histone Demethylase 1 in RANKL-Mediated Osteoclast Differentiation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
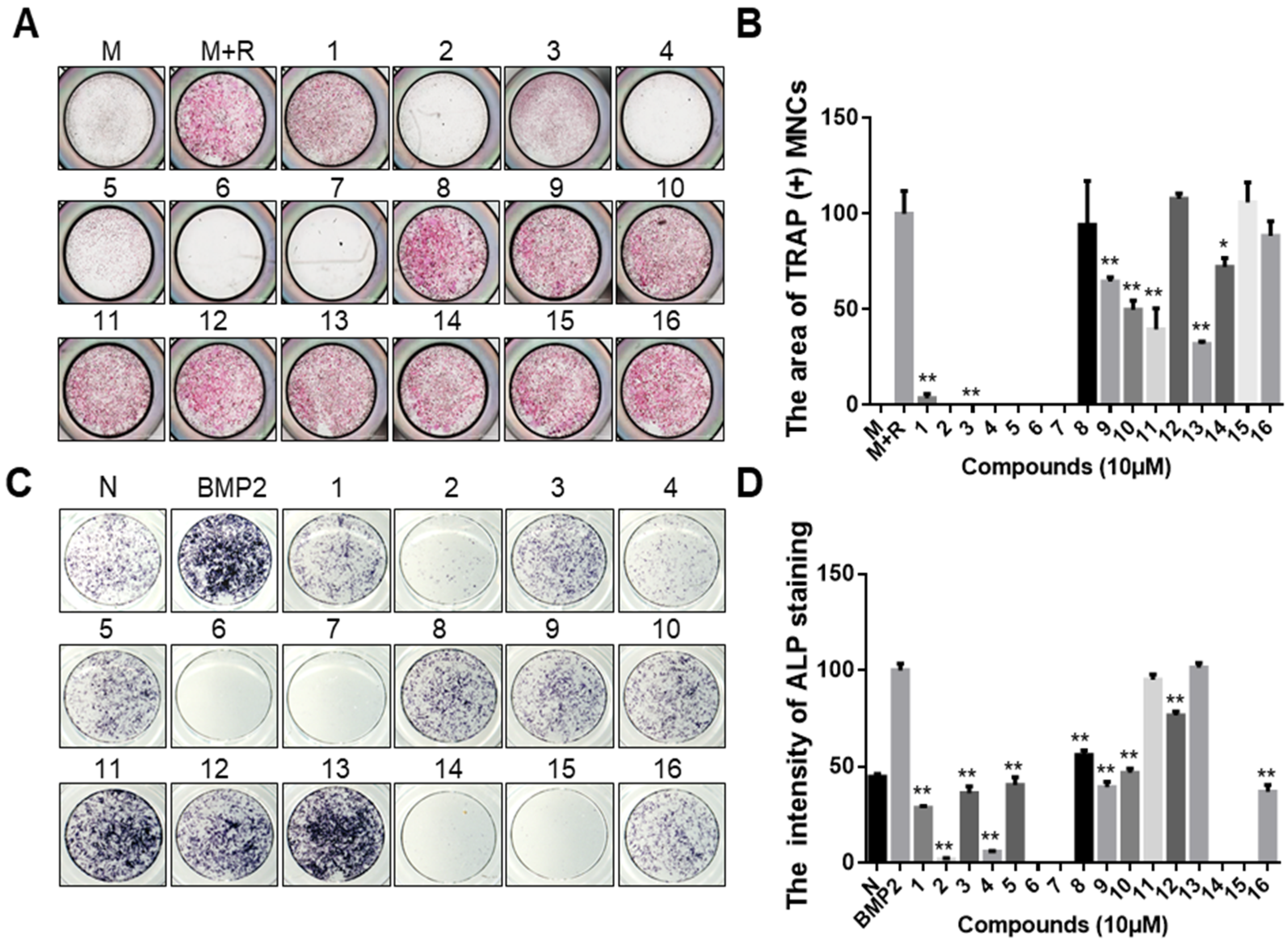
2.1. Identification of Anti-Osteoporosis Candidates from Epigenetic Modulator Inhibitors
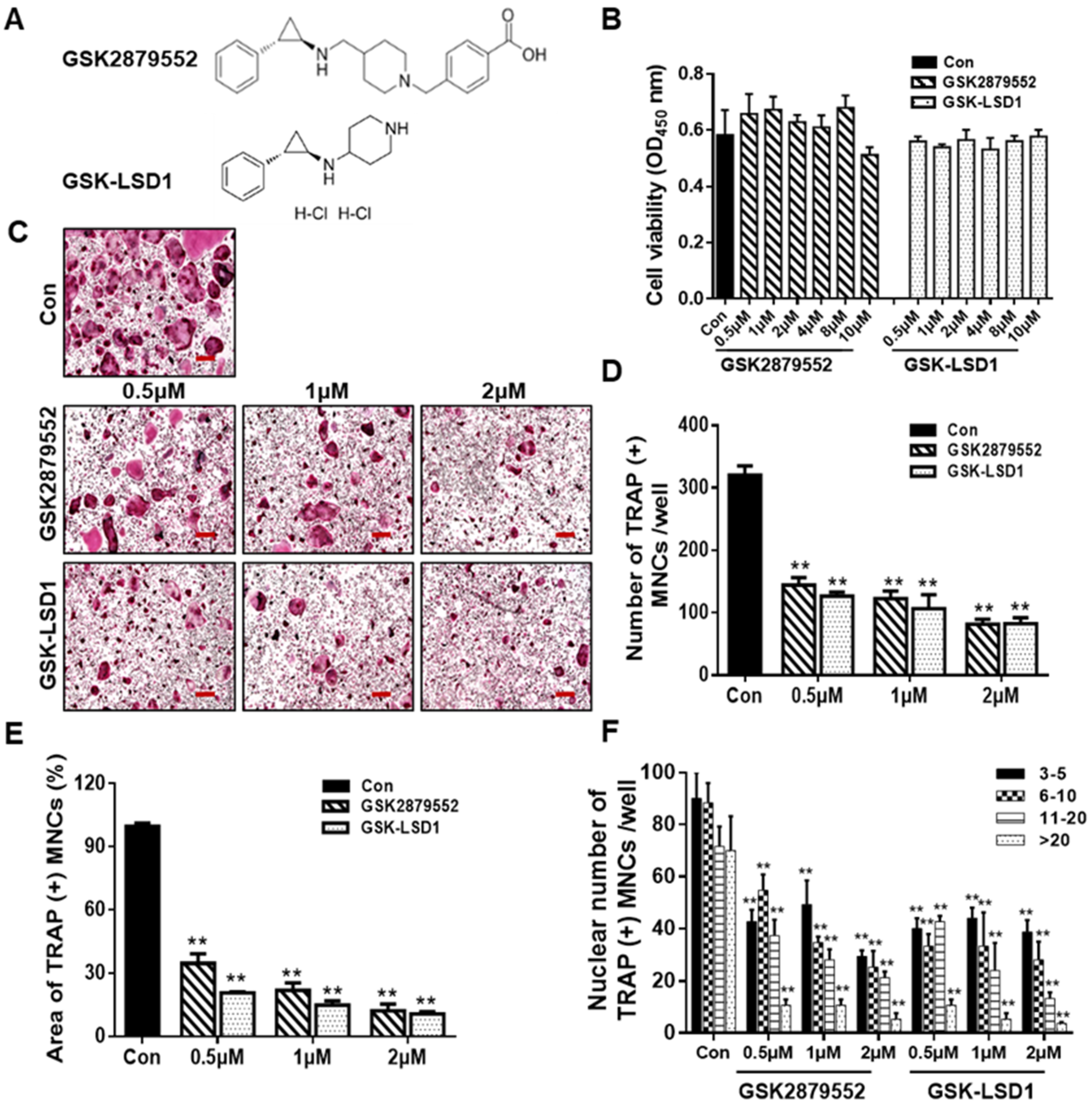
2.2. LSD1 Inhibitors Prevent RANKL-Stimulated Osteoclast Differentiation in a Dose-Dependent Manner
2.3. Suppression of RANKL-Stimulated F-Actin Rings and Bone Resorption by LSD1 Inhibitors
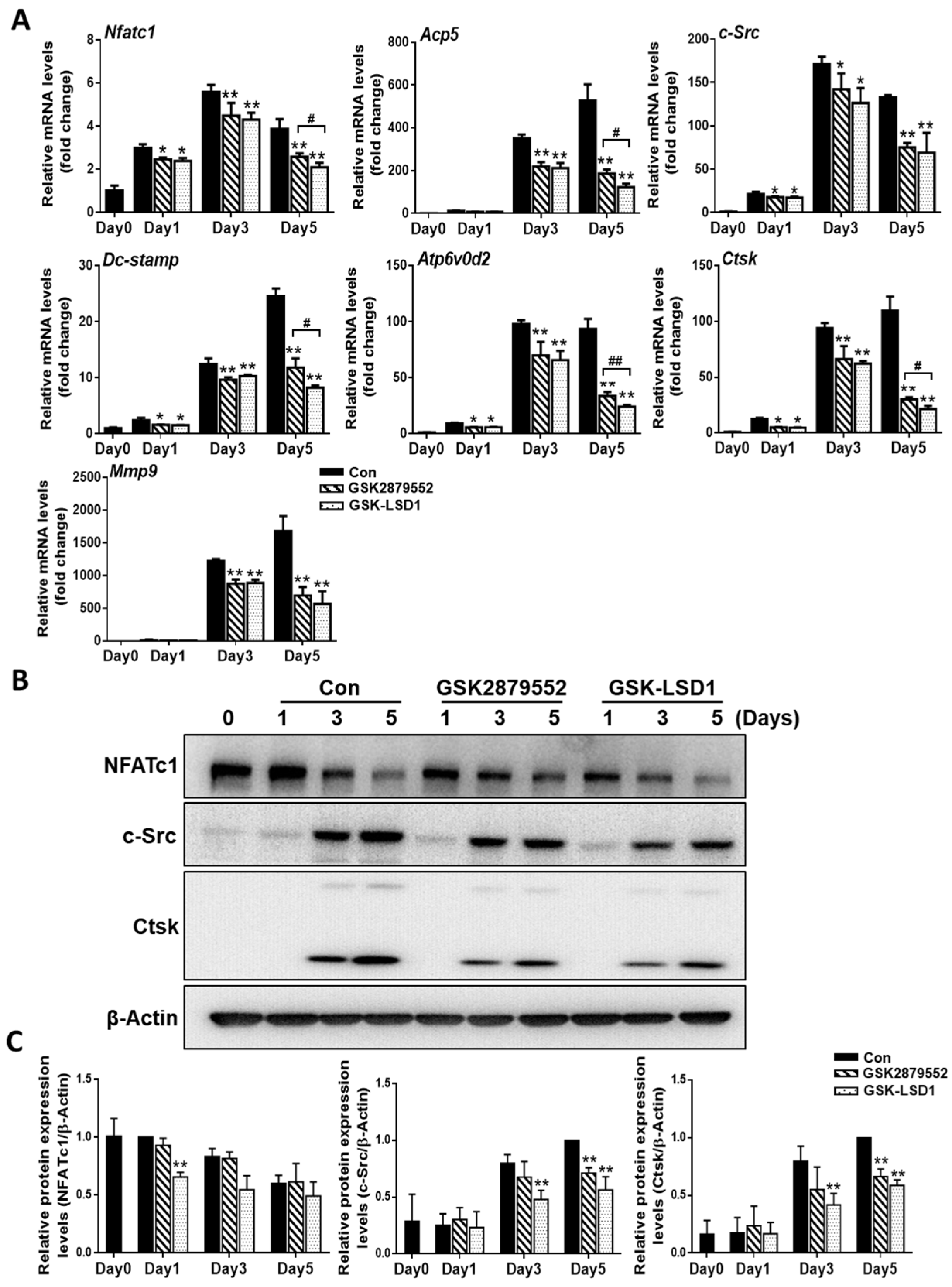
2.4. LSD1 Inhibitors Reduce the Expression of Osteoclast-Specific Genes and Proteins
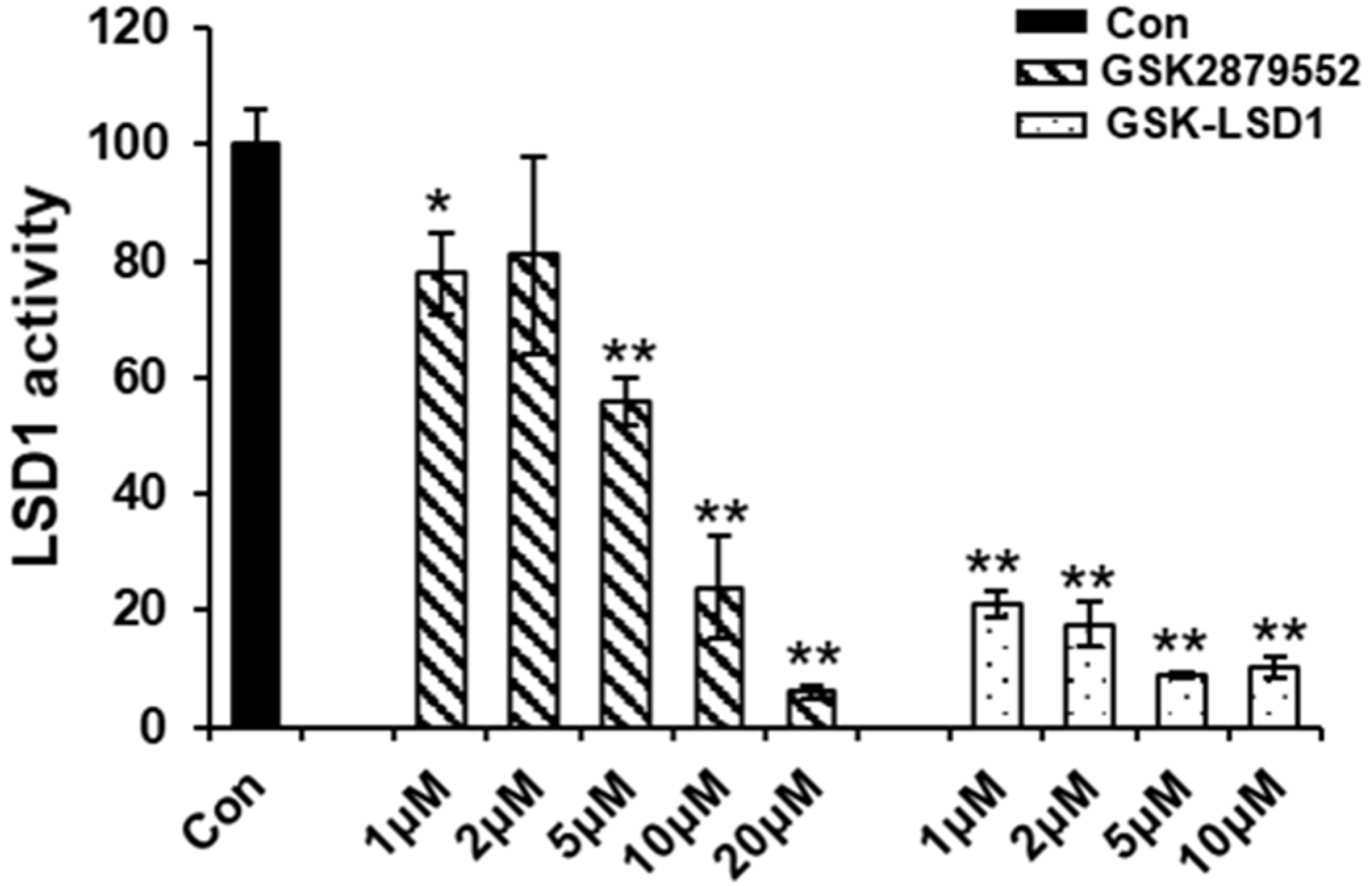
2.5. LSD1 Inhibitors Reduce the Demethylation Activity of LSD1
2.6. Knockout of LSD1 in Raw 264.7 Cells Suppresses RANKL-Induced Osteoclast Differentiation
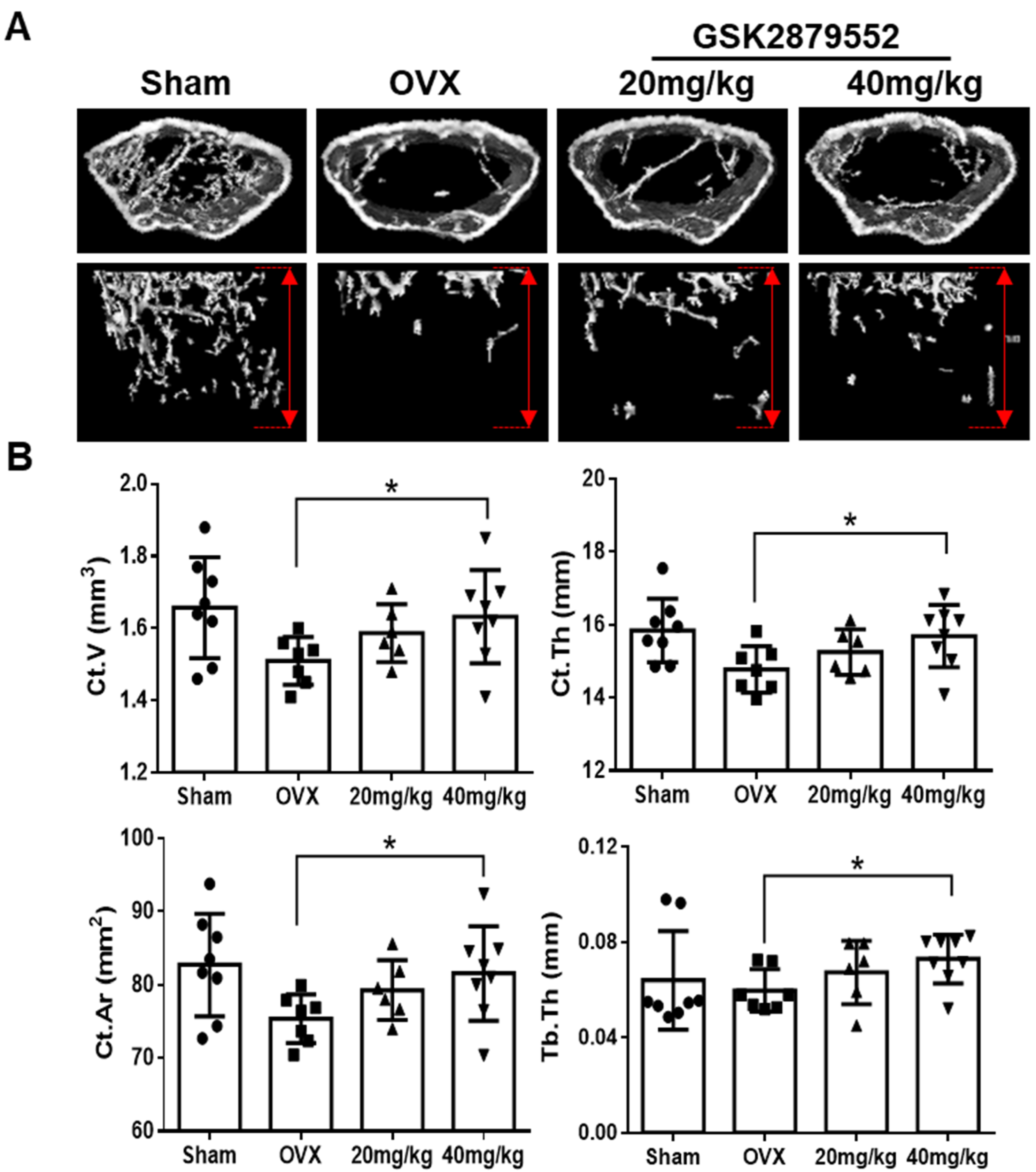
2.7. GSK2879552 Prevents Cortical Bone Loss in Ovariectomized (OVX) Mice
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Osteoclast and Osteoblast Culture
4.3. TRAP Staining Assay
4.4. ALP Staining
4.5. Cytotoxicity Assay
4.6. F-Actin Ring Staining
4.7. Bone Resorption Assay
4.8. Real-Time PCR Assay
4.9. Western Blot Assay
4.10. LSD1 Activity Analysis
4.11. Generation of LSD1-Knockout Raw 264.7 Cells by the CRISPR-Cas9 System
4.12. OVX-Induced Osteoporosis Mice Model and Micro-Computed Tomography
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanaka, Y.; Nakayamada, S.; Okada, Y. Osteoblasts and Osteoclasts in Bone Remodeling and Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, A.A.; Al Mamun, A. Current Advances in Regulation of Bone Homeostasis. FASEB Bioadv. 2020, 2, 668–679. [Google Scholar] [CrossRef]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 Expression Determines Its Essential Role in Bone Homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential Role of P38 Mitogen-Activated Protein Kinase in Cathepsin K Gene Expression during Osteoclastogenesis through Association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef]
- Karsenty, G.; Wagner, E.F. Reaching a Genetic and Molecular Understanding of Skeletal Development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef]
- Miyazaki, T.; Tanaka, S.; Sanjay, A.; Baron, R. The Role of C-Src Kinase in the Regulation of Osteoclast Function. Mod. Rheumatol. 2006, 16, 68–74. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An Operational Definition of Epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef]
- Astleford, K.; Campbell, E.; Norton, A.; Mansky, K.C. Epigenetic Regulators Involved in Osteoclast Differentiation. Int. J. Mol. Sci. 2020, 21, 7080. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, W.; Yang, X.; Na, L.; Chen, L.; Liu, G. The Roles of Epigenetics Regulation in Bone Metabolism and Osteoporosis. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jing, L.; Li, M.; He, L.; Guo, Z. Regulation of Histone Arginine Methylation/Demethylation by Methylase and Demethylase. Mol. Med. Rep. 2019, 19, 3963–3971. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, W.-G. Advances in histone methyltransferases and histone demethylases. Ai Zheng 2008, 27, 1018–1025. [Google Scholar]
- Zhao, L.; Duan, Y.-T.; Lu, P.; Zhang, Z.-J.; Zheng, X.-K.; Wang, J.-L.; Feng, W.-S. Epigenetic Targets and Their Inhibitors in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 2395–2419. [Google Scholar] [CrossRef]
- Wei, X.; Yi, X.; Zhu, X.-H.; Jiang, D.-S. Histone Methylation and Vascular Biology. Clin. Epigenet. 2020, 12, 30. [Google Scholar] [CrossRef]
- Cai, S.F.; Levine, R.L. Genetic and Epigenetic Determinants of AML Pathogenesis. Semin. Hematol. 2019, 56, 84–89. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Sun, H.-L.; Liang, H.; Li, K.; Fan, Q.-M.; Zhao, Q.-H. Dynamic and Distinct Histone Modifications of Osteogenic Genes during Osteogenic Differentiation. J. Biochem. 2015, 158, 445–457. [Google Scholar] [CrossRef]
- Perillo, B.; Tramontano, A.; Pezone, A.; Migliaccio, A. LSD1: More than Demethylation of Histone Lysine Residues. Exp. Mol. Med. 2020, 52, 1936–1947. [Google Scholar] [CrossRef]
- Nicholson, T.B.; Chen, T. LSD1 Demethylates Histone and Non-Histone Proteins. Epigenetics 2009, 4, 129–132. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. P53 Is Regulated by the Lysine Demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J.; Lee, W.; Yim, H.Y.; Ahn, J.-Y.; Park, S.W.; Shin, H.-J.R.; Yu, R.; Won, K.-J.; Bae, J.-S.; et al. PKCα-LSD1-NF-ΚB-Signaling Cascade Is Crucial for Epigenetic Control of the Inflammatory Response. Mol. Cell 2018, 69, 398–411.e6. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-A.; Zhu, J.; Yennawar, N.; Eek, P.; Tan, S. Crystal Structure of the LSD1/CoREST Histone Demethylase Bound to Its Nucleosome Substrate. Mol. Cell 2020, 78, 903–914.e4. [Google Scholar] [CrossRef] [PubMed]
- Amente, S.; Lania, L.; Majello, B. The Histone LSD1 Demethylase in Stemness and Cancer Transcription Programs. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Musri, M.M.; Carmona, M.C.; Hanzu, F.A.; Kaliman, P.; Gomis, R.; Párrizas, M. Histone Demethylase LSD1 Regulates Adipogenesis. J. Biol. Chem. 2010, 285, 30034–30041. [Google Scholar] [CrossRef]
- Choi, J.; Jang, H.; Kim, H.; Kim, S.-T.; Cho, E.-J.; Youn, H.-D. Histone Demethylase LSD1 Is Required to Induce Skeletal Muscle Differentiation by Regulating Myogenic Factors. Biochem. Biophys. Res. Commun. 2010, 401, 327–332. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Majello, B. Epigenetic Regulation of Epithelial to Mesenchymal Transition by the Lysine-Specific Demethylase LSD1/KDM1A. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 905–910. [Google Scholar] [CrossRef]
- Sun, J.; Ermann, J.; Niu, N.; Yan, G.; Yang, Y.; Shi, Y.; Zou, W. Histone Demethylase LSD1 Regulates Bone Mass by Controlling WNT7B and BMP2 Signaling in Osteoblasts. Bone Res. 2018, 6, 14. [Google Scholar] [CrossRef]
- Rummukainen, P.; Tarkkonen, K.; Dudakovic, A.; Al-Majidi, R.; Nieminen-Pihala, V.; Valensisi, C.; Hawkins, R.D.; van Wijnen, A.J.; Kiviranta, R. Lysine-Specific Demethylase 1 (LSD1) Epigenetically Controls Osteoblast Differentiation. PLoS ONE 2022, 17, e0265027. [Google Scholar] [CrossRef]
- Astleford-Hopper, K.; Bradley, E.; Mansky, K.C. Female LSD1 Conditional Knockout Mice Have an Increased Bone Mass. bioRxiv 2022. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal Transduct. Target Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, B.-R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Alsaqer, S.F.; Tashkandi, M.M.; Kartha, V.K.; Yang, Y.-T.; Alkheriji, Y.; Salama, A.; Varelas, X.; Kukuruzinska, M.; Monti, S.; Bais, M.V. Inhibition of LSD1 Epigenetically Attenuates Oral Cancer Growth and Metastasis. Oncotarget 2017, 8, 73372–73386. [Google Scholar] [CrossRef] [PubMed]
- Chellaiah, M.A. Regulation of Actin Ring Formation by Rho GTPases in Osteoclasts. J. Biol. Chem. 2005, 280, 32930–32943. [Google Scholar] [CrossRef]
- Sl, T. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Miyamoto, T. Regulators of Osteoclast Differentiation and Cell-Cell Fusion. Keio J. Med. 2011, 60, 101–105. [Google Scholar] [CrossRef]
- Zhao, Q. Osteoclast Differentiation and Gene Regulation. Front. Biosci. 2007, 12, 2519. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Sanjay, A.; Neff, L.; Tanaka, S.; Horne, W.C.; Baron, R. Src Kinase Activity Is Essential for Osteoclast Function. J. Biol. Chem. 2004, 279, 17660–17666. [Google Scholar] [CrossRef]
- Lotinun, S.; Kiviranta, R.; Matsubara, T.; Alzate, J.A.; Neff, L.; Lüth, A.; Koskivirta, I.; Kleuser, B.; Vacher, J.; Vuorio, E.; et al. Osteoclast-Specific Cathepsin K Deletion Stimulates S1P-Dependent Bone Formation. J. Clin. Investig. 2013, 123, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Murata, K.; Ito, S.; Suzuki, A.; Terao, C.; Ishie, S.; Umemoto, A.; Murotani, Y.; Nishitani, K.; Yoshitomi, H.; et al. Role of Lysine-Specific Demethylase 1 in Metabolically Integrating Osteoclast Differentiation and Inflammatory Bone Resorption Through Hypoxia-Inducible Factor 1α and E2F1. Arthritis Rheumatol. 2022, 74, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Bartell, S.M.; Han, L.; Kim, H.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Chambliss, K.L.; Shaul, P.W.; Roberson, P.K.; Weinstein, R.S.; et al. Non-Nuclear-Initiated Actions of the Estrogen Receptor Protect Cortical Bone Mass. Mol. Endocrinol. 2013, 27, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ding, M.; Cho, E.; Seong, J.; Lee, S.; Lee, T. 2-NPPA Mitigates Osteoclastogenesis via Reducing TRAF6-Mediated c-Fos Expression. Front. Pharmacol. 2021, 11, 599081. [Google Scholar] [CrossRef]
- Chen, Z.; Cho, E.; Lee, J.; Lee, S.; Lee, T.-H. Inhibitory Effects of N-[2-(4-Acetyl-1-Piperazinyl) Phenyl]-2-(2-Chlorophenoxy) Acetamide on Osteoclast Differentiation In Vitro via the Downregulation of TRAF6. Int. J. Mol. Sci. 2019, 20, 5196. [Google Scholar] [CrossRef]
- Cho, E.; Chen, Z.; Ding, M.; Seong, J.; Lee, S.; Min, S.H.; Choi, D.K.; Lee, T.-H. PMSA Prevents Osteoclastogenesis and Estrogen-Dependent Bone Loss in Mice. Bone 2021, 142, 115707. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, M.; Chen, Z.; Cho, E.; Park, S.-W.; Lee, T.-H. Crucial Role of Lysine-Specific Histone Demethylase 1 in RANKL-Mediated Osteoclast Differentiation. Int. J. Mol. Sci. 2023, 24, 3605. https://doi.org/10.3390/ijms24043605
Ding M, Chen Z, Cho E, Park S-W, Lee T-H. Crucial Role of Lysine-Specific Histone Demethylase 1 in RANKL-Mediated Osteoclast Differentiation. International Journal of Molecular Sciences. 2023; 24(4):3605. https://doi.org/10.3390/ijms24043605
Chicago/Turabian StyleDing, Mina, Zhihao Chen, Eunjin Cho, Sang-Wook Park, and Tae-Hoon Lee. 2023. "Crucial Role of Lysine-Specific Histone Demethylase 1 in RANKL-Mediated Osteoclast Differentiation" International Journal of Molecular Sciences 24, no. 4: 3605. https://doi.org/10.3390/ijms24043605
APA StyleDing, M., Chen, Z., Cho, E., Park, S.-W., & Lee, T.-H. (2023). Crucial Role of Lysine-Specific Histone Demethylase 1 in RANKL-Mediated Osteoclast Differentiation. International Journal of Molecular Sciences, 24(4), 3605. https://doi.org/10.3390/ijms24043605