
The Journey of Cancer Cells to the Brain: Challenges and Opportunities
 , , , ,
, , , ,
Abstract
:1. Introduction
- (1)
- There is a slight variation among BM patients based on the source of the primary cancer (Table 1). Notably, it was reported that lung cancer is responsible for 80% of reported BM cases, while melanoma, breast cancer, and kidney cancer are responsible for 3.8, 3.7, and 3% of the BM cases, respectively [2,3]. However, in another study, it was reported that secondary brain tumors are most likely to derive from breast cancer, followed by lung carcinomas, renal cell carcinomas, and melanomas [4]. The reasons behind these discrepancies could be related to the age of patients, for example, it was shown that kidney/renal tumors and melanomas are the most frequent in children [3]. Gender could also play a significant role in determining the origin of the BMs cells; it was reported that the primary cancers most often responsible for BM are of the breast in women and of the lung in both genders (Table 1);
- (2)
- The number of metastatic foci has been shown to be inversely proportional to prognosis, with a worse outcome predicted based on the detection of one or more metastatic sites [5]. Due to constant improvements in diagnostic and therapeutic strategies, the median survival among patients with metastatic disease of the brain has been reported to have increased [6];
- (3)
- The type of treatment influences the survival rate, as it has been shown that patients undergoing specified therapy, including surgery, chemotherapy, or radiotherapy, have a higher chance of surviving longer [7];
- (4)
- Further investigation is needed to pinpoint the relationship between BM location and survival. Patients suffering from BMs in the frontal lobe have a poorer prognosis compared to those with non-frontal lobe BMs (4.9 and 10 months, respectively). Furthermore, metastasis infiltrating the eloquent cortex (containing speech and sensory fields) in relation to non-eloquent areas is associated with higher median survival (21.2 months vs. 18 months). Interestingly, several studies have revealed that 85% of metastasis cases are reported in the cerebrum, including the posterior areas of the two hemispheres and the anterior border zone between the anterior and middle cerebral arteries. Conversely, only 10% to 15% of metastasis cases are detected in the cerebellum, followed by 3% in the brainstem. However, studies comparing the survival of patients based on the BM locations in these regions seem to be scarce [8,9].
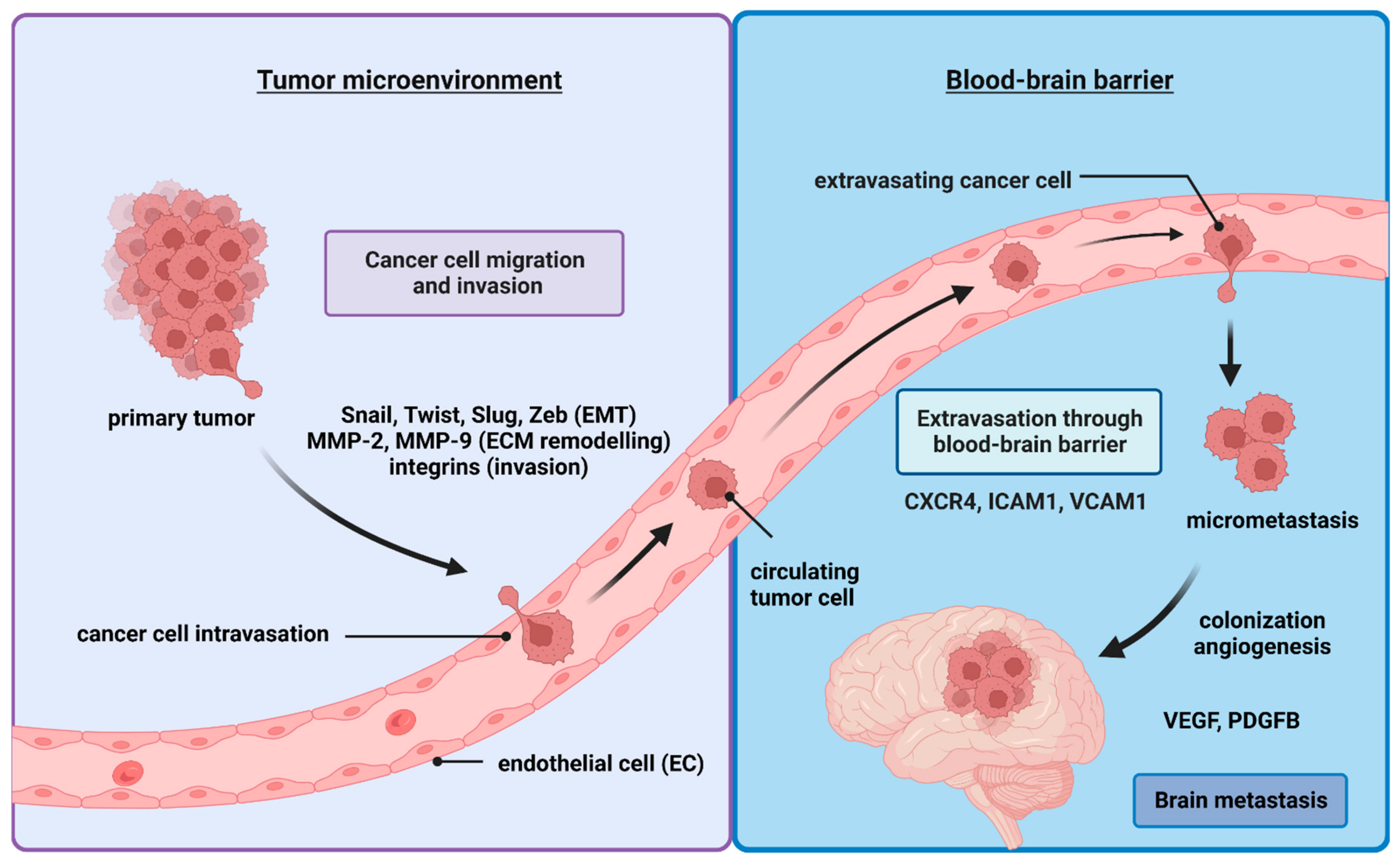
2. The Metastatic Cells Journey to the Brain
2.1. Epithelial–Mesenchymal Transition (EMT)
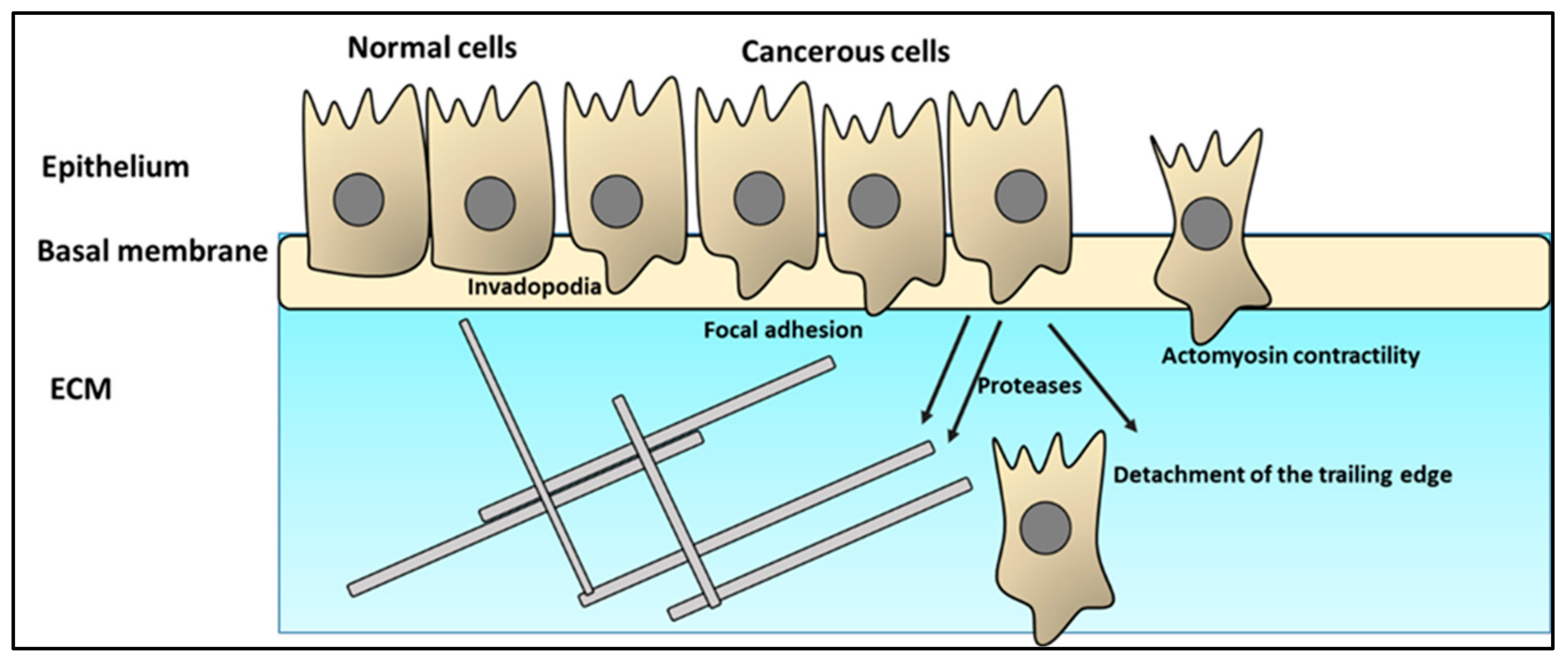
2.2. Cancer Cell Infiltration of the ECM
2.3. Intravasation
- (i)
- Differences in function between blood vessels and lymphatic vessels. The lymphatic system’s main functions include transferring various macromolecules, fluids, and immune cells to the blood and maintaining plasma volume [57]. The composition of the lymph fluid is almost identical to that of the interstitial tissue fluids, which promote the survival of migrating tumor cells [55];
- (ii)
- Blood and lymphatic vessels differ considerably in their structure [55]. While blood circulation constitutes a closed system, the lymphatic system flows in one direction from the peripheral tissues to the blood. To achieve its function, the lymphatic system uses lymphatic capillaries and pre-collectors, followed by lymphatic vessels and trunks that reach the bloodstream. Lymphatic vessels are threefold the size of blood vessels, with an incomplete, discontinuous basal lamina, absence of pericytes, and smooth muscles. These differences in structure mean that the migrating cells face lower mechanical resistance in traveling through lymphatic vessels compared to through blood vessels. Thus, less energy is needed by the cancer cells to travel through lymphatic vessels. Lymphatic vessels are leakier, thus, supporting cancer cells to spread more [55,58];
- (iii)
- The role of ECM: cancer cells migrating through the blood and lymphatic vessels use ECM to their advantage. Endostatin, tumstatin, canstatin, arresten, hexastatin, and type IV and type XVIII ECM collagens all have a significant influence on the creation of both blood and lymphatic vessels [55,59]. Integrin 91, an ECM receptor, has been linked to the development of lymphangiogenesis [60]. Similarly, low-molecular-weight hyaluronan promotes lymphangiogenesis by interacting with its lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1), promoting lymphatic endothelial cell proliferation and tube formation. Lymphangiogenesis and tumor invasion are tightly connected to MT1-MMP-mediated proMMP-2 activation and the production of ECM1 and EMILIN1, an elastic microfibril-associated protein [61,62];
- (iv)
- Expression of various genes and receptors specific to blood or lymphatic vessels, as well as cross-talk between lymphatic and blood vessels, could also contribute to the decision of cancer cells when choosing their route.
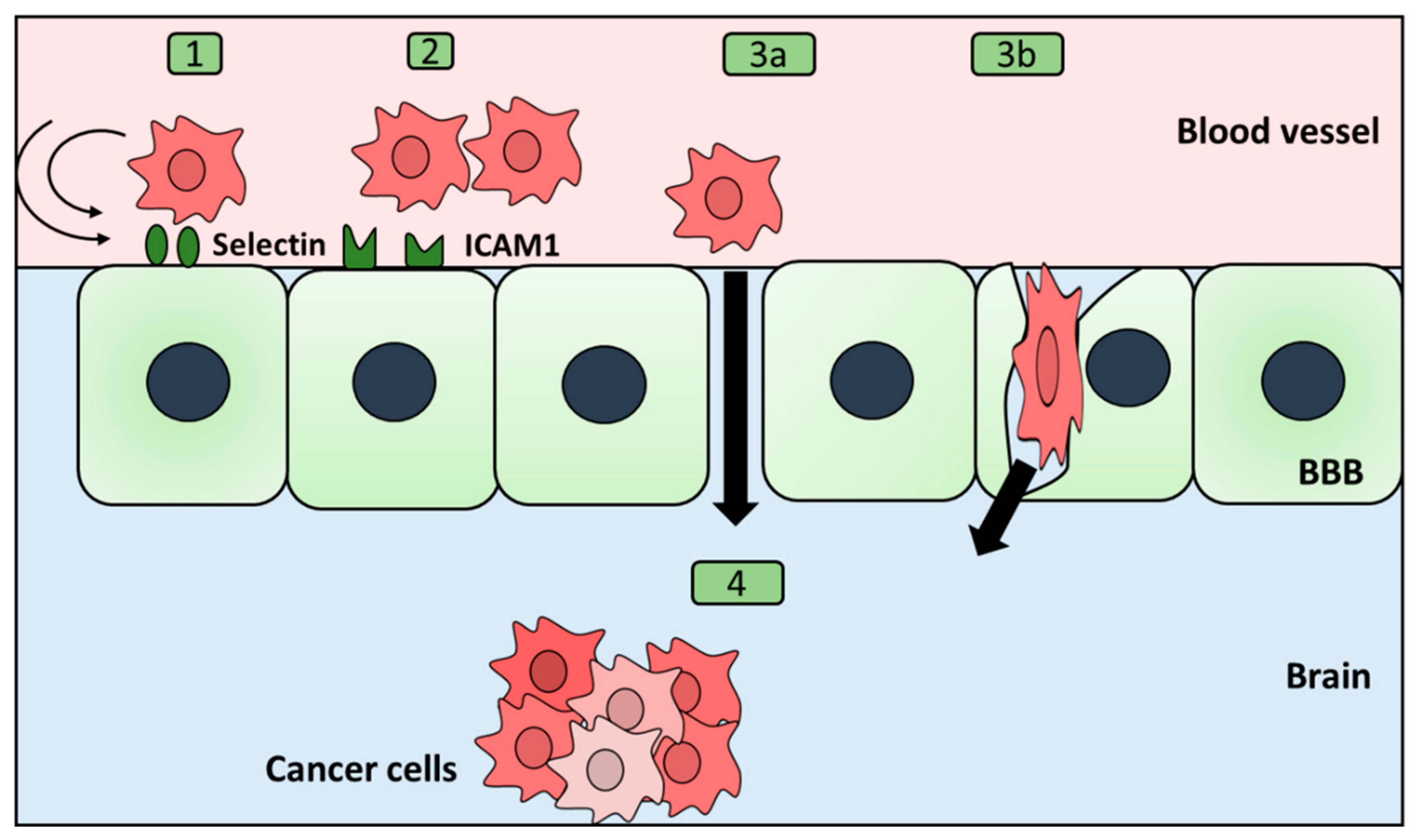
2.4. Extravasation through the Blood–Brain Barrier
- BBB permeability
2.5. Mesenchymal-to-Epithelial Transition (MET)
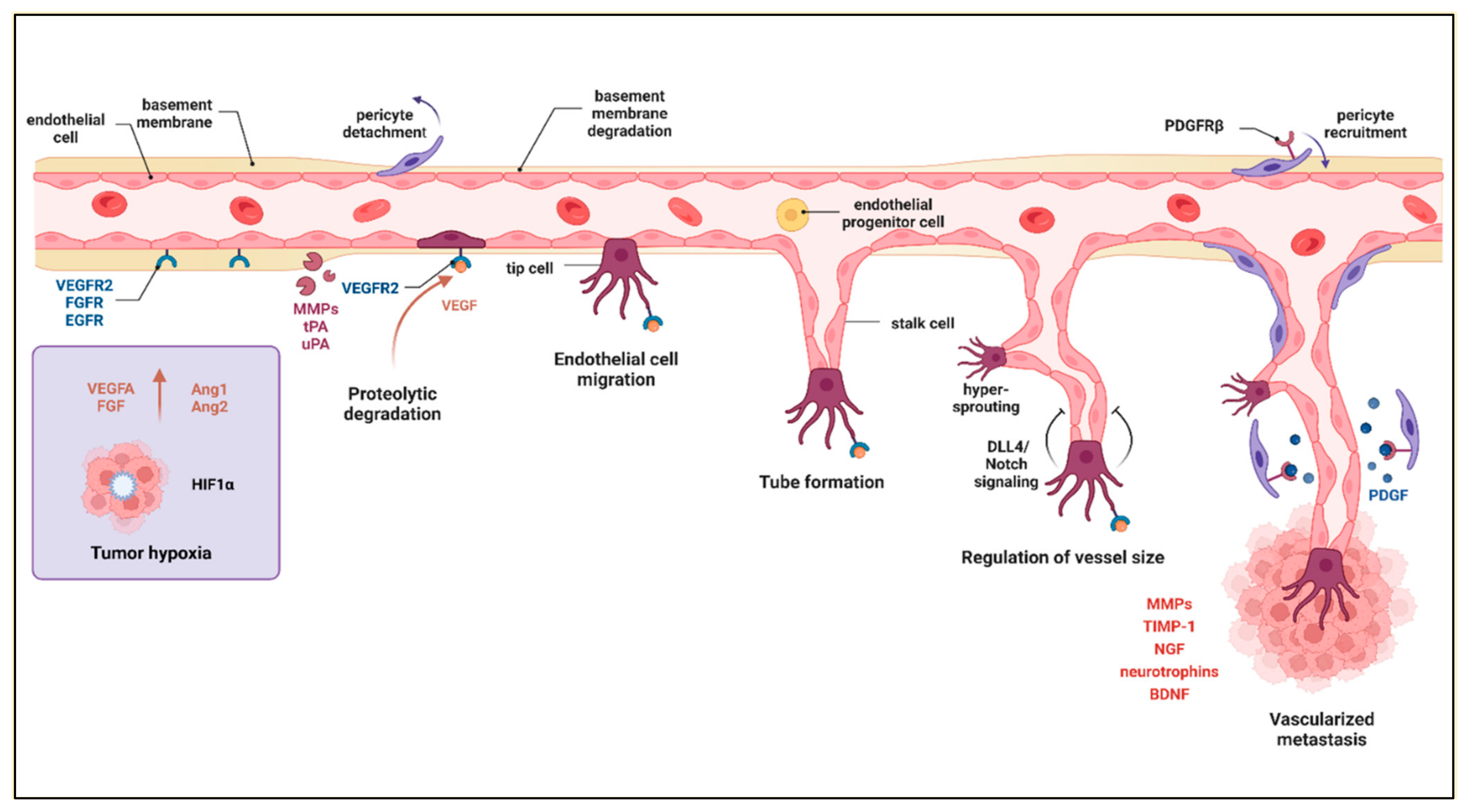
2.6. Angiogenesis
3. Survival of Cancer Cells in the Metastatic Environment
- (i)
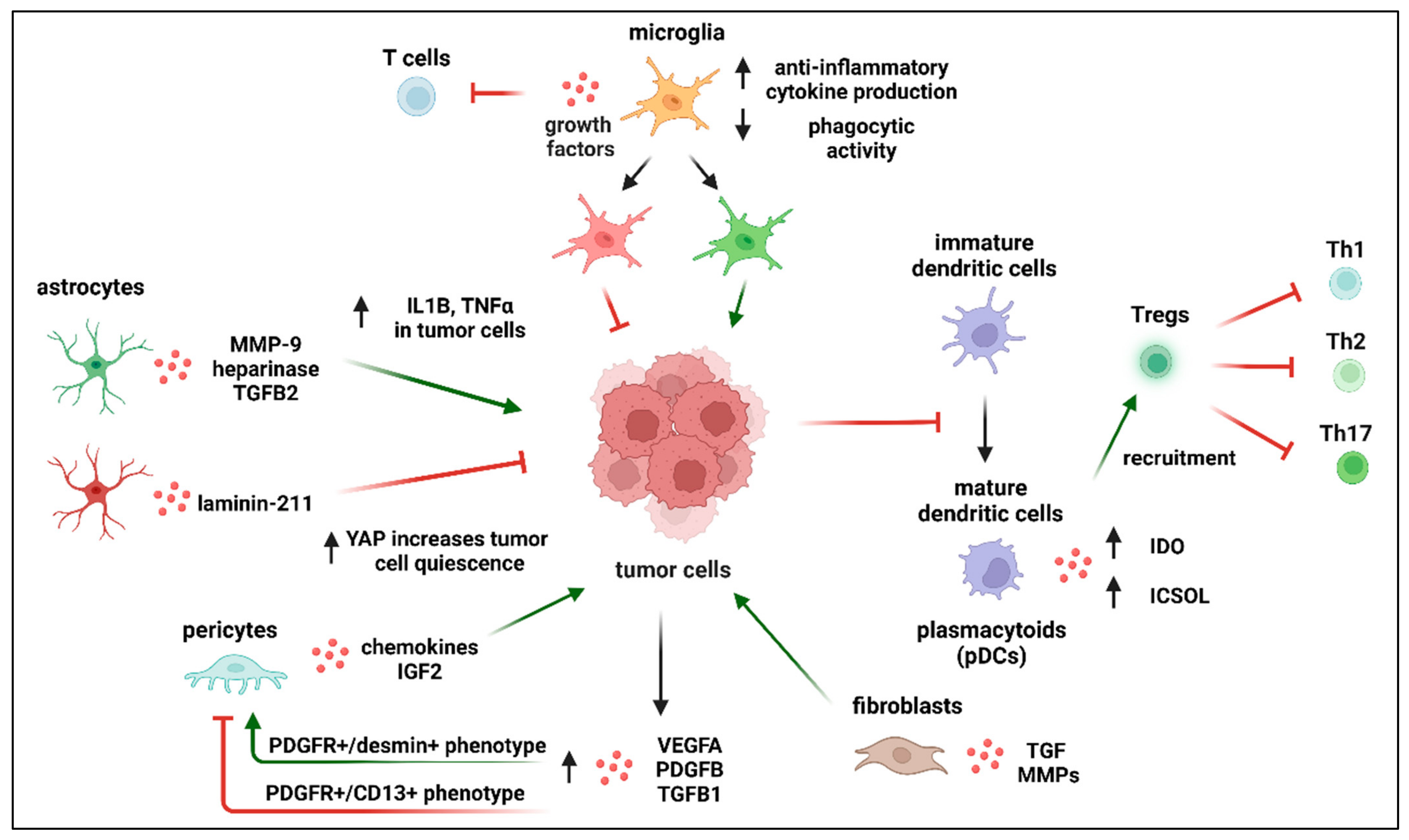
- Activation of microglia in the areas surrounding metastatic sites has been widely documented [149,150]. These activated microglia are characterized by increased anti-inflammatory cytokine production, decreased phagocytic activity, growth factor release, a chemo-attractive effect on peripheral monocytes, and inhibition of T cell proliferation within the tumor microenvironment. However, they exhibit the upregulation of proteins that support tumor cell adhesions such as: cellular adhesion molecules, CAMs, LFA-1, ALCAM, and E-selectin. It has been demonstrated that metastatic lung cancer cells produce IL6, which is able to induce anti-inflammatory action of microglia by switching JAK2/STAT3 signaling [151]. Furthermore, macrophages “cooperate” with cancer cells in a paracrinne manner, generating a positive feedback loop. Macrophages, by expressing EGF, promote protrusion formation in cancer cells, therefore enhancing cancer invasion. Simultaneously, cancer cells produce colony-stimulating factor-1 (CSF-1) that strenghtens EGF expression in macrophages. EGF in turn promotes CSF-1 expression in cancer cells [152];
- (ii)
- In glioblastoma, the most aggressive form of solid, primary tumor of the CNS, microglia contribute significantly to the total tumor mass, indicating that they play an important role in tumor progression within the CNS.
- (i)
- (ii)
- The observation that microglia represent a heterogeneous combination of different groups of subpopulations that differ in their inflammatory profiles considerably. These subpopulations contribute differently to the tumor microenvironment [155]. Those immune cells with a pro-inflammatory phenotype have been described as exerting an antitumorigenic effect, while those with an anti-inflammatory profile have shown tumor-supporting activity. Furthermore, the exact role of certain microglial subpopulations is still not clear. For example, disease-associated microglia (DAM) deep transcriptomic analysis revealed an upregulated profile of several microglial genes such as Lgals3, Trem2, NOS2, and COX1 [156]. However, how these different pathways interact with the invading cells is yet to be determined;
- (iii)
- Transition between pro- and anti-inflammatory states of the same subpopulation of microglia. The processes underlying the phenotypic switch between these two states and the potential coexistence of both identities during metastasis formation in the brain are poorly understood [157];
- (iv)
- Comparability of the phenotypes of microglia and bone-marrow-derived macrophages (BMDMs) infiltrating the brain. Microglia develop from embryonic yolk sac progenitor cells and remain in the CNS, but bone-marrow-derived macrophages (BMDMs) penetrate the brain during metastasis [158,159]. Up to now, no unique markers that can distinguish between the activities of microglia and BMDM have been discovered.
4. Future Prospects and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smedby, K.E.; Brandt, L.; Bäcklund, M.L.; Blomqvist, P. Brain Metastases Admissions in Sweden between 1987 and 2006. Br. J. Cancer 2009, 101, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Leibold, A.T.; Monaco, G.N.; Dey, M. The Role of the Immune System in Brain Metastasis. Curr. Neurobiol. 2019, 10, 33–48. [Google Scholar] [PubMed]
- Singh, R.; Stoltzfus, K.C.; Chen, H.; Louie, A.V.; Lehrer, E.J.; Horn, S.R.; Palmer, J.D.; Trifiletti, D.M.; Brown, P.D.; Zaorsky, N.G. Epidemiology of Synchronous Brain Metastases. Neuro-Oncol. Adv. 2020, 2, vdaa041. [Google Scholar] [CrossRef] [PubMed]
- Sacks, P.; Rahman, M. Epidemiology of Brain Metastases. Neurosurg. Clin. N. Am. 2020, 31, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Maniwa, T.; Ohmura, A.; Hiroshima, T.; Ike, A.; Kimura, T.; Nakamura, H.; Nakatsuka, S.I.; Okami, J.; Higashiyama, M. Number of Metastatic Lymph Nodes and Zones as Prognostic Factors in Non-Small-Cell Lung Cancer. Interact. Cardiovasc. Thorac. Surg. 2020, 31, 305–314. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Mesko, S.; Li, J.; Cagney, D.; Aizer, A.; Lin, N.U.; Nesbit, E.; Kruser, T.J.; Chan, J.; Braunstein, S.; et al. Survival in Patients With Brain Metastases: Summary Report on the Updated Diagnosis-Specific Graded Prognostic Assessment and Definition of the Eligibility Quotient. J. Clin. Oncol. 2020, 38, 3773–3784. [Google Scholar] [CrossRef]
- Stelzer, K. Epidemiology and Prognosis of Brain Metastases. Surg. Neurol. Int. 2013, 4, S192–S202. [Google Scholar] [CrossRef]
- Tsao, M.N.; Xu, W.; Wong, R.K.S.; Lloyd, N.; Laperriere, N.; Sahgal, A.; Rakovitch, E.; Chow, E. Whole Brain Radiotherapy for the Treatment of Newly Diagnosed Multiple Brain Metastases. Cochrane Database Syst. Rev. 2018, 1, CD003869. [Google Scholar] [CrossRef]
- Kancharla, P.; Ivanov, A.; Chan, S.; Ashamalla, H.; Huang, R.Y.; Yanagihara, T.K. The Effect of Brain Metastasis Location on Clinical Outcomes: A Review of the Literature. Neuro-Oncol. Adv. 2019, 1, vdz017. [Google Scholar] [CrossRef]
- Margarido, A.S.; Uceda-Castro, R.; Hahn, K.; de Bruijn, R.; Kester, L.; Hofland, I.; Lohuis, J.; Seinstra, D.; Broeks, A.; Jonkers, J.; et al. Epithelial-to-Mesenchymal Transition Drives Invasiveness of Breast Cancer Brain Metastases. Cancers 2022, 14, 3115. [Google Scholar] [CrossRef]
- Jeevan, D.S.; Cooper, J.B.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Molecular Pathways Mediating Metastases to the Brain via Epithelial-to-Mesenchymal Transition: Genes, Proteins, and Functional Analysis. Anticancer Res. 2016, 36, 523–532. [Google Scholar]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; Den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin–ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef]
- Xuan, B.; Ghosh, D.; Jiang, J.; Shao, R.; Dawson, M.R. Vimentin Filaments Drive Migratory Persistence in Polyploidal Cancer Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 26756–26765. [Google Scholar] [CrossRef]
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A Long Nuclear-Retained Non-Coding RNA Regulates Synaptogenesis by Modulating Gene Expression. EMBO J. 2010, 29, 3082–3093. [Google Scholar] [CrossRef]
- Yang, M.H.; Hu, Z.Y.; Xu, C.; Xie, L.Y.; Wang, X.Y.; Chen, S.Y.; Li, Z.G. MALAT1 Promotes Colorectal Cancer Cell Proliferation/Migration/Invasion via PRKA Kinase Anchor Protein 9. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 166–174. [Google Scholar] [CrossRef]
- Cai, X.; Liu, Y.; Yang, W.; Xia, Y.; Yang, C.; Yang, S.; Liu, X. Long Noncoding RNA MALAT1 as a Potential Therapeutic Target in Osteosarcoma. J. Orthop. Res. 2016, 34, 932–941. [Google Scholar] [CrossRef]
- Tang, Y.; He, Y.; Zhang, P.; Wang, J.; Fan, C.; Yang, L.; Xiong, F.; Zhang, S.; Gong, Z.; Nie, S.; et al. LncRNAs Regulate the Cytoskeleton and Related Rho/ROCK Signaling in Cancer Metastasis. Mol. Cancer 2018, 17, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Meshkat, B.I.; Liechty, K.W.; Xu, J. Lncrna Malat1 Modulates Tgf-Β1-Induced Emt in Keratinocyte. Int. J. Mol. Sci. 2021, 22, 11816. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Q.; Chang, L.; Wei, C.; Bei, H.; Yin, Y.; Chen, M.; Wang, H.; Liang, J.; Wu, Y. LncRNA MALAT1 Modulates Ox-LDL Induced EndMT through the Wnt/β-Catenin Signaling Pathway. Lipids Health Dis. 2019, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Piao, H.L.; Kim, B.J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N.; et al. Long Noncoding RNA MALAT1 Suppresses Breast Cancer Metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.N.; Appel, B. Oligodendrocytes Express Synaptic Proteins That Modulate Myelin Sheath Formation. Nat. Commun. 2019, 10, 4125. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Gires, O. EpCAM (CD326) Finding Its Role in Cancer. Br. J. Cancer 2007, 96, 417–423. [Google Scholar] [CrossRef]
- Duan, S.; Luo, X.; Zeng, H.; Zhan, X.; Yuan, C. SNORA71B Promotes Breast Cancer Cells across Blood–Brain Barrier by Inducing Epithelial-Mesenchymal Transition. Breast Cancer 2020, 27, 1072–1081. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Metheny-Barlow, L.; Lo, H.W. EGFR and HER2 Signaling in Breast Cancer Brain Metastasis. Front. Biosci. Elit. 2016, 8, 245–263. [Google Scholar]
- Schmitz, K.J.; Grabellus, F.; Callies, R.; Otterbach, F.; Wohlschlaeger, J.; Levkau, B.; Kimmig, R.; Schmid, K.W.; Baba, H.A. High Expression of Focal Adhesion Kinase (P125FAK) in Node-Negative Breast Cancer Is Related to Overexpression of HER-2/Neu and Activated Akt Kinase but Does Not Predict Outcome. Breast Cancer Res. 2005, 7, R194–R203. [Google Scholar] [CrossRef]
- Brennan, K.; McSherry, E.A.; Hudson, L.; Kay, E.W.; Hill, A.D.K.; Young, L.S.; Hopkins, A.M. Junctional Adhesion Molecule-A Is Co-Expressed with HER2 in Breast Tumors and Acts as a Novel Regulator of HER2 Protein Degradation and Signaling. Oncogene 2013, 32, 2799–2804. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted Therapeutic Options and Future Perspectives for HER2-Positive Breast Cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef]
- Gupta, P.; Srivastava, S.K. HER2 Mediated de Novo Production of TGFβ Leads to SNAIL Driven Epithelial-to-Mesenchymal Transition and Metastasis of Breast Cancer. Mol. Oncol. 2014, 8, 1532–1547. [Google Scholar] [CrossRef]
- Hsu, J.C.C.; Reid, D.W.; Hoffman, A.M.; Sarkar, D.; Nicchitta, C.V. Oncoprotein AEG-1 Is an Endoplasmic Reticulum RNA-Binding Protein Whose Interactome Is Enriched in Organelle Resident Protein-Encoding MRNAs. RNA 2018, 24, 688–703. [Google Scholar] [CrossRef]
- Manna, D.; Sarkar, D. Multifunctional Role of Astrocyte Elevated Gene-1 (Aeg-1) in Cancer: Focus on Drug Resistance. Cancers 2021, 13, 1792. [Google Scholar] [CrossRef]
- Zhou, Z.; Deng, H.; Yan, W.; Luo, M.; Tu, W.; Xia, Y.; He, J.; Han, P.; Fu, Y.; Tian, D. AEG-1 Promotes Anoikis Resistance and Orientation Chemotaxis in Hepatocellular Carcinoma Cells. PLoS ONE 2014, 9, e100372. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. Mechanisms of AEG-1 and CXCR4 gene expression regulating the epithelial-mesenchymal transition pathway involved in brain metastases of breast cancer. J. Balk. Union Oncol. 2017, 22, 953–957. [Google Scholar]
- Kim, W.K.; Kwon, Y.; Jang, M.; Park, M.; Kim, J.; Cho, S.; Jang, D.G.; Lee, W.-B.; Jung, S.H.; Choi, H.J.; et al. β-catenin activation down-regulates cell-cell junction-related genes and induces epithelial-to-mesenchymal transition in colorectal cancers. Sci. Rep. 2019, 9, 18440. [Google Scholar] [CrossRef]
- Garrod, D.; Chidgey, M. Desmosome Structure, Composition and Function. Biochim. Biophys. Acta Bio. 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Moll, R. Molecular diversity of cytokeratins: Significance for cell and tumor differentiation. Acta Histochem. Suppl. 1991, 41, 117–127. [Google Scholar]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Proteolytic interstitial cell migration: A five-step process. Cancer Metastasis Rev. 2009, 28, 129–135. [Google Scholar] [CrossRef]
- Klemke, R.L. Trespassing Cancer Cells: “fingerprinting” Invasive Protrusions Reveals Metastatic Culprits. Curr. Opin. Cell Biol. 2012, 24, 662669. [Google Scholar] [CrossRef] [PubMed]
- Dorfleutner, A.; Stehlik, C.; Zhang, J.; Gallick, G.E.; Flynn, D.C. AFAP-110 is required for actin stress fiber formation and cell adhesion in MDA-MB-231 breast cancer cells. J. Cell Physiol. 2007, 213, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The “ins” and “Outs” of Podosomes and Invadopodia: Characteristics, Formation and Function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Saykali, B.A.; El-Sibai, M. Invadopodia, Regulation, and Assembly in Cancer Cell Invasion. Cell Commun. Adhes. 2014, 21, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the Scaffold to Improve Focal Adhesion Kinase–Targeted Cancer Therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar] [CrossRef]
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef]
- Barney, L.E.; Hall, C.L.; Schwartz, A.D.; Parks, A.N.; Sparages, C.; Galarza, S.; Platt, M.O.; Mercurio, A.M.; Peyton, S.R. Tumor cell–organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 2020, 6, eaaz4157. [Google Scholar] [CrossRef]
- Saad, S.; Gottlieb, D.J.; Bradstock, K.F.; Overall, C.M.; Bendall, L.J. Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Res. 2002, 62, 283–289. [Google Scholar]
- Rawlings, N.D.; Barrett, A.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2011, 40, D343–D350. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, I.; Cantelli, G.; Bruce, F.; Sanz-Moreno, V. Rho, ROCK and actomyosin contractility in metastasis as drug targets. F1000Research 2016, 5, 783. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.-T. The Role of Tumor Stroma in Cancer Progression and Prognosis: Emphasis on Carcinoma-Associated Fibroblasts and Non-small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells. In Advances in Cancer Research; ScienceDirect: Amsterdam, The Netherlands, 2015; pp. 95–139. ISBN 9780128023167. [Google Scholar]
- Chiang, S.P.H.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. 2016, 39, 397–410. [Google Scholar] [CrossRef]
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018, 359, 1408–1411. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; von der Weid, P. Lymphatic system: An active pathway for immune protection. Semin. Cell Dev. Biol. 2015, 38, 83–89. [Google Scholar] [CrossRef]
- Ganss, R. Tumour vessel remodelling: New opportunities in cancer treatment. Vasc. Biol. 2020, 2, R35–R43. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix: Biosynthesis, Remodeling, and Functions during Vascular Morphogenesis and Neovessel Stabilization. Circ. Res. 2005, 8, 245–263. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef]
- Danussi, C.; Spessotto, P.; Petrucco, A.; Wassermann, B.; Sabatelli, P.; Montesi, M.; Doliana, R.; Bressan, G.M.; Colombatti, A. Emilin1 Deficiency Causes Structural and Functional Defects of Lymphatic Vasculature. Mol. Cell Biol. 2008, 28, 4026–4039. [Google Scholar] [CrossRef]
- Wu, Q.; Li, X.; Yang, H.; Lu, C.; You, J.; Zhang, Z. Extracellular matrix protein 1 is correlated to carcinogenesis and lymphatic metastasis of human gastric cancer. World J. Surg. Oncol. 2014, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Krog, B.L.; Henry, M.D. Biomechanics of the Circulating Tumor Cell Microenvironment. Adv. Exp. Med. Biol. 2018, 1092, 209–233. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-J.; Sung, J.Y.; Kim, S.-H.; Yun, U.-J.; Kim, H.; Jang, E.-J.; Yoo, H.-E.; Hong, E.K.; Goh, S.-H.; Moon, A.; et al. Fibronectin regulates anoikis resistance via cell aggregate formation. Cancer Lett. 2021, 508, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Maeshiro, M.; Shinriki, S.; Liu, R.; Nakachi, Y.; Komohara, Y.; Fujiwara, Y.; Ohtsubo, K.; Yoshida, R.; Iwamoto, K.; Nakayama, H.; et al. Colonization of distant organs by tumor cells generating circulating homotypic clusters adaptive to fluid shear stress. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Yoon, S.R.; Kim, T.-D.; Choi, I. Understanding of molecular mechanisms in natural killer cell therapy. Exp. Mol. Med. 2015, 47, e141. [Google Scholar] [CrossRef] [Green Version]
- Trikha, M.; Zhou, Z.; Timar, J.; Raso, E.; Kennel, M.; Emmell, E.; Nakada, M.T. Multiple Roles for Platelet GPIIb/IIIa and Avβ3 Integrins in Tumor Growth, Angiogenesis, and Metastasis. Cancer Res. 2002, 62, 2824–2833. [Google Scholar]
- Egan, K.; Cooke, N.; Kenny, D. Living in shear: Platelets protect cancer cells from shear induced damage. Clin. Exp. Metastasis 2014, 31, 697–704. [Google Scholar] [CrossRef]
- Ward, M.P.; Kane, L.E.; Norris, L.A.; Mohamed, B.M.; Kelly, T.; Bates, M.; Clarke, A.; Brady, N.; Martin, C.M.; Brooks, R.D.; et al. Platelets, Immune Cells and the Coagulation Cascade; Friend or Foe of the Circulating Tumour Cell? Mol. Cancer 2021, 20, 1–17. [Google Scholar] [CrossRef]
- Anvari, S.; Osei, E.; Maftoon, N. Interactions of platelets with circulating tumor cells contribute to cancer metastasis. Sci. Rep. 2021, 11, 15477. [Google Scholar] [CrossRef]
- Maurer, S.; Ferrari de Andrade, L. NK Cell Interaction with Platelets and Myeloid Cells in the Tumor Milieu. Front. Immunol. 2020, 11, 608849. [Google Scholar] [CrossRef]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-Derived MHC Class I Confers a Pseudonormal Phenotype to Cancer Cells That Subverts the Antitumor Reactivity of Natural Killer Immune Cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, X.H.-F.; Massagué, J. Macrophage Binding to Receptor VCAM-1 Transmits Survival Signals in Breast Cancer Cells that Invade the Lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef]
- Konstantopoulos, K.; Thomas, S.N. Cancer Cells in Transit: The Vascular Interactions of Tumor Cells. Annu. Rev. Biomed. Eng. 2009, 11, 177–202. [Google Scholar] [CrossRef]
- Lee, B.-C.; Lee, T.-H.; Avraham, S.; Avraham, H.K. Involvement of the Chemokine Receptor CXCR4 and Its Ligand Stromal Cell-Derived Factor 1α in Breast Cancer Cell Migration Through Human Brain Microvascular Endothelial Cells. Mol. Cancer Res. 2004, 2, 327–338. [Google Scholar] [CrossRef]
- Lorger, M.; Felding-Habermann, B. Capturing Changes in the Brain Microenvironment during Initial Steps of Breast Cancer Brain Metastasis. Am. J. Pathol. 2010, 176, 2958–2971. [Google Scholar] [CrossRef]
- Herman, H.; Fazakas, C.; Haskó, J.; Molnár, K.; Mészáros, Á.; Nyúl-Tóth, Á.; Szabó, G.; Erdélyi, F.; Ardelean, A.; Hermenean, A.; et al. Paracellular and transcellular migration of metastatic cells through the cerebral endothelium. J. Cell. Mol. Med. 2019, 23, 2619–2631. [Google Scholar] [CrossRef] [Green Version]
- Abidine, Y.; Constantinescu, A.; Laurent, V.M.; Rajan, V.S.; Michel, R.; Laplaud, V.; Duperray, A.; Verdier, C. Mechanosensitivity of Cancer Cells in Contact with Soft Substrates Using AFM. Biophys. J. 2018, 114, 1165–1175. [Google Scholar] [CrossRef]
- Winkler, F. The brain metastatic niche. J. Mol. Med. 2015, 93, 1213–1220. [Google Scholar] [CrossRef]
- Kienast, Y.; Von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.F.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2009, 16, 116–122. [Google Scholar] [CrossRef]
- Lu, W.; Bucana, C.D.; Schroit, A.J. Pathogenesis and vascular integrity of breast cancer brain metastasis. Int. J. Cancer 2006, 120, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The Vascular Basement Membrane as “Soil” in Brain Metastasis. PLoS ONE 2009, 4, e5857. [Google Scholar] [CrossRef] [PubMed]
- Maraveyas, A.; Johnson, M.; Xiao, Y.P.; Noble, S. Malignant melanoma as a target malignancy for the study of the anti-metastatic properties of the heparins. Cancer Metastasis Rev. 2010, 29, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.M.; Duperray, A.; Rajan, V.S.; Verdier, C.; Laurent, V.M.; Duperray, A.; Rajan, V.S.; Verdier, C. Atomic Force Microscopy Reveals a Role for Endothelial Cell ICAM-1 Expression in Bladder Cancer Cell Adherence. PLoS ONE 2014, 9, e98034. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Yang, S.-H.; Lee, J.E.; Cho, D.G.; Kim, H.K.; Kim, S.H.; Kim, I.S.; Hong, J.T.; Sung, J.H.; Son, B.C.; et al. E-cadherin as a predictive marker of brain metastasis in non-small-cell lung cancer, and its regulation by pioglitazone in a preclinical model. J. Neuro-Oncol. 2012, 109, 219–227. [Google Scholar] [CrossRef]
- Yoshimasu, T.; Sakurai, T.; Oura, S.; Hirai, I.; Tanino, H.; Kokawa, Y.; Naito, Y.; Okamura, Y.; Ota, I.; Tani, N.; et al. Increased expression of integrin alpha3beta1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Sci. 2004, 95, 142–148. [Google Scholar] [CrossRef]
- Mickael, M.-E.; Kubick, N.; Klimovich, P.; Flournoy, P.; Bieńkowska, I.; Sacharczuk, M. Paracellular and Transcellular Leukocytes Diapedesis Are Divergent but Interconnected Evolutionary Events. Genes 2021, 12, 254. [Google Scholar] [CrossRef]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef]
- Nakajima, M.; Katayama, K.-I.; Tamechika, I.; Hayashi, K.; Amano, Y.; Uehata, M.; Goto, N.; Kondo, T. WF-536 inhibits metastatic invasion by enhancing the host cell barrier and inhibiting tumour cell motility. Clin. Exp. Pharmacol. Physiol. 2003, 30, 457–463. [Google Scholar] [CrossRef]
- Godinho-Pereira, J.; Garcia, A.; Figueira, I.; Malhó, R.; Brito, M. Behind Brain Metastases Formation: Cellular and Molecular Alterations and Blood–Brain Barrier Disruption. Int. J. Mol. Sci. 2021, 22, 7057. [Google Scholar] [CrossRef]
- Khuon, S.; Liang, L.; Dettman, R.W.; Sporn, P.H.S.; Wysolmerski, R.B.; Chew, T.-L. Myosin light chain kinase mediates transcellular intravasation of breast cancer cells through the underlying endothelial cells: A three-dimensional FRET study. J. Cell Sci. 2010, 123, 431–440. [Google Scholar] [CrossRef]
- Figueira, I.; Galego, S.; Custódio-Santos, T.; Vicente, R.; Molnár, K.; Haskó, J.; Malhó, R.; Videira, M.; Wilhelm, I.; Krizbai, I.; et al. Picturing Breast Cancer Brain Metastasis Development to Unravel Molecular Players and Cellular Crosstalk. Cancers 2021, 13, 910. [Google Scholar] [CrossRef]
- Blethen, K.E.; Arsiwala, T.A.; Fladeland, R.A.; Sprowls, S.A.; Panchal, D.M.; Adkins, C.E.; Kielkowski, B.N.; Earp, L.E.; Glass, M.J.; Pritt, T.A.; et al. Modulation of the blood-tumor barrier to enhance drug delivery and efficacy for brain metastases. Neuro-Oncol. Adv. 2021, 3, v133–v143. [Google Scholar] [CrossRef]
- Allen, B.D.; Limoli, C.L. Breaking barriers: Neurodegenerative repercussions of radiotherapy induced damage on the blood-brain and blood-tumor barrier. Free Radic. Biol. Med. 2021, 178, 189–201. [Google Scholar] [CrossRef]
- Bernatz, S.; Ilina, E.I.; Devraj, K.; Harter, P.N.; Mueller, K.; Kleber, S.; Braun, Y.; Penski, C.; Renner, C.; Halder, R.; et al. Impact of Docetaxel on blood-brain barrier function and formation of breast cancer brain metastases. J. Exp. Clin. Cancer Res. 2019, 38, 434. [Google Scholar] [CrossRef]
- Takaishi, M.; Tarutani, M.; Takeda, J.; Sano, S. Mesenchymal to Epithelial Transition Induced by Reprogramming Factors Attenuates the Malignancy of Cancer Cells. PLoS ONE 2016, 11, e0156904. [Google Scholar] [CrossRef]
- Das, S.; Becker, B.N.; Hoffmann, F.M.; Mertz, J.E. Complete reversal of epithelial to mesenchymal transition requires inhibition of both ZEB expression and the Rho pathway. BMC Cell Biol. 2009, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Palen, K.; Weber, J.; Dwinell, M.B.; Johnson, B.D.; Ramchandran, R.; Gershan, J.A. E-cadherin re-expression shows in vivo evidence for mesenchymal to epithelial transition in clonal metastatic breast tumor cells. Oncotarget 2016, 7, 43363–43375. [Google Scholar] [CrossRef]
- Drees, F.; Pokutta, S.; Yamada, S.; Nelson, W.J.; Weis, W.I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 2005, 123, 903–915. [Google Scholar] [CrossRef]
- Kvokačková, B.; Remšík, J.; Jolly, M.; Souček, K. Phenotypic Heterogeneity of Triple-Negative Breast Cancer Mediated by Epithelial–Mesenchymal Plasticity. Cancers 2021, 13, 2188. [Google Scholar] [CrossRef]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Demmer, J.; Zhou, C.; Hubbard, M.J. Molecular cloning of ERp29, a novel and widely expressed resident of the endoplasmic reticulum. FEBS Lett. 1997, 402, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Brecker, M.; Khakhina, S.; Schubert, T.J.; Thompson, Z.; Rubenstein, R.C. The Probable, Possible, and Novel Functions of ERp. Front. Physiol. 2020, 11, 574339. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, E.; Baryshev, M.; Backlund, M.; Sharipo, A.; Mkrtchian, S. Genomic organization and promoter characterization of the gene encoding a putative endoplasmic reticulum chaperone, ERp. Gene 2002, 285, 127–139. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, J.C.; Sayer, R.J.; Lucocq, J.; Hubbard, M.J. ERp29, a general endoplasmic reticulum marker, is highly expressed throughout the brain. J. Comp. Neurol. 2004, 477, 29–42. [Google Scholar] [CrossRef]
- Bambang, I.F.; Xu, S.; Zhou, J.; Salto-Tellez, M.; Sethi, S.K.; Zhang, D. Overexpression of endoplasmic reticulum protein 29 regulates mesenchymal–epithelial transition and suppresses xenograft tumor growth of invasive breast cancer cells. Lab. Investig. 2009, 89, 1229–1242. [Google Scholar] [CrossRef]
- Oberstein, A.; Shenk, T. Cellular responses to human cytomegalovirus infection: Induction of a mesenchymal-to-epithelial transition (MET) phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, E8244–E8253. [Google Scholar] [CrossRef] [Green Version]
- Taher, C.; de Boniface, J.; Mohammad, A.-A.; Religa, P.; Hartman, J.; Yaiw, K.-C.; Frisell, J.; Rahbar, A.; Söderberg-Naucler, C. High Prevalence of Human Cytomegalovirus Proteins and Nucleic Acids in Primary Breast Cancer and Metastatic Sentinel Lymph Nodes. PLoS ONE 2013, 8, e56795. [Google Scholar] [CrossRef]
- Taher, C.; Frisk, G.; Fuentes, S.; Religa, P.; Costa, H.; Assinger, A.; Vetvik, K.K.; Bukholm, I.R.; Yaiw, K.-C.; Smedby, K.E.; et al. High Prevalence of Human Cytomegalovirus in Brain Metastases of Patients with Primary Breast and Colorectal Cancers. Transl. Oncol. 2014, 7, 732–740. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A.; Dammacco, F. The Role of the Vascular Phase in Solid Tumor Growth: A Historical Review. Neoplasia 1999, 1, 293–302. [Google Scholar] [CrossRef]
- Reijneveld, J.C.; Voest, E.E.; Taphoorn, M.J.B. Angiogenesis in malignant primary and metastatic brain tumors. J. Neurol. 2000, 247, 597–608. [Google Scholar] [CrossRef]
- Lorger, M.; Krueger, J.S.; O’Neal, M.; Staflin, K.; Felding-Habermann, B. Activation of tumor cell integrin αvβ3 controls angiogenesis and metastatic growth in the brain. Proc. Natl. Acad. Sci. USA 2009, 106, 10666–10671. [Google Scholar] [CrossRef]
- Pepper, M.S. Role of the Matrix Metalloproteinase and Plasminogen Activator–Plasmin Systems in Angiogenesis. Arter. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef]
- Wong, A.P.; Cortez, S.L.; Baricos, W.H. Role of plasmin and gelatinase in extracellular matrix degradation by cultured rat mesangial cells. Am. J. Physiol. Physiol. 1992, 263, F1112–F1118. [Google Scholar] [CrossRef]
- Gupta, M.K.; Qin, R.Y. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Shibasaki, F. Hypoxia-Inducible Factor as an Angiogenic Master Switch. Front. Pediatr. 2015, 3, 33. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of Glioma-Associated Neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Chen, L.-T.; Xu, S.-D.; Xu, H.; Zhang, J.-F.; Ning, J.-F.; Wang, S.-F. MicroRNA-378 is associated with non-small cell lung cancer brain metastasis by promoting cell migration, invasion and tumor angiogenesis. Med. Oncol. 2011, 29, 1673–1680. [Google Scholar] [CrossRef]
- Hua, Z.; Lv, Q.; Ye, W.; Wong, C.-K.A.; Cai, G.; Gu, D.; Ji, Y.; Zhao, C.; Wang, J.; Yang, B.B.; et al. MiRNA-Directed Regulation of VEGF and Other Angiogenic Factors under Hypoxia. PLoS ONE 2006, 1, e116. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Ilhan-Mutlu, A.; Dinhof, C.; Magerle, M.; Hackl, M.; Widhalm, G.; Hainfellner, J.A.; Dieckmann, K.; Pichler, J.; Hutterer, M.; et al. Differential role of angiogenesis and tumour cell proliferation in brain metastases according to primary tumour type: Analysis of 639 cases. Neuropathol. Appl. Neurobiol. 2014, 41, e41–e55. [Google Scholar] [CrossRef] [PubMed]
- Salgado, K.B.; Toscani, N.V.; Silva, L.L.M.; Hilbig, A.; Barbosa-Coutinho, L.M. Immunoexpression of endoglin in brain metastasis secondary to malignant melanoma: Evaluation of angiogenesis and comparison with brain metastasis secondary to breast and lung carcinomas. Clin. Exp. Metastasis 2007, 24, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Rojiani, M.V.; Ghoshal-Gupta, S.; Kutiyanawalla, A.; Mathur, S.; Rojiani, A.M. TIMP-1 Overexpression in Lung Carcinoma Enhances Tumor Kinetics and Angiogenesis in Brain Metastasis. J. Neuropathol. Exp. Neurol. 2015, 74, 293–304. [Google Scholar] [CrossRef]
- Fisher, C.; Gilbertson-Beadling, S.; Powers, E.; Petzold, G.; Poorman, R.; Mitchell, M. Interstitial Collagenase Is Required for Angiogenesis in Vitro. Dev. Biol. 1994, 162, 499–510. [Google Scholar] [CrossRef]
- Lee, M.-H.; Atkinson, S.; Rapti, M.; Handsley, M.; Curry, V.; Edwards, D.; Murphy, G. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)-1 against native membrane type 1 matrix metalloproteinase (MT1-MMP) in a cell-based environment. Cancer Lett. 2010, 290, 114–122. [Google Scholar] [CrossRef]
- Krygier, S.; Djakiew, D. Neurotrophin receptor p75NTR suppresses growth and nerve growth factor-mediated metastasis of human prostate cancer cells. Int. J. Cancer 2001, 98, 1–7. [Google Scholar] [CrossRef]
- Nakamura, K.; Martin, K.C.; Jackson, J.K.; Beppu, K.; Woo, C.-W.; Thiele, C.J. Brain-Derived Neurotrophic Factor Activation of TrkB Induces Vascular Endothelial Growth Factor Expression via Hypoxia-Inducible Factor-1α in Neuroblastoma Cells. Cancer Res. 2006, 66, 4249–4255. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel Co-Option in Primary Human Tumors and Metastases: An Obstacle to Effective Anti-Angiogenic Treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Li, L.; Hanahan, D. Hijacking the Neuronal NMDAR Signaling Circuit to Promote Tumor Growth and Invasion. Cell 2013, 153, 86–100. [Google Scholar] [CrossRef]
- Deutsch, S.I.; Tang, A.H.; Burket, J.A.; Benson, A.D. NMDA Receptors on the Surface of Cancer Cells: Target for Chemotherapy? Biomed. Pharmacother. 2014, 68, 493–496. [Google Scholar] [CrossRef]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc. Natl. Acad. Sci. USA 2014, 111, 984–989. [Google Scholar] [CrossRef]
- Dai, J.; Cimino, P.J.; Gouin, K.H.; Grzelak, C.A.; Barrett, A.; Lim, A.R.; Long, A.; Weaver, S.; Saldin, L.T.; Uzamere, A.; et al. Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat. Cancer 2021, 3, 25–42. [Google Scholar] [CrossRef]
- Marchetti, D.; Li, J.; Shen, R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res. 2000, 60, 4767–4770. [Google Scholar]
- Hanibuchi, M.; Kim, S.-J.; Fidler, I.J.; Nishioka, Y. The molecular biology of lung cancer brain metastasis: An overview of current comprehensions and future perspectives. J. Med. Investig. 2014, 61, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Kobayashi, A.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Wilber, A.; Mo, Y.Y.; Moore, B.E.; et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med. 2013, 5, 384–396. [Google Scholar] [CrossRef]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. npj Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’Ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 2014, 137, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Haskó, J.; Fazakas, C.; Molnár, K.; Mészáros, A.; Patai, R.; Szabó, G.; Erdélyi, F.; Nyúl-Tóth, Á.; Győri, F.; Kozma, M.; et al. Response of the neurovascular unit to brain metastatic breast cancer cells. Acta Neuropathol. Commun. 2019, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Mészáros, A.; Fazakas, C.; Kozma, M.; Győri, F.; Reisz, Z.; Tiszlavicz, L.; Farkas, A.E.; Nyúl-Tóth, Á.; Haskó, J.; et al. Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol. Oncol. 2020, 14, 2040–2057. [Google Scholar] [CrossRef]
- Ippolitov, D.; Arreza, L.; Munir, M.N.; Hombach-Klonisch, S. Brain Microvascular Pericytes—More than Bystanders in Breast Cancer Brain Metastasis. Cells 2022, 11, 1263. [Google Scholar] [CrossRef]
- Menter, D.G.; Herrmann, J.L.; Nicolson, G.L. The role of trophic factors and autocrine/paracrine growth factors in brain metastasis. Clin. Exp. Metastasis 1995, 13, 67–88. [Google Scholar] [CrossRef]
- Poggi, A.; Musso, A.; Dapino, I.; Zocchi, M.R. Mechanisms of tumor escape from immune system: Role of mesenchymal stromal cells. Immunol. Lett. 2014, 159, 55–72. [Google Scholar] [CrossRef]
- Soto, M.S.; Sibson, N.R. The Multifarious Role of Microglia in Brain Metastasis. Front. Cell. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef] [Green Version]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol. 2018, 9, 697. [Google Scholar] [CrossRef]
- Jin, Y.; Kang, Y.; Wang, M.; Wu, B.; Su, B.; Yin, H.; Tang, Y.; Li, Q.; Wei, W.; Mei, Q.; et al. Targeting polarized phenotype of microglia via IL6/JAK2/STAT3 signaling to reduce NSCLC brain metastasis. Signal Transduct. Target. Ther. 2022, 7, 52. [Google Scholar] [CrossRef]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages Promote the Invasion of Breast Carcinoma Cells via a Colony-Stimulating Factor-1/Epidermal Growth Factor Paracrine Loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef]
- Louie, E.; Chen, X.F.; Coomes, A.; Ji, K.; Tsirka, S.; I Chen, E. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 2012, 32, 4064–4077. [Google Scholar] [CrossRef]
- Hwang, S.-Y.; Yoo, B.-C.; Jung, J.-W.; Oh, E.-S.; Hwang, J.-S.; Shin, J.-A.; Kim, S.-Y.; Cha, S.-H.; Han, I.-O. Induction of glioma apoptosis by microglia-secreted molecules: The role of nitric oxide and cathepsin B. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1656–1668. [Google Scholar] [CrossRef]
- Caponegro, M.D.; Oh, K.; Madeira, M.M.; Radin, D.; Sterge, N.; Tayyab, M.; Moffitt, R.A.; Tsirka, S.E. A distinct microglial subset at thetumor–stromainterface of glioma. Glia 2021, 69, 1767–1781. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Andreou, K.E.; Soto, M.S.; Allen, D.; Economopoulos, V.; De Bernardi, A.; Larkin, J.R.; Sibson, N.R. Anti-inflammatory Microglia/Macrophages As a Potential Therapeutic Target in Brain Metastasis. Front. Oncol. 2017, 7, 251. [Google Scholar] [CrossRef]
- Ginhoux, F.; Prinz, M. Origin of Microglia: Current Concepts and Past Controversies. Cold Spring Harb. Perspect. Biol. 2015, 7, a020537. [Google Scholar] [CrossRef]
- You, H.; Baluszek, S.; Kaminska, B. Supportive roles of brain macrophages in CNS metastases and assessment of new approaches targeting their functions. Theranostics 2020, 10, 2949–2964. [Google Scholar] [CrossRef]
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.-J.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 1268. [Google Scholar] [CrossRef]
- Bhaumik, S.; Mickael, M.E.; Moran, M.; Spell, M.; Basu, R. RORγt Promotes Foxp3 Expression by Antagonizing the Effector Program in Colonic Regulatory T Cells. J. Immunol. 2021, 207, 2027–2038. [Google Scholar] [CrossRef]
- Marciscano, A.E.; Anandasabapathy, N. The role of dendritic cells in cancer and anti-tumor immunity. Semin. Immunol. 2021, 52, 101481. [Google Scholar] [CrossRef] [PubMed]
- Mestrallet, G.; Sone, K.; Bhardwaj, N. Strategies to overcome DC dysregulation in the tumor microenvironment. Front. Immunol. 2022, 13, 5878. [Google Scholar] [CrossRef] [PubMed]
- Thepmalee, C.; Panya, A.; Junking, M.; Chieochansin, T.; Yenchitsomanus, P.-T. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum. Vaccines Immunother. 2018, 14, 1423–1431. [Google Scholar] [CrossRef]
- Mickael, M.E.; Bhaumik, S.; Chakraborti, A.; Umfress, A.A.; van Groen, T.; Macaluso, M.; Totenhagen, J.; Sorace, A.G.; Bibb, J.A.; Standaert, D.G.; et al. RORγt-Expressing Pathogenic CD4+ T Cells Cause Brain Inflammation during Chronic Colitis. J. Immunol. 2022, 208, 2054–2066. [Google Scholar] [CrossRef]
- Luo, L.; Liu, P.; Zhao, K.; Zhao, W.; Zhang, X. The Immune Microenvironment in Brain Metastases of Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 698844. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Whitley, S.K.; Bhaumik, S.; Zindl, C.L.; Schoeb, T.R.; Benveniste, E.N.; Pear, W.S.; Hatton, R.; Weaver, C.T. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell–iTreg cell balance. Nat. Immunol. 2015, 16, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.; Łazarczyk, M.; Kubick, N.; Klimovich, P.; Gurba, A.; Paszkiewicz, J.; Teodorowicz, P.; Kocki, T.; Horbańczuk, J.O.; Manda, G.; et al. Investigation of the Molecular Evolution of Treg Suppression Mechanisms Indicates a Convergent Origin. Curr. Issues Mol. Biol. 2023, 45, 628–648. [Google Scholar] [CrossRef]
- Edwar-Mickael, M.; Kubick, N. CD4+ Tregs may be essential for solving astrocyte glial scar deadlock. Neural Regen. Res. 2021, 16, 2563. [Google Scholar] [CrossRef]
- Kubick, N.; Klimovich, P.; Flournoy, P.; Bieńkowska, I.; Łazarczyk, M.; Sacharczuk, M.; Bhaumik, S.; Mickael, M.-E.; Basu, R. Interleukins and Interleukin Receptors Evolutionary History and Origin in Relation to CD4+ T Cell Evolution. Genes 2021, 12, 813. [Google Scholar] [CrossRef]
- Mickael, M.-E.; Bieńkowska, I.; Sacharczuk, M. An Update on the Evolutionary History of Bregs. Genes 2022, 13, 890. [Google Scholar] [CrossRef]
- Kubick, N.; Henckell Flournoy, P.C.; Klimovich, P.; Manda, G.; Mickael, M.E. What Has Single-Cell RNA Sequencing Revealed about Microglial Neuroimmunology? Immun. Inflamm. Dis. 2020, 8, 825–839. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Rate of Brain Metastasis (%) | Rate of Survival by Month |
|---|---|---|
| Colorectal cancer | 0.27 | 3–17 |
| Breast | 0.41 | 3–36 |
| Lung | 12 | 7–46 |
| Kidney | 1.48 | 4–35 |
| Melanoma | 0.65 | 5–34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łazarczyk, M.; Mickael, M.E.; Skiba, D.; Kurzejamska, E.; Ławiński, M.; Horbańczuk, J.O.; Radziszewski, J.; Fraczek, K.; Wolinska, R.; Paszkiewicz, J.; et al. The Journey of Cancer Cells to the Brain: Challenges and Opportunities. Int. J. Mol. Sci. 2023, 24, 3854. https://doi.org/10.3390/ijms24043854
Łazarczyk M, Mickael ME, Skiba D, Kurzejamska E, Ławiński M, Horbańczuk JO, Radziszewski J, Fraczek K, Wolinska R, Paszkiewicz J, et al. The Journey of Cancer Cells to the Brain: Challenges and Opportunities. International Journal of Molecular Sciences. 2023; 24(4):3854. https://doi.org/10.3390/ijms24043854
Chicago/Turabian StyleŁazarczyk, Marzena, Michel Edwar Mickael, Dominik Skiba, Ewa Kurzejamska, Michał Ławiński, Jarosław Olav Horbańczuk, Jakub Radziszewski, Karolina Fraczek, Renata Wolinska, Justyna Paszkiewicz, and et al. 2023. "The Journey of Cancer Cells to the Brain: Challenges and Opportunities" International Journal of Molecular Sciences 24, no. 4: 3854. https://doi.org/10.3390/ijms24043854