
SRSF3-Mediated Ki67 Exon 7-Inclusion Promotes Head and Neck Squamous Cell Carcinoma Progression via Repressing AKR1C2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
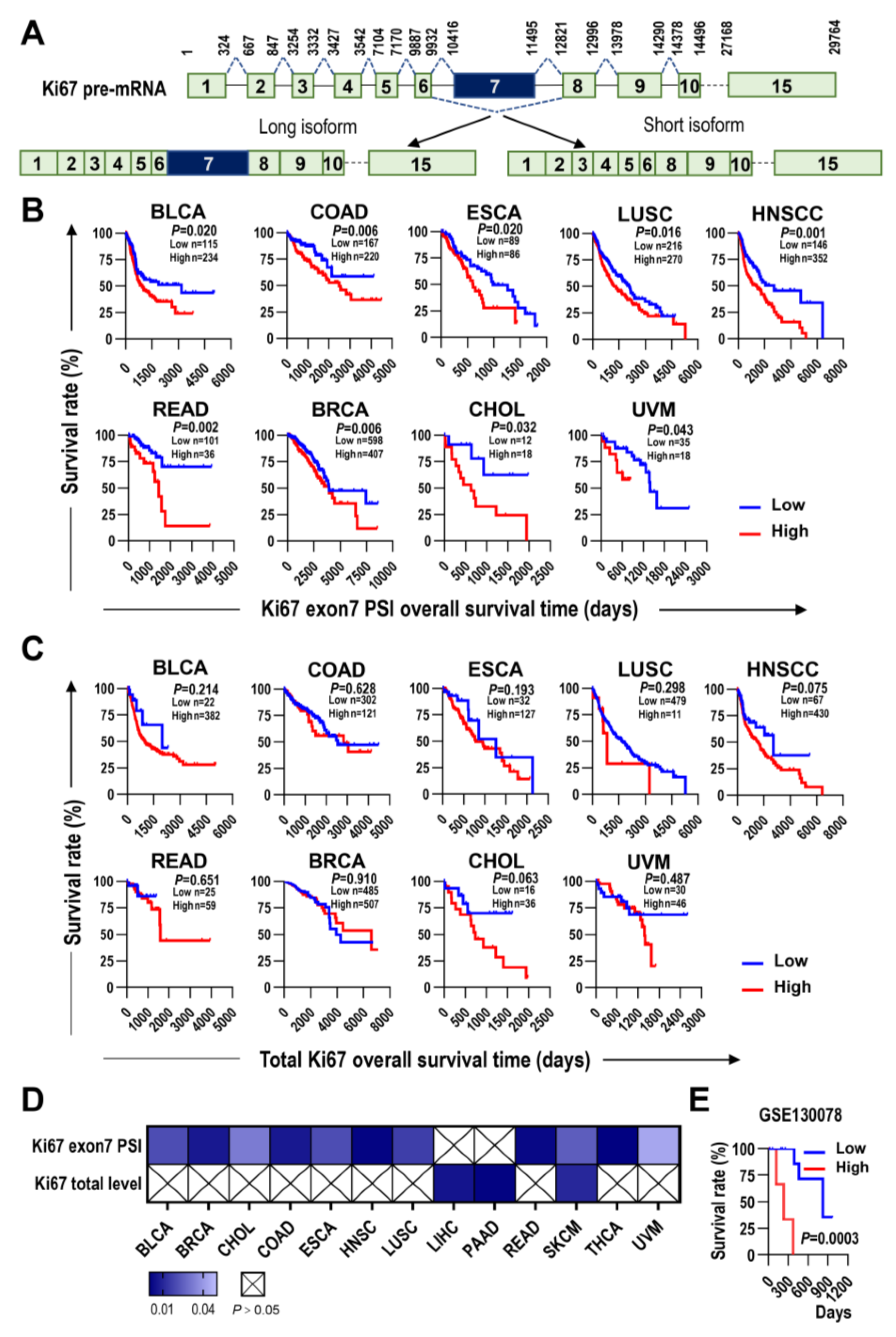
2.1. The Inclusion Level of Ki67 Exon 7 Is Superior to Ki67 Total Transcription Level in Overall Survival Prediction of Multiple Types of Cancer
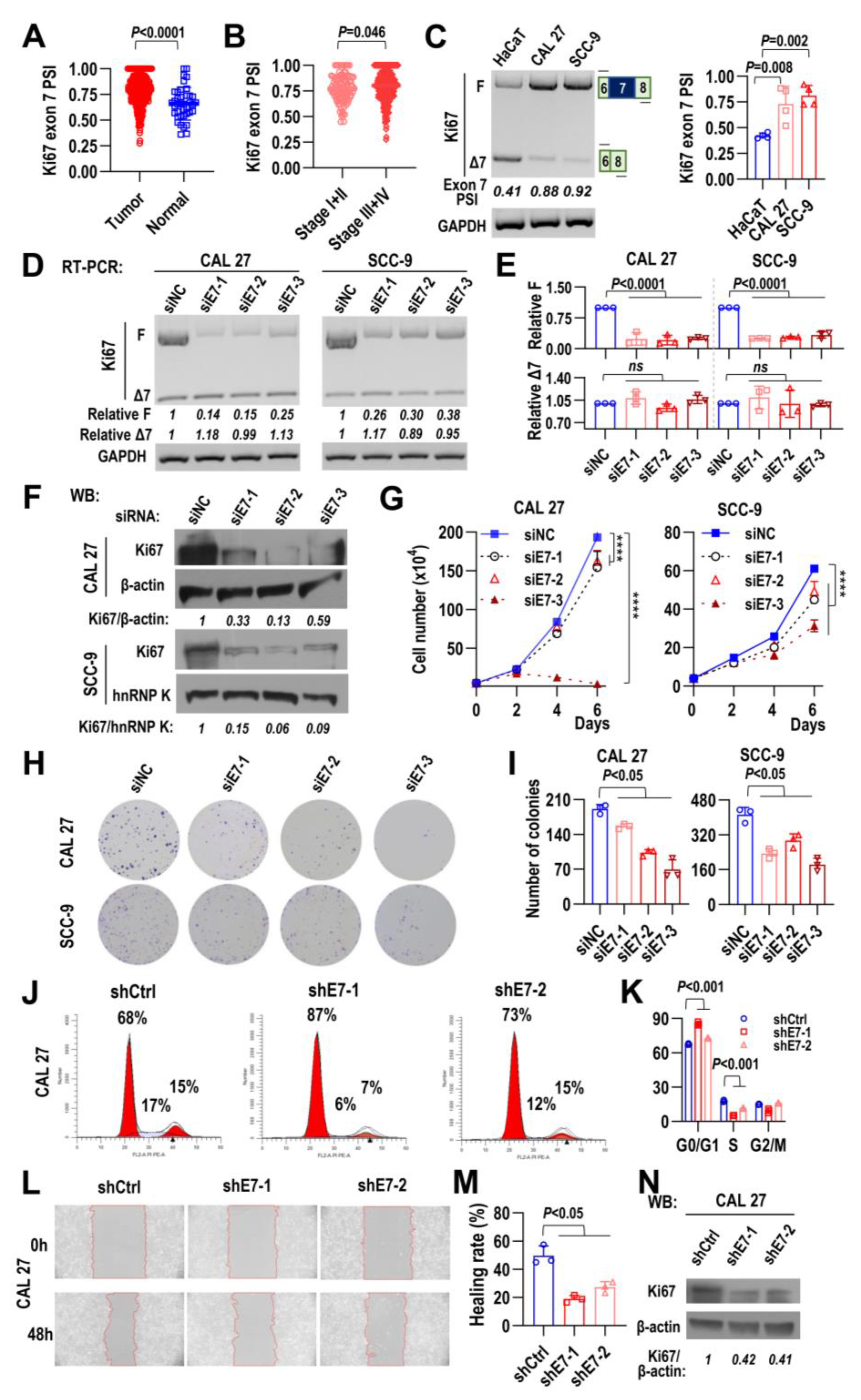
2.2. Ki67 Exon 7 Inclusion Is Required for HNSCC Cell Proliferation, Cell Cycle Progression, and Cell Migration
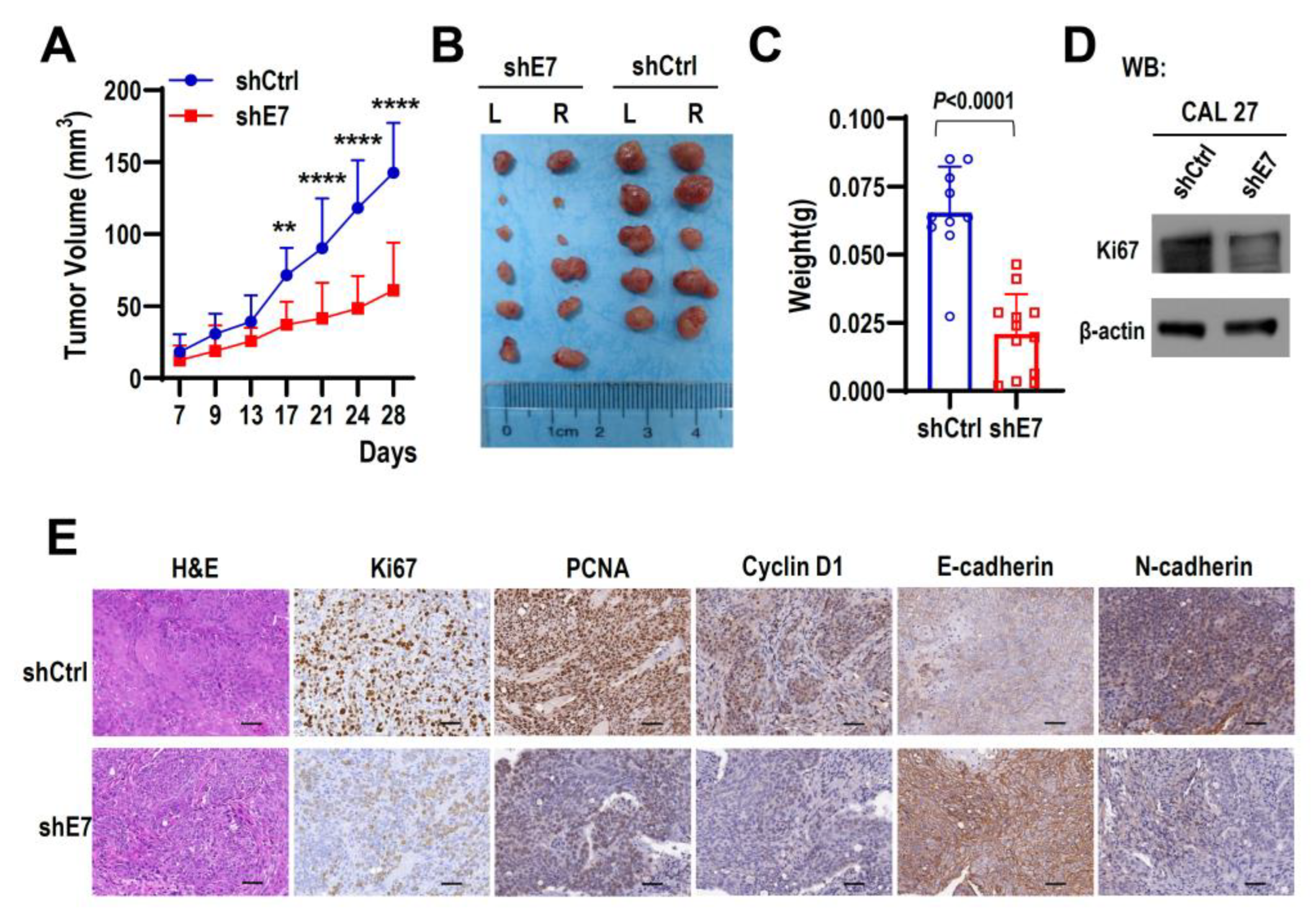
2.3. Ki67 Exon 7 Is Essential for Tumorigenicity and Of HNSCC Cells In Vivo
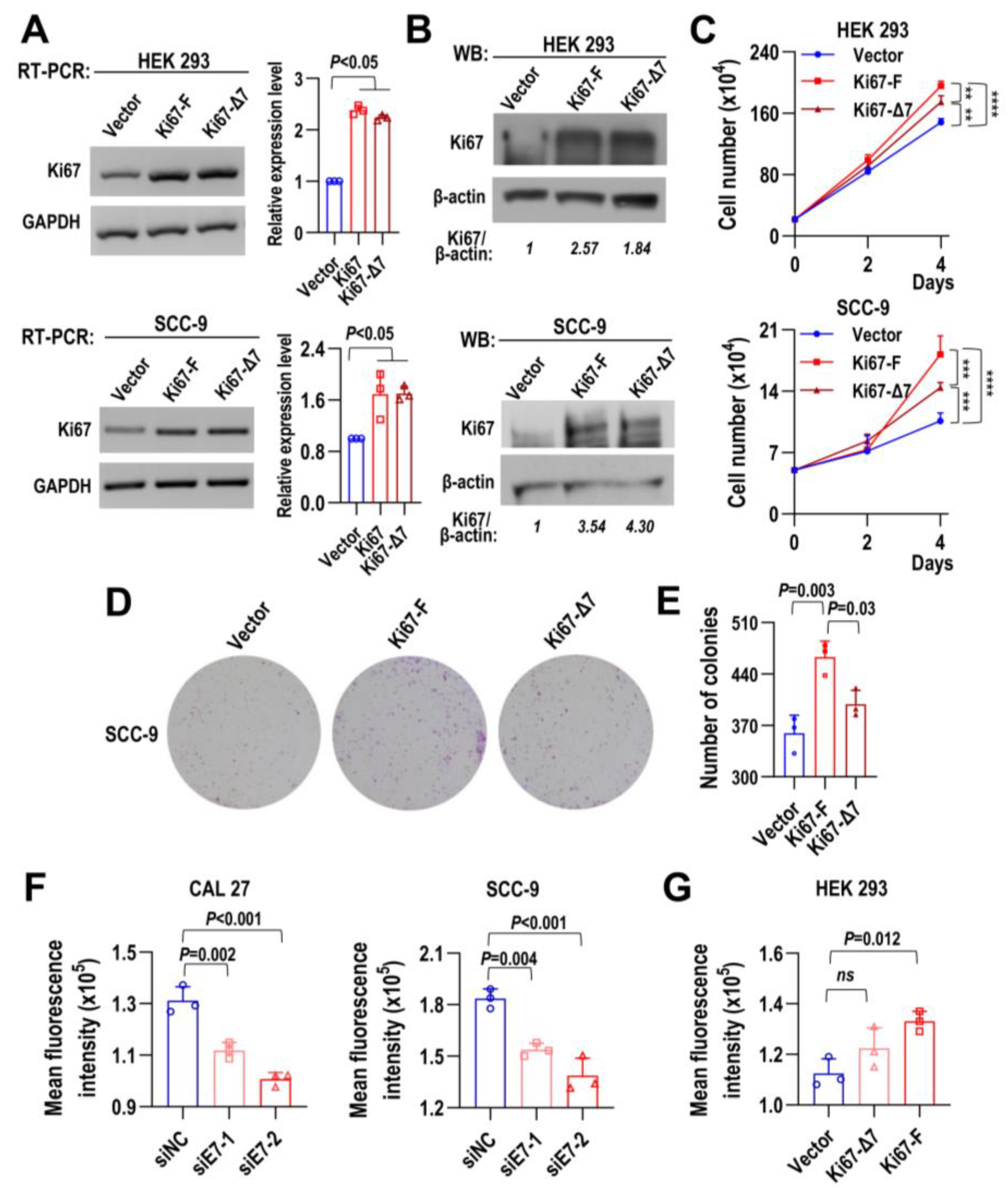
2.4. Full-Length Ki67 with Exon 7 Promotes Cell Proliferation and Colony Formation
2.5. Full-Length Ki67 with Exon 7 Increases Intracellular ROS Production
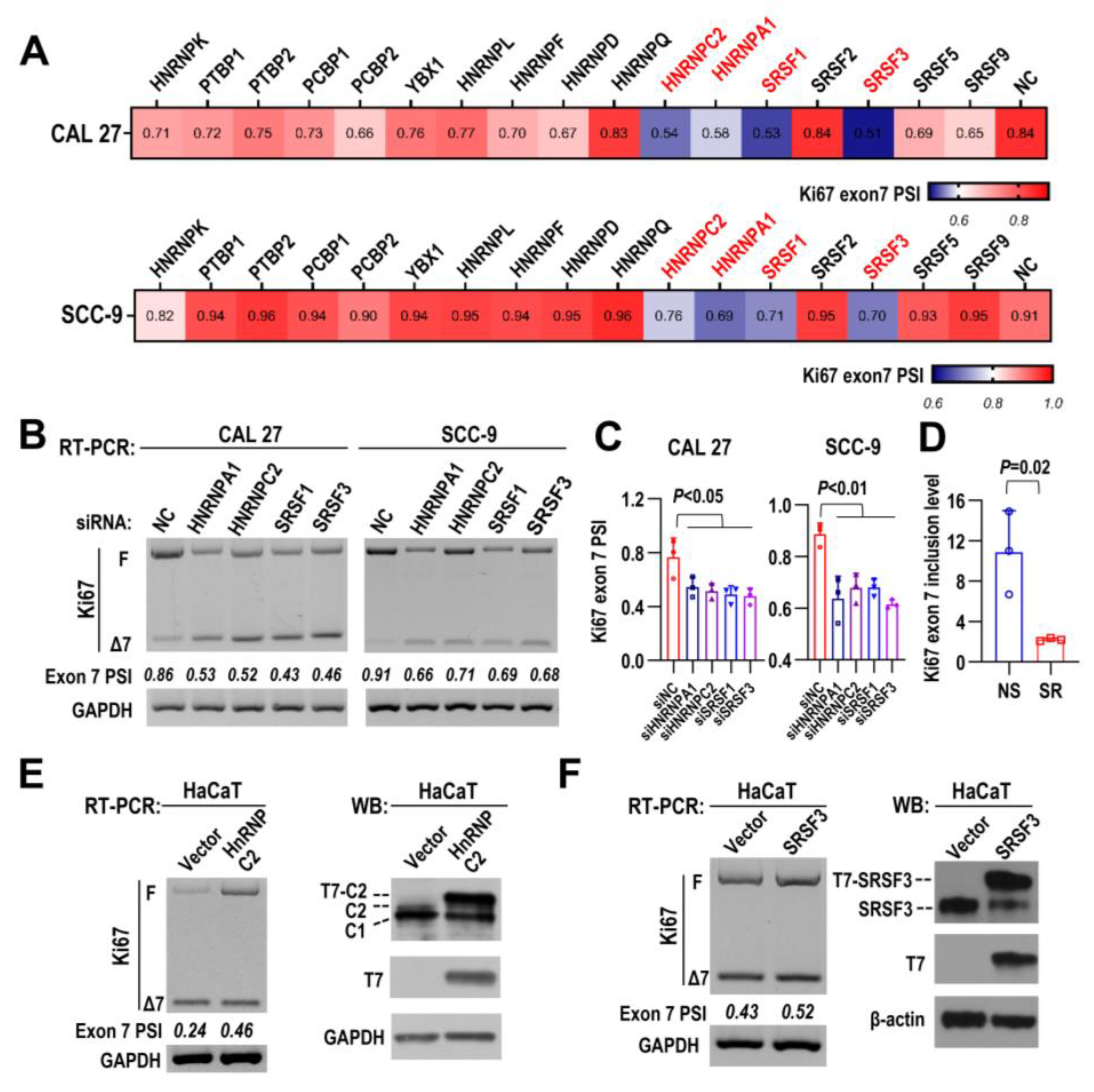
2.6. HNRNPC2 and SRSF3 Are Responsible for Ki67 Exon 7 Inclusion
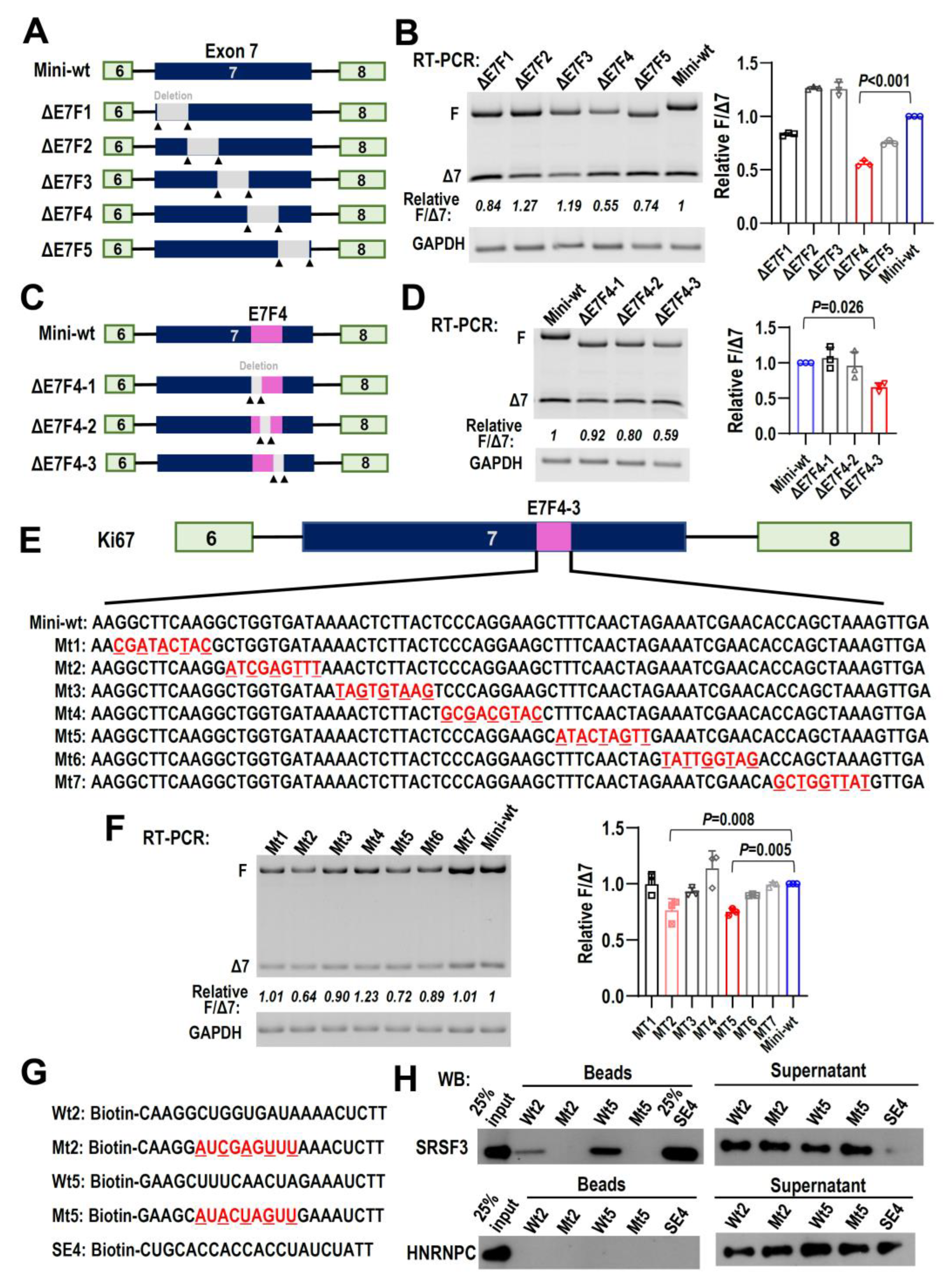
2.7. Splicing Factor SRSF3 Interacts with Motifs in Ki67 Exon 7
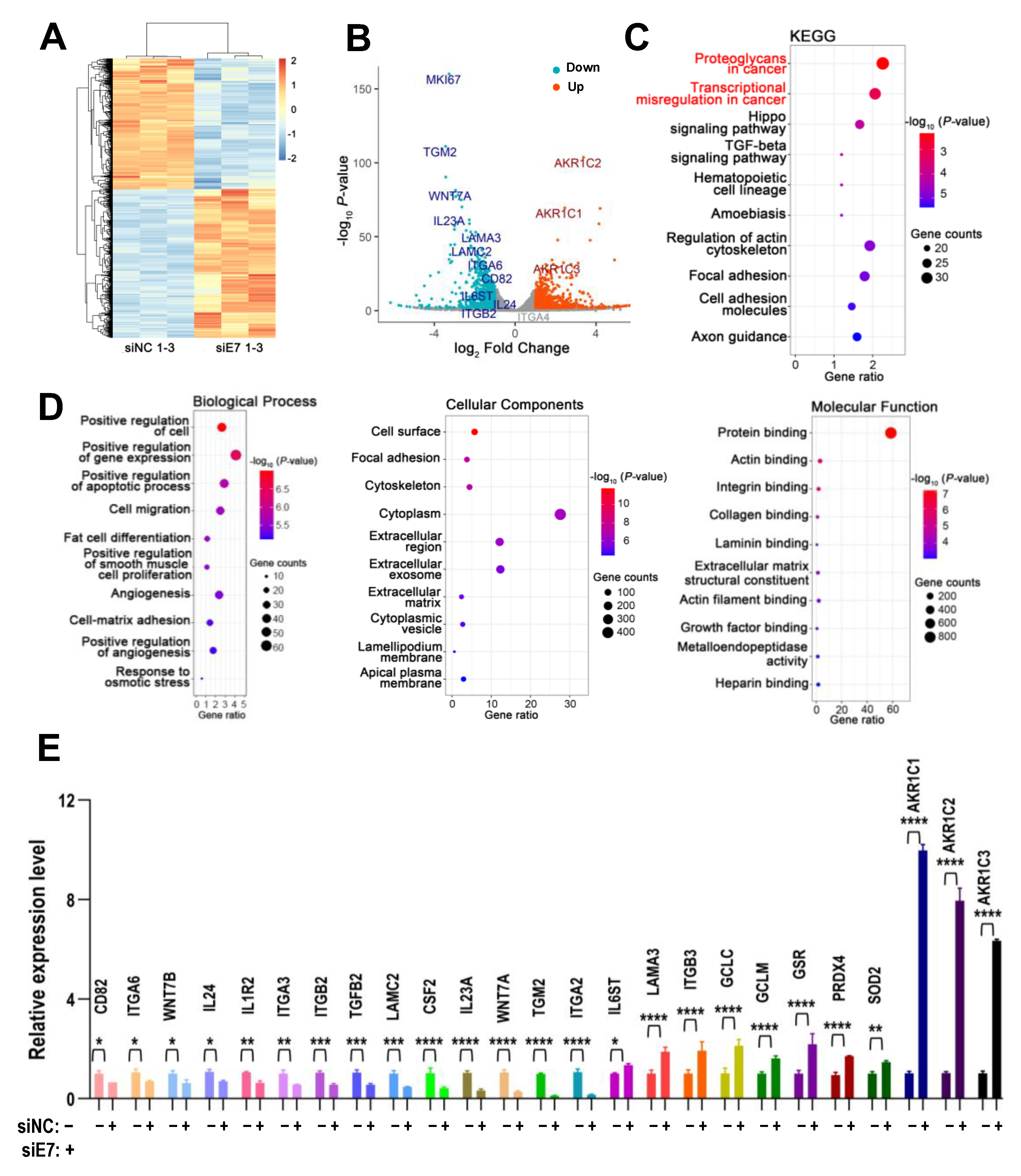
2.8. Cellular Targets of Ki67 Exon 7-Included Isoform in HNSCC Cells
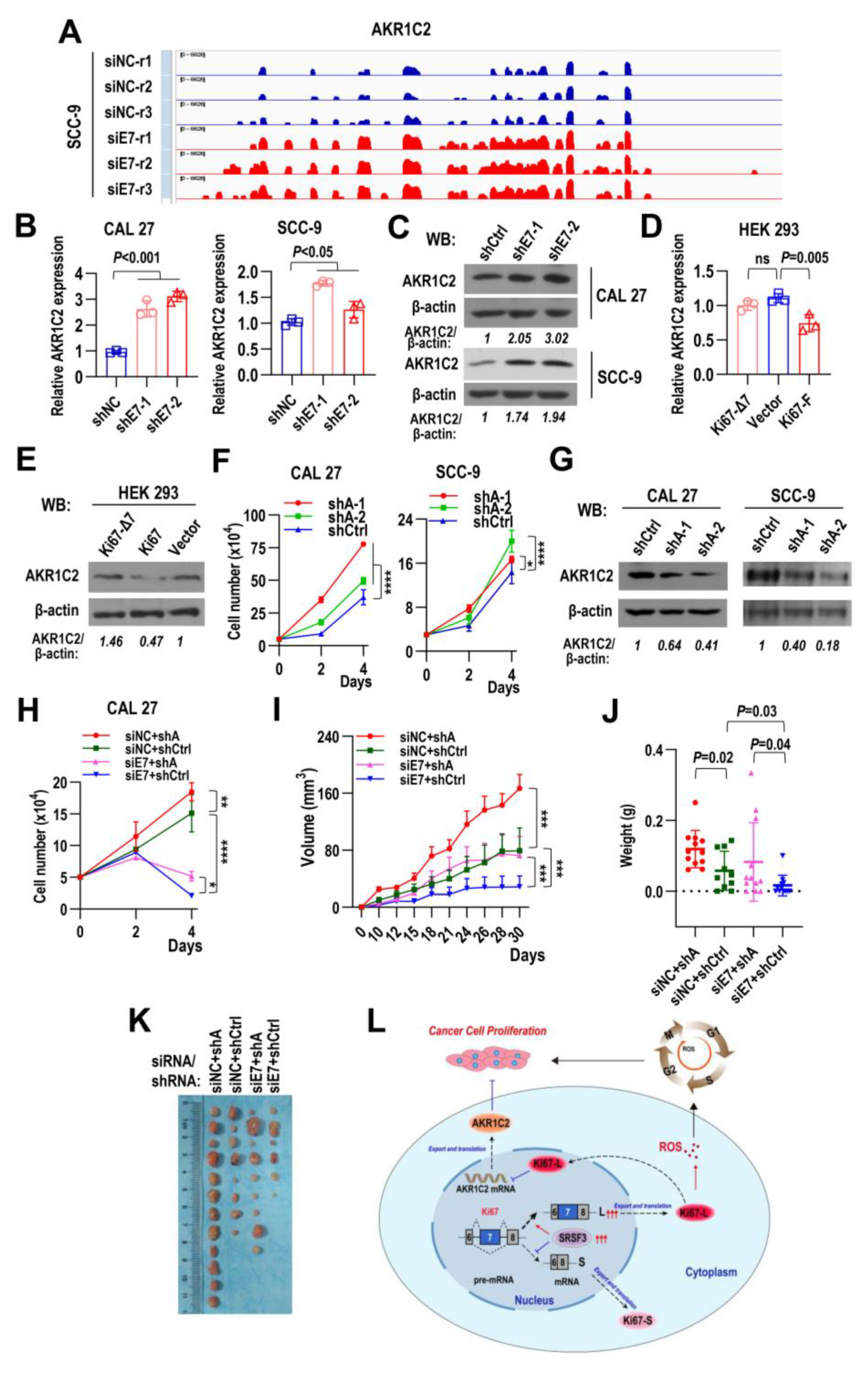
2.9. Ki67 Exon 7-Included Isoform Promotes Tumorigenesis by Inhibiting AKR1C2 Expression
3. Discussion
4. Materials and Methods
4.1. TCGA Datasets
4.2. GEO Dataset
4.3. Cell Culture
4.4. siRNA and Transfection
4.5. Plasmids and Transfection
4.6. Minigene Assay
4.7. Semiquantitative Reverse Transcription PCR (RT-PCR) and Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
4.8. RNA-seq
4.9. Western Blot
4.10. Colony Formation
4.11. Wound-Healing Migration Assay
4.12. Cell Cycle Analysis
4.13. Detection of ROS
4.14. RNA Pull-Down Assay
4.15. Tumorigenicity in Nude Mice
4.16. Histological and Immunohistological Analyses
4.17. Overall Survival Analysis
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schluter, C.; Duchrow, M.; Wohlenberg, C.; Becker, M.H.; Key, G.; Flad, H.D.; Gerdes, J. The Cell Proliferation-Associated Antigen of Antibody Ki-67: A Very Large, Ubiquitous Nuclear Protein with Numerous Repeated Elements, Representing a New Kind of Cell Cycle-Maintaining Proteins. J. Cell Biol. 1993, 123, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Duchrow, M.; Schluter, C.; Wohlenberg, C.; Flad, H.D.; Gerdes, J. Molecular Characterization of the Gene Locus of the Human Cell Proliferation-Associated Nuclear Protein Defined by Monoclonal Antibody Ki-67. Cell Prolif. 1996, 29, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Darzynkiewicz, Z. Cell Cycle Dependent Expression and Stability of the Nuclear Protein Detected by Ki-67 Antibody in Hl-60 Cells. Cell Prolif. 1992, 25, 31–40. [Google Scholar] [CrossRef]
- Scholzen, T.; Gerdes, J. The Ki-67 Protein: From the Known and the Unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Booth, D.G.; Takagi, M.; Sanchez-Pulido, L.; Petfalski, E.; Vargiu, G.; Samejima, K.; Imamoto, N.; Ponting, C.P.; Tollervey, D.; Earnshaw, W.C.; et al. Ki-67 Is a Pp1-Interacting Protein That Organises the Mitotic Chromosome Periphery. Elife 2014, 3, e01641. [Google Scholar] [CrossRef]
- Cuylen, S.; Blaukopf, C.; Politi, A.Z.; Müller-Reichert, T.; Neumann, B.; Poser, I.; Ellenberg, J.; Hyman, A.A.; Gerlich, D.W. Ki-67 Acts as a Biological Surfactant to Disperse Mitotic Chromosomes. Nature 2016, 535, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The Cell Proliferation Antigen Ki-67 Organises Heterochromatin. Elife 2016, 5, e13722. [Google Scholar] [CrossRef]
- Menon, S.S.; Guruvayoorappan, C.; Sakthivel, K.M.; Rasmi, R.R. Ki-67 Protein as a Tumour Proliferation Marker. Clin. Chim Acta 2019, 491, 39–45. [Google Scholar] [CrossRef]
- Andre, F.; Arnedos, M.; Goubar, A.; Ghouadni, A.; Delaloge, S. Ki67—No Evidence for Its Use in Node-Positive Breast Cancer. Nat. Rev. Clin. Oncol. 2015, 12, 296–301. [Google Scholar] [CrossRef]
- Ács, B.; Zámbó, V.; Vízkeleti, L.; Szász, A.M.; Madaras, L.; Szentmártoni, G.; Tőkés, T.; Molnár, B.; Molnár, I.A.; Vári-Kakas, S.; et al. Ki-67 as a Controversial Predictive and Prognostic Marker in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Diagn. Pathol. 2017, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, S.C.Y.; O Nielsen, T.; Zabaglo, L.; Arun, I.; Badve, S.S.; Bane, A.L.; Bartlett, J.M.S.; Borgquist, S.; Chang, M.C.; Dodson, A.; et al. Analytical Validation of a Standardized Scoring Protocol for Ki67: Phase 3 of an International Multicenter Collaboration. NPJ Breast Cancer 2016, 2, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Manley, J.L. Mechanisms of Alternative Splicing Regulation: Insights from Molecular and Genomics Approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-Mrna Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.H.; Broll, R.; Bruch, H.-P.; Finniss, S.; Bogler, O.; Duchrow, M. Proliferation Marker Pki-67 Occurs in Different Isoforms with Various Cellular Effects. J. Cell Biochem. 2004, 91, 1280–1292. [Google Scholar] [CrossRef]
- Chierico, L.; Rizzello, L.; Guan, L.; Joseph, A.S.; Lewis, A.; Battaglia, G. The Role of the Two Splice Variants and Extranuclear Pathway on Ki-67 Regulation in Non-Cancer and Cancer Cells. PLoS ONE 2017, 12, e0171815. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. Srp20 Is a Proto-Oncogene Critical for Cell Proliferation and Tumor Induction and Maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef]
- Sun, Y.; Yan, L.; Guo, J.; Shao, J.; Jia, R. Downregulation of Srsf3 by Antisense Oligonucleotides Sensitizes Oral Squamous Cell Carcinoma and Breast Cancer Cells to Paclitaxel Treatment. Cancer Chemother. Pharmacol. 2019, 84, 1133–1143. [Google Scholar] [CrossRef]
- Peiqi, L.; Zhaozhong, G.; Yaotian, Y.; Jun, J.; Jihua, G.; Rong, J. Expression of Srsf3 Is Correlated with Carcinogenesis and Progression of Oral Squamous Cell Carcinoma. Int. J. Med. Sci. 2016, 13, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial Reactive Oxygen Species and Cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Rashid, K.; AlAmodi, A.A.; Agha, M.V.; Akhtar, S.; Hakeem, I.; Raza, S.S.; Uddin, S. Reactive Oxygen Species (Ros) in Cancer Pathogenesis and Therapy: An Update on the Role of Ros in Anticancer Action of Benzophenanthridine Alkaloids. Biomed. Pharmacother. 2021, 143, 112142. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox Control of the Cell Cycle in Health and Disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.C.; Joughin, B.A.; Van De Kooij, B.; Lim, D.C.; Lauffenburger, D.A.; Yaffe, M.B. Ros and Oxidative Stress Are Elevated in Mitosis During Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst 2019, 8, 163–167.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- YYou, B.-H.; Yoon, J.-H.; Kang, H.; Lee, E.K.; Lee, S.K.; Nam, J.-W. Heres, a Lncrna That Regulates Canonical and Noncanonical Wnt Signaling Pathways Via Interaction with Ezh2. Proc. Natl. Acad. Sci. USA 2019, 116, 24620–24629. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, M.; Jia, R.; Yang, Y.; Zhu, J.; Zheng, Z.-M. A Genome Landscape of Srsf3-Regulated Splicing Events and Gene Expression in Human Osteosarcoma U2os Cells. Nucleic Acids Res. 2016, 44, 1854–1870. [Google Scholar] [CrossRef]
- Abubakar, M.; Orr, N.; Daley, F.; Coulson, P.; Ali, H.R.; Blows, F.; Benitez, J.; Milne, R.; Brenner, H.; Stegmaier, C.; et al. Prognostic Value of Automated Ki67 Scoring in Breast Cancer: A Centralised Evaluation of 8088 Patients from 10 Study Groups. Breast Cancer Res. 2016, 18, 104. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Jia, Y.; Dong, B.; Tang, L.; Liu, Y.; Du, H.; Yuan, P.; Dong, P.; Ji, J. Apoptosis and Ki 67 Index Correlate with Preoperative Chemotherapy Efficacy and Better Predict the Survival of Gastric Cancer Patients with Combined Therapy. Cancer Chemother. Pharmacol. 2014, 73, 885–893. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Leung, S.C.Y.; Rimm, D.L.; Dodson, A.; Acs, B.; Badve, S.; Denkert, C.; Ellis, M.J.; Fineberg, S.; Flowers, M.; et al. Assessment of Ki67 in Breast Cancer: Updated Recommendations from the International Ki67 in Breast Cancer Working Group. J. Natl. Cancer Inst. 2021, 113, 808–819. [Google Scholar] [CrossRef]
- Stanton, K.J.; A Sidner, R.; A Miller, G.; Cummings, O.W.; Schmidt, C.; Howard, T.J.; Wiebke, E.A. Analysis of Ki-67 Antigen Expression, DNA Proliferative Fraction, and Survival in Resected Cancer of the Pancreas. Am. J. Surg. 2003, 186, 486–492. [Google Scholar] [CrossRef]
- Harris, L.N.; Ismaila, N.; McShane, L.M.; Andre, F.; Collyar, D.E.; Gonzalez-Angulo, A.M.; Hammond, E.H.; Kuderer, N.M.; Liu, M.C.; Mennel, R.G.; et al. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women with Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 1134–1150. [Google Scholar] [CrossRef]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 Is a Promising Molecular Target in the Diagnosis of Cancer (Review). Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Shen, J.; Jia, J.; Jia, R. Inclusion of Hnrnp L Alternative Exon 7 Is Associated with Good Prognosis and Inhibited by Oncogene Srsf3 in Head and Neck Squamous Cell Carcinoma. Biomed. Res. Int 2019, 2019, 9612425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penning, T.M.; Wangtrakuldee, P.; Auchus, R.J. Structural and Functional Biology of Aldo-Keto Reductase Steroid-Transforming Enzymes. Endocrine Rev. 2019, 40, 447–475. [Google Scholar] [CrossRef] [PubMed]
- Wenners, A.; Hartmann, F.; Jochens, A.; Roemer, A.M.; Alkatout, I.; Klapper, W.; Van Mackelenbergh, M.; Mundhenke, C.; Jonat, W.; Bauer, M. Stromal Markers Akr1c1 and Akr1c2 Are Prognostic Factors in Primary Human Breast Cancer. Int J. Clin. Oncol. 2016, 21, 548–556. [Google Scholar] [CrossRef]
- Jin, Y.-X.; Zhou, X.-F.; Chen, Y.-Y.; Jin, W.-X.; Wang, Y.-H.; Ye, D.-R.; Sun, Y.-H.; Li, Y.-F.; Wang, Q.-X.; Zhang, X.-H.; et al. Up-Regulated Akr1c2 Is Correlated with Favorable Prognosis in Thyroid Carcinoma. J. Cancer 2019, 10, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- MacCallum, D.E.; Hall, P.A. Biochemical Characterization of Pki67 with the Identification of a Mitotic-Specific Form Associated with Hyperphosphorylation and Altered DNA Binding. Exp. Cell Res. 1999, 252, 186–198. [Google Scholar] [CrossRef]
- Lou, H.; Du, S.; Ji, Q.; Stolz, A. Induction of AKR1C2 by Phase II Inducers: Identification of a Distal Consensus Antioxidant Response Element Regulated by NRF2. Mol. Pharmacol. 2006, 69, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Kopacz, A.; Kloska, D.; Klimczyk, D.; Kopec, M.; Jozkowicz, A.; Piechota-Polanczyk, A. Nrf2 Transcriptional Activity Governs Intestine Development. Int. J. Mol. Sci. 2022, 23, 175. [Google Scholar] [CrossRef]
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A Ros-Dependent Mechanism Promotes Cdk2 Phosphorylation to Drive Progression through S Phase. Dev. Cell 2022, 57, 1712–1727.e9. [Google Scholar] [CrossRef] [PubMed]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The Influence of Reactive Oxygen Species on Cell Cycle Progression in Mammalian Cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Jia, R.; Bian, Z. Srsf5 Functions as a Novel Oncogenic Splicing Factor and Is Upregulated by Oncogene Srsf3 in Oral Squamous Cell Carcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liu, X.; Tao, M.; Kruhlak, M.; Guo, M.; Meyers, C.; Baker, C.C.; Zheng, Z.-M. Control of the Papillomavirus Early-to-Late Switch by Differentially Expressed Srp20. J. Virol. 2009, 83, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, X.; Liu, M.; Li, D.; Li, Z.; Guo, J.; Jia, R. Ran and Ybx1 Are Required for Cell Proliferation and Il-4 Expression and Linked to Poor Prognosis in Oral Squamous Cell Carcinoma. Exp. Cell Res. 2021, 406, 112767. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Jia, J.; Jia, R. Ptbp1 and Ptbp2 Impaired Autoregulation of Srsf3 in Cancer Cells. Sci. Rep. 2015, 5, 14548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Guo, J.; Che, X.; Jia, R. Pcbp1 Inhibits the Expression of Oncogenic Stat3 Isoform by Targeting Alternative Splicing of Stat3 Exon 23. Int. J. Biol. Sci. 2019, 15, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Srivastava, G.P.; Xu, D.; Schulz, L.C.; Roberts, R.M. A Link between Sin1 (Mapkap1) and Poly(Rc) Binding Protein 2 (Pcbp2) in Counteracting Environmental Stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11673–11678. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Zhang, S.; Liu, M.; Zhang, Y.; Liu, Y.; Fan, M.; Guo, J. Hnrnp L Is Important for the Expression of Oncogene Srsf3 and Oncogenic Potential of Oral Squamous Cell Carcinoma Cells. Sci. Rep. 2016, 6, 35976. [Google Scholar] [CrossRef]
- Lund, N.; Milev, M.P.; Wong, R.; Sanmuganantham, T.; Woolaway, K.; Chabot, B.; Abou Elela, S.; Mouland, A.J.; Cochrane, A. Differential Effects of Hnrnp D/Auf1 Isoforms on Hiv-1 Gene Expression. Nucleic Acids Res. 2012, 40, 3663–3675. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, D.; Ku, L.; Osterhout, D.J.; Feng, Y. The Selective Rna-Binding Protein Quaking I (Qki) Is Necessary and Sufficient for Promoting Oligodendroglia Differentiation. J. Biol. Chem. 2007, 282, 23553–23560. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, X.; Zhang, J.; Hu, J.; Gorczynski, R.M. Alternative Splicing of Cd200 Is Regulated by an Exonic Splicing Enhancer and Sf2/Asf. Nucleic Acids Res. 2010, 38, 6684–6696. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Lin, C.; Huang, Q.; Jia, J.; Guo, J.; Jia, R. SRSF3-Mediated Ki67 Exon 7-Inclusion Promotes Head and Neck Squamous Cell Carcinoma Progression via Repressing AKR1C2. Int. J. Mol. Sci. 2023, 24, 3872. https://doi.org/10.3390/ijms24043872
Liu M, Lin C, Huang Q, Jia J, Guo J, Jia R. SRSF3-Mediated Ki67 Exon 7-Inclusion Promotes Head and Neck Squamous Cell Carcinoma Progression via Repressing AKR1C2. International Journal of Molecular Sciences. 2023; 24(4):3872. https://doi.org/10.3390/ijms24043872
Chicago/Turabian StyleLiu, Miaomiao, Can Lin, Qiwei Huang, Jun Jia, Jihua Guo, and Rong Jia. 2023. "SRSF3-Mediated Ki67 Exon 7-Inclusion Promotes Head and Neck Squamous Cell Carcinoma Progression via Repressing AKR1C2" International Journal of Molecular Sciences 24, no. 4: 3872. https://doi.org/10.3390/ijms24043872