
Basic Methods of Cell Cycle Analysis

Abstract
1. Introduction
2. Growth Models
2.1. Exponential Growth Model
2.2. Logistic Growth Model
2.3. Monod Kinetics Model
2.4. Allee Effect Model
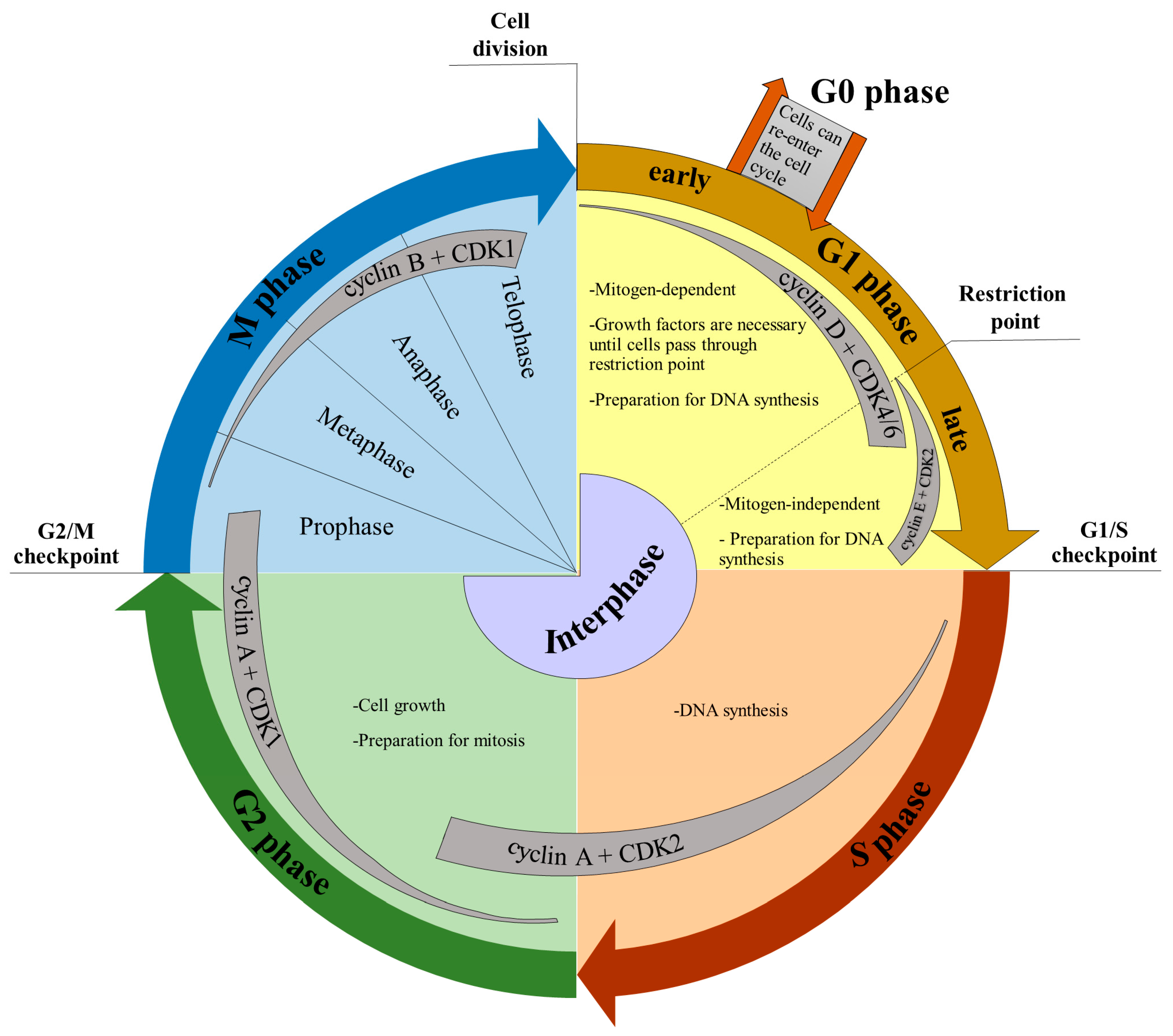
3. Cell Cycle and Markers of the Cell Cycle Phases
4. Determination of Fractions of Cells in Distinct Phases of the Cell Cycle
5. From the Fractions to the Cell Cycle Phase Length
- The weak ergodic assumption—If the distribution of cells among different states does not change over time, then the proportion observed in any state is proportional to the time each cell spends, on average, in that state.
- Strong ergodic assumption—If all cells are going through an identical cycle of events, then the proportion of cells in any cycle stage observed in the population at a single time point is the same as the proportion of time spent in that cycle stage as a single cell progresses through the cycle [52].
6. Approaches Based on Kinetic Analysis
6.1. Approaches Based on DNA Labeling
6.1.1. Marker Nucleosides
6.1.2. Mitotic Window Approach
6.1.3. Continuous Labeling Method
6.1.4. Double Labeling Approach
6.2. Methods Based on Time Lapse Microscopy
7. Conclusions and Future Perspectives
8. Notes
- Below are listed some of the commonly used markers of senescent cells. However, none of the senescence-associated markers are exclusive and typical for senescent cells; therefore, usually at least two different markers are detected
- (a)
- (b)
- Senescent cells contain increased numbers of lysosomes [99,100]. This finding resulted in the development of a technique based on the detection of activity of the senescence-associated (SA) β-galactosidase at pH 6 [100]. Unfortunately, a significant delay between the senescence entry and SA β-galactosidase-derived staining was also previously reported [101]. Moreover, intense SA β-galactosidase labeling could also reflect an alteration in lysosomal number or activity in non-proliferating cells [102] or in terminally differentiated cells such as neurons [103]. Furthermore, endogenous SA β-galactosidase activity was found also in confluent non-transformed fibroblast culture [104].
- (c)
- (d)
- Below is a list of freeware software that can be used for image analysis
- (a)
- ImageJ/FIJI—ImageJ is widely used software for processing and analyzing scientific images; FIJI is a “batteries-included” distribution of ImageJ and ImageJ2, which includes many useful plugins contributed by the community. It can be downloaded from https://imagej.net/downloads (accessed on 9 January 2023) [110].
- (b)
- CellProfilerTM—CellProfiler is software allowing for automatic processing and analysis of sets of images according to the instructions in the form of user-defined pipelines. It can be downloaded from https://cellprofiler.org (accessed on 9 January 2023) [111].
- (c)
- CellProfiler AnalystTM—CellProfiler Analyst allows data to be explored from CellProfiler through interactive visualizations that link to images, classifying complex or subtle phenotypes using machine learning. It can be downloaded from https://cellprofileranalyst.org (accessed on 9 January 2023) [112].
- (d)
- Ilastik—ilastik is software that applies machine learning algorithms to easily segment, classify, track, and count the cells or other experimental data. It can be downloaded from https://www.ilastik.org/download.html (accessed on 9 January 2023) [113].
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, L.-M.; Margulis, V.; Lotan, Y. Prognostic Role of Cell Cycle and Proliferative Markers in Clear Cell Renal Cell Carcinoma. Urol. Clin. N. Am. 2015, 43, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Traganos, F.; Darzynkiewicz, Z. Growth Imbalance and Altered Expression of Cyclin-B1, Cyclin-a, Cyclin-E, and Cyclin-D3 in Molt-4 Cells Synchronized in the Cell-Cycle by Inhibitors of DNA-Replication. Cell Growth Differ. 1995, 6, 1485–1493. [Google Scholar]
- Fang, H.-S.; Lang, M.-F.; Sun, J. New Methods for Cell Cycle Analysis. Chin. J. Anal. Chem. 2019, 47, 1293–1301. [Google Scholar] [CrossRef]
- Eastman, A.E.; Guo, S. The palette of techniques for cell cycle analysis. FEBS Lett. 2020, 594, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Charlebois, D.A.; Balázsi, G. Modeling cell population dynamics. Silico Biol. 2019, 13, 21–39. [Google Scholar] [CrossRef]
- Corso, A. MA 138–Calculus 2 with Life Science Applications Solving Differential Equations. Available online: https://www.ms.uky.edu/~ma138/Spring18/Lectures/Lecture_12-13.pdf (accessed on 14 December 2022).
- Vogels, M.; Zoeckler, R.; Stasiw, D.M.; Cerny, L.C. P. F. Verhulst’s “notice sur la loi que la populations suit dans son accroissement” from correspondence mathematique et physique. Ghent, vol. X, 1838. J. Biol. Phys. 1975, 3, 183–192. [Google Scholar] [CrossRef]
- Verhulst, P.F. Notice sur la loi que la population suit dans son accroissement. Corresp. Math. Phys. 1838, 10, 113–121. [Google Scholar]
- Lerma, M.A. 3.4. The Logistic Equation. Available online: https://sites.math.northwestern.edu/~mlerma/courses/math214-2-03f/notes/c2-all.pdf (accessed on 14 December 2022).
- Monod, J. The growth of bacterial cultures. Annu. Rev. Microbiol. 1949, 3, 371–394. [Google Scholar] [CrossRef]
- Korolev, K.S.; Xavier, J.B.; Gore, J. Turning ecology and evolution against cancer. Nat. Rev. Cancer 2014, 14, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Courchamp, F.; Clutton-Brock, T.; Grenfell, B. Inverse density dependence and the Allee effect. Trends Ecol. Evol. 1999, 14, 405–410. [Google Scholar] [CrossRef]
- Winter, A.; Richter, A.; Eikeset, A.M. Implications of Allee effects for fisheries management in a changing climate: Evidence from Atlantic cod. Ecol. Appl. 2020, 30, e01994. [Google Scholar] [CrossRef]
- Neufeld, Z.; Von Witt, W.; Lakatos, D.; Wang, J.; Hegedus, B.; Czirok, A. The role of Allee effect in modelling post resection recurrence of glioblastoma. PLoS Comput. Biol. 2017, 13, e1005818. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol. Biol. 2014, 14, 10. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Pardee, A.B. The Restriction Point of the Cell Cycle. Cell Cycle 2002, 1, 102–109. [Google Scholar] [CrossRef]
- Pardee, A.B. A Restriction Point for Control of Normal Animal Cell Proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef]
- Wang, Y.M.; Ji, P.; Liu, J.S.; Broaddus, R.R.; Xue, F.X.; Zhang, W. Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy. Mol. Cancer 2009, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Oki, T.; Nishimura, K.; Kitaura, J.; Togami, K.; Maehara, A.; Izawa, K.; Sakaue-Sawano, A.; Niida, A.; Miyano, S.; Aburatani, H.; et al. A novel cell-cycle-indicator, mVenus-p27K−, identifies quiescent cells and visualizes G0–G1 transition. Sci. Rep. 2014, 4, 4012. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; Zuffo, R.D.; Matti, V.; D’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Mikuła-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Książek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2019, 77, 213–229. [Google Scholar] [CrossRef]
- McKinnon, K.M. Flow Cytometry: An Overview. Curr. Protoc. Immunol. 2018, 120, 5.1.1–5.1.11. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Juan, G.; Bedner, E. Determining Cell Cycle Stages by Flow Cytometry. Curr. Protoc. Cell Biol. 1999, 1, 8.4.1–8.4.18. [Google Scholar] [CrossRef]
- Frydrych, I. Cell cycle analysis by flow cytometry. In Laboratory Techniques in Cellular and Molecular Medicine, 1st ed.; Agrawal, K., Bouchal, J., Das, V., Drábek, J., Džubák, P., Hajdúch, M., Koberna, K., Ligasová, A., Mistrík, M., de Sanctis, J.B., Eds.; Palacký University Olomouc: Olomouc, Czech Republic, 2021; pp. 193–200. [Google Scholar] [CrossRef]
- Jayat, C.; Ratinaud, M.-H. Cell cycle analysis by flow cytometry: Principles and applications. Biol. Cell 1993, 78, 15–25. [Google Scholar] [CrossRef]
- Baisch, H.; Beck, H.-P.; Christensen, I.J.; Hartmann, N.R.; Fried, J.; Dean, P.N.; Gray, J.W.; Jett, J.H.; Johnston, D.A.; White, R.A.; et al. A comparison of mathematical methods for the analysis of DNA histograms obtained by flow cytometry. Cell Prolif. 1982, 15, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.N. A simplified method of DNA distribution analysis. Cell Prolif. 1980, 13, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.N.; Jett, J.H. Mathematical analysis of DNA distributions derived from flow microfluorometry. J. Cell Biol. 1974, 60, 523–527. [Google Scholar] [CrossRef]
- Jett, J.H.; Gurley, L.R. An Improved Sum-Of-Normals Technique For Cell Cycle Distribution Analysis of Flow Cytometric Dna Histograms. Cell Prolif. 1981, 14, 413–423. [Google Scholar] [CrossRef]
- Watson, J.V.; Chambers, S.H.; Smith, P.J. A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry 1987, 8, 1–8. [Google Scholar] [CrossRef]
- Liboska, R.; Ligasová, A.; Strunin, D.; Rosenberg, I.; Koberna, K. Most Anti-BrdU Antibodies React with 2′-Deoxy-5-Ethynyluridine—The Method for the Effective Suppression of This Cross-Reactivity. PLoS ONE 2012, 7, e51679. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of Cell Cycle by Flow Cytometry. In Checkpoint Controls and Cancer: Volume 2: Activation and Regulation Protocols; Schönthal, A.H., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 301–311. [Google Scholar]
- Hendzel, M.J.; Wei, Y.; Mancini, M.A.; Van Hooser, A.; Ranalli, T.; Brinkley, B.R.; Bazett-Jones, D.P.; Allis, C.D. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 1997, 106, 348–360. [Google Scholar] [CrossRef]
- Shapiro, H.M. Flow cytometric estimation of DNA and RNA content in intact cells stained with hoechst 33342 and pyronin Y. Cytometry 1981, 2, 143–150. [Google Scholar] [CrossRef]
- Qiu, L.; Liu, M.; Pan, K. A Triple Staining Method for Accurate Cell Cycle Analysis Using Multiparameter Flow Cytometry. Molecules 2013, 18, 15412–15421. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. Elife 2016, 5, e13722. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, M.L.; Zheng, L.-X.; Baldwin, A.; Ohta, T.; Hurt, M.M.; Marzluff, W.F. Stem-Loop Binding Protein, the Protein That Binds the 3′ End of Histone mRNA, Is Cell Cycle Regulated by Both Translational and Posttranslational Mechanisms. Mol. Cell. Biol. 2000, 20, 4188–4198. [Google Scholar] [CrossRef]
- Mitchison, J.M. The Biology of the Cell Cycle; Cambridge University Press: London, UK, 1971; p. 313. [Google Scholar]
- Wheeler, R.J. Analyzing the dynamics of cell cycle processes from fixed samples through ergodic principles. Mol. Biol. Cell 2015, 26, 3898–3903. [Google Scholar] [CrossRef]
- Zeuthen, E. Synchrony in Cell Division and Growth, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1964. [Google Scholar]
- Schorl, C.; Sedivy, J.M. Analysis of cell cycle phases and progression in cultured mammalian cells. Methods 2007, 41, 143–150. [Google Scholar] [CrossRef]
- Pereira, P.D.; Serra-Caetano, A.; Cabrita, M.; Bekman, E.; Braga, J.; Rino, J.; Santus, R.; Filipe, P.L.; Sousa, A.E.; Ferreira, J.A. Quantification of cell cycle kinetics by EdU (5-ethynyl-2′-deoxyuridine)-coupled-fluorescence-intensity analysis. Oncotarget 2017, 8, 40514–40532. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.; Futran, M.; Hidalgo, D.; Pop, R.; Iyer, D.R.; Scully, R.; Rhind, N.; Socolovsky, M. Global increase in replication fork speed during a p57KIP2-regulated erythroid cell fate switch. Sci. Adv. 2017, 3, e1700298. [Google Scholar] [CrossRef]
- Martynoga, B.; Morrison, H.; Price, D.; Mason, J. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 2005, 283, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Bialic, M.; Nachar, B.A.A.; Koźlak, M.; Coulon, V.; Schwob, E. Measuring S-Phase Duration from Asynchronous Cells Using Dual EdU-BrdU Pulse-Chase Labeling Flow Cytometry. Genes 2022, 13, 408. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.X.; I Fakhreddin, R.; Shimerov, H.K.; Kedziora, K.M.; Kumar, R.J.; Perez, J.; Limas, J.C.; Grant, G.D.; Cook, J.G.; Gupta, G.P.; et al. Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol. Syst. Biol. 2019, 15, e8604. [Google Scholar] [CrossRef]
- Ligasová, A.; Koberna, K. DNA Replication: From Radioisotopes to Click Chemistry. Molecules 2018, 23, 3007. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.H.; Woods, P.S.; Hughes, W.L. The organization and duplication of chromosomes as revealed by autoradiographic studies using tritium-labeled thymidinee. Proc. Natl. Acad. Sci. USA 1957, 43, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Visser, D.; Frisch, D.; Huang, B. Synthesis of 5-chlorodeoxyuridine and a comparative study of 5-halodeoxyuridines in E. Coli. Biochem. Pharmacol. 1960, 5, 157–164. [Google Scholar] [CrossRef]
- Prusoff, W.H. Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim. Biophys. Acta 1959, 32, 295–296. [Google Scholar] [CrossRef]
- Aten, J.A.; Bakker, P.J.M.; Stap, J.; Boschman, G.A.; Veenhof, C.H.N. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. J. 1992, 24, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ligasová, A.; Konečný, P.; Frydrych, I.; Koberna, K. Cell cycle profiling by image and flow cytometry: The optimised protocol for the detection of replicational activity using 5-Bromo-2′-deoxyuridine, low concentration of hydrochloric acid and exonuclease III. PLoS ONE 2017, 12, e0175880. [Google Scholar] [CrossRef]
- Ligasová, A.; Konečný, P.; Frydrych, I.; Koberna, K. Looking for ugly ducklings: The role of the stability of BrdU-antibody complex and the improved method of the detection of DNA replication. PLoS ONE 2017, 12, e0174893. [Google Scholar] [CrossRef]
- Ligasová, A.; Strunin, D.; Liboska, R.; Rosenberg, I.; Koberna, K. Atomic Scissors: A New Method of Tracking the 5-Bromo-2′-Deoxyuridine-Labeled DNA In Situ. PLoS ONE 2012, 7, e52584. [Google Scholar] [CrossRef] [PubMed]
- Ageno, M.; Dore, E.; Frontali, C. The Alkaline Denaturation of DNA. Biophys. J. 1969, 9, 1281–1311. [Google Scholar] [CrossRef]
- Dimitrova, D.S.; Berezney, R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 2002, 115, 4037–4051. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.A.; Pombo, A. Replicon Clusters Are Stable Units of Chromosome Structure: Evidence That Nuclear Organization Contributes to the Efficient Activation and Propagation of S Phase in Human Cells. J. Cell Biol. 1998, 140, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Barbie, D.A.; Classon, M.; Dyson, N.; Harlow, E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000, 14, 2855–2868. [Google Scholar] [CrossRef]
- Koberna, K.; Ligasová, A.; Malínský, J.; Pliss, A.; Siegel, A.J.; Cvačková, Z.; Fidlerová, H.; Mašata, M.; Fialová, M.; Raška, I.; et al. Electron microscopy of DNA replication in 3-D: Evidence for similar-sized replication foci throughout S-phase. J. Cell. Biochem. 2004, 94, 126–138. [Google Scholar] [CrossRef]
- Dolbeare, F.; Gray, J.W. Use of restriction endonucleases and exonuclease III to expose halogenated pyrimidines for immunochemical staining. Cytometry 1988, 9, 631–635. [Google Scholar] [CrossRef]
- Fox, M.; Arndt-Jovin, D.; Jovin, T.; Baumann, P.; Robert-Nicoud, M. Spatial and temporal distribution of DNA replication sites localized by immunofluorescence and confocal microscopy in mouse fibroblasts. J. Cell Sci. 1991, 99, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Darzynkiewicz, Z. Labelling DNA strand breaks with BrdUTP. Detection of apoptosis and cell proliferation. Cell Prolif. 1995, 28, 571–579. [Google Scholar] [CrossRef]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef]
- Kohlmeier, F.; Maya-Mendoza, A.; Jackson, D.A. EdU induces DNA damage response and cell death in mESC in culture. Chromosom. Res. 2013, 21, 87–100. [Google Scholar] [CrossRef]
- Ligasová, A.; Strunin, D.; Friedecký, D.; Adam, T.; Koberna, K. A Fatal Combination: A Thymidylate Synthase Inhibitor with DNA Damaging Activity. PLoS ONE 2015, 10, e0117459. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.H.; Rahman, M.; Levkoff, L.H.; Millette, S.; Martin-Carreras, T.; Dunbar, E.M.; Reynolds, B.A.; Laywell, E.D. Ethynyldeoxyuridine (EdU) suppresses in vitro population expansion and in vivo tumor progression of human glioblastoma cells. J. Neuro-Oncol. 2011, 105, 485–498. [Google Scholar] [CrossRef]
- Zhao, H.; Halicka, H.D.; Li, J.; Biela, E.; Berniak, K.; Dobrucki, J.; Darzynkiewicz, Z. DNA damage signaling, impairment of cell cycle progression, and apoptosis triggered by 5-ethynyl-2′-deoxyuridine incorporated into DNA. Cytom. Part A 2013, 83, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.H.; Levkoff, L.H.; Marshall, G.P.; Caldeira, M.; Steindler, D.A.; Reynolds, B.A.; Laywell, E.D. Bromodeoxyuridine Induces Senescence in Neural Stem and Progenitor Cells. Stem Cells 2008, 26, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Quastler, H.; Sherman, F. Cell population kinetics in the intestinal epithelium of the mouse. Exp. Cell Res. 1959, 17, 420–438. [Google Scholar] [CrossRef]
- Stanners, C.; Till, J. DNA synthesis in individual L-strain mouse cells. Biochim. Biophys. Acta 1960, 37, 406–419. [Google Scholar] [CrossRef]
- Maga, G.; Hübscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116, 3051–3060. [Google Scholar] [CrossRef]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2010, 107, 1127–1140. [Google Scholar] [CrossRef]
- Madsen, P.; Celis, J.E. S-phase patterns of cyclin (PCNA) antigen staining resemble topographical patterns of DNA synthesis. A role for cyclin in DNA replication? FEBS Lett. 1985, 193, 5–11. [Google Scholar] [CrossRef]
- Dowling, M.R.; Kan, A.; Heinzel, S.; Zhou, J.H.S.; Marchingo, J.M.; Wellard, C.J.; Markham, J.F.; Hodgkin, P.D. Stretched cell cycle model for proliferating lymphocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 6377–6382. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Conlon, M.; Wells, A.; Cao, J.Y.; Dixon, S.J. Systematic Quantification of Population Cell Death Kinetics in Mammalian Cells. Cell Syst. 2017, 4, 600–610.e6. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087163. [Google Scholar] [CrossRef] [PubMed]
- Schmid, I.; Krall, W.J.; Uittenbogaart, C.H.; Braun, J.; Giorgi, J.V. Dead cell discrimination with 7-amino-actinomcin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 1992, 13, 204–208. [Google Scholar] [CrossRef]
- Icha, J.; Weber, M.; Waters, J.C.; Norden, C. Phototoxicity in live fluorescence microscopy, and how to avoid it. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Skylaki, S.; Hilsenbeck, O.; Schroeder, T. Challenges in long-term imaging and quantification of single-cell dynamics. Nat. Biotechnol. 2016, 34, 1137–1144. [Google Scholar] [CrossRef]
- Doan, M.; Vorobjev, I.; Rees, P.; Filby, A.; Wolkenhauer, O.; Goldfeld, A.E.; Lieberman, J.; Barteneva, N.; Carpenter, A.; Hennig, H. Diagnostic Potential of Imaging Flow Cytometry. Trends Biotechnol. 2018, 36, 649–652. [Google Scholar] [CrossRef]
- Behbehani, G.K. Cell Cycle Analysis by Mass Cytometry. Methods Mol. Biol. 2017, 1686, 105–124. [Google Scholar] [CrossRef]
- Schneider, E.L.; Mitsui, Y. The relationship between in vitro cellular aging and in vivo human age. Proc. Natl. Acad. Sci. USA 1976, 73, 3584–3588. [Google Scholar] [CrossRef]
- Sherwood, S.W.; Rush, D.; Ellsworth, J.L.; Schimke, R.T. Defining cellular senescence in IMR-90 cells: A flow cytometric analysis. Proc. Natl. Acad. Sci. USA 1988, 85, 9086–9090. [Google Scholar] [CrossRef]
- Adewoye, A.B.; Tampakis, D.; Follenzi, A.; Stolzing, A. Multiparameter flow cytometric detection and quantification of senescent cells in vitro. Biogerontology 2020, 21, 773–786. [Google Scholar] [CrossRef]
- Perillo, N.L.; Walford, R.L.; Newman, M.A.; Effros, R.B. Human T lymphocytes possess a limited in vitro life span. Exp. Gerontol. 1989, 24, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Lipetz, J.; Cristofalo, V.J. Ultrastructural changes accompanying the aging of human diploid cells in culture. J. Ultrastruct. Res. 1972, 39, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Al-Baker, E.; Dropcova, S.; Denyer, S.; Ostad, N.; Lloyd, A.; Kill, I.R.; Faragher, R.G. Different Kinetics of Senescence in Human Fibroblasts and Peritoneal Mesothelial Cells. Exp. Cell Res. 1997, 236, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.E.; Akimov, S.S.; Hass, R.; Zelenin, A.V.; Prudovsky, I.A. Endogenous β-Galactosidase Activity in Continuously Nonproliferating Cells. Exp. Cell Res. 1998, 243, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated β-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is β-Galactosidase Staining a Marker of Senescence in Vitro and in Vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.V.; Demidenko, Z.N.; Bukreeva, E.I.; Pospelov, V.A.; Gudkov, A.; Blagosklonny, M.V. Pseudo-DNA damage response in senescent cells. Cell Cycle 2009, 8, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Evans, M.K. Techniques to Induce and Quantify Cellular Senescence. J. Vis. Exp. 2017, 123, e55533. [Google Scholar] [CrossRef]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16ink4a. Cell Cycle 2011, 10, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 1–11. [Google Scholar] [CrossRef]
- Dao, D.; Fraser, A.N.; Hung, J.; Ljosa, V.; Singh, S.; Carpenter, A.E. CellProfiler Analyst: Interactive data exploration, analysis and classification of large biological image sets. Bioinformatics 2016, 32, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. Ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method (Suitable for Estimation) | Principle | Common Markers | Notes | Ref. |
|---|---|---|---|---|
| Mitotic window (G2 and S phases) | Analysis of the signal from the replicated chromatin in mitotic cells. | EdU, BrdU | Time consuming. Variability of G2 and S phases can be addressed. | [54] |
| Continuous labeling based on cell number of the labeled cells (sum of G2, M, and G1 phases) | Analysis of the number of labeled cells after continuous labeling of replicated chromatin. | EdU, BrdU | Time consuming. Long incubation time with the potentially toxic marker nucleoside. BrdU is less toxic than EdU. | [54] |
| Continuous labeling based on the signal intensity of the labeled cells (S phase) | Analysis of the signal intensity after continuous labeling of replicated chromatin. | EdU | The toxicity of EdU should be tested before its use. | [55] |
| Double labeling (S phase, generation time) | Analysis of the number of labeled cells after labeling of replicated chromatin by two consecutive pulses with the different replication marker nucleosides. | EdU, BrdU, IdU | Fast. The sensitivity to the presence of G1 and senescence cells. | [56,57] |
| Double labelling with chase period (S phase) | Analysis of the number of labeled cells after labeling of replicated chromatin by two consecutive pulses with different replication marker nucleosides separated by different chase periods. | EdU, BrdU | Nocodazole treatment is required. | [58] |
| Time lapse microscopy (all phases) | Analysis of living cells expressing fluorescently labeled reporters at regular time interval | Fluorescently labeled PCNA, Ctd1, Geminin | Time consuming. Special equipment is necessary. Phototoxicity can affect the results. Variability of different cell cycle stages can be addressed. Mortality rate can be measured. | [49,59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ligasová, A.; Frydrych, I.; Koberna, K. Basic Methods of Cell Cycle Analysis. Int. J. Mol. Sci. 2023, 24, 3674. https://doi.org/10.3390/ijms24043674
Ligasová A, Frydrych I, Koberna K. Basic Methods of Cell Cycle Analysis. International Journal of Molecular Sciences. 2023; 24(4):3674. https://doi.org/10.3390/ijms24043674
Chicago/Turabian StyleLigasová, Anna, Ivo Frydrych, and Karel Koberna. 2023. "Basic Methods of Cell Cycle Analysis" International Journal of Molecular Sciences 24, no. 4: 3674. https://doi.org/10.3390/ijms24043674
APA StyleLigasová, A., Frydrych, I., & Koberna, K. (2023). Basic Methods of Cell Cycle Analysis. International Journal of Molecular Sciences, 24(4), 3674. https://doi.org/10.3390/ijms24043674