

New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins


{kind=link}
{kind=link}
{kind=link}
Abstract
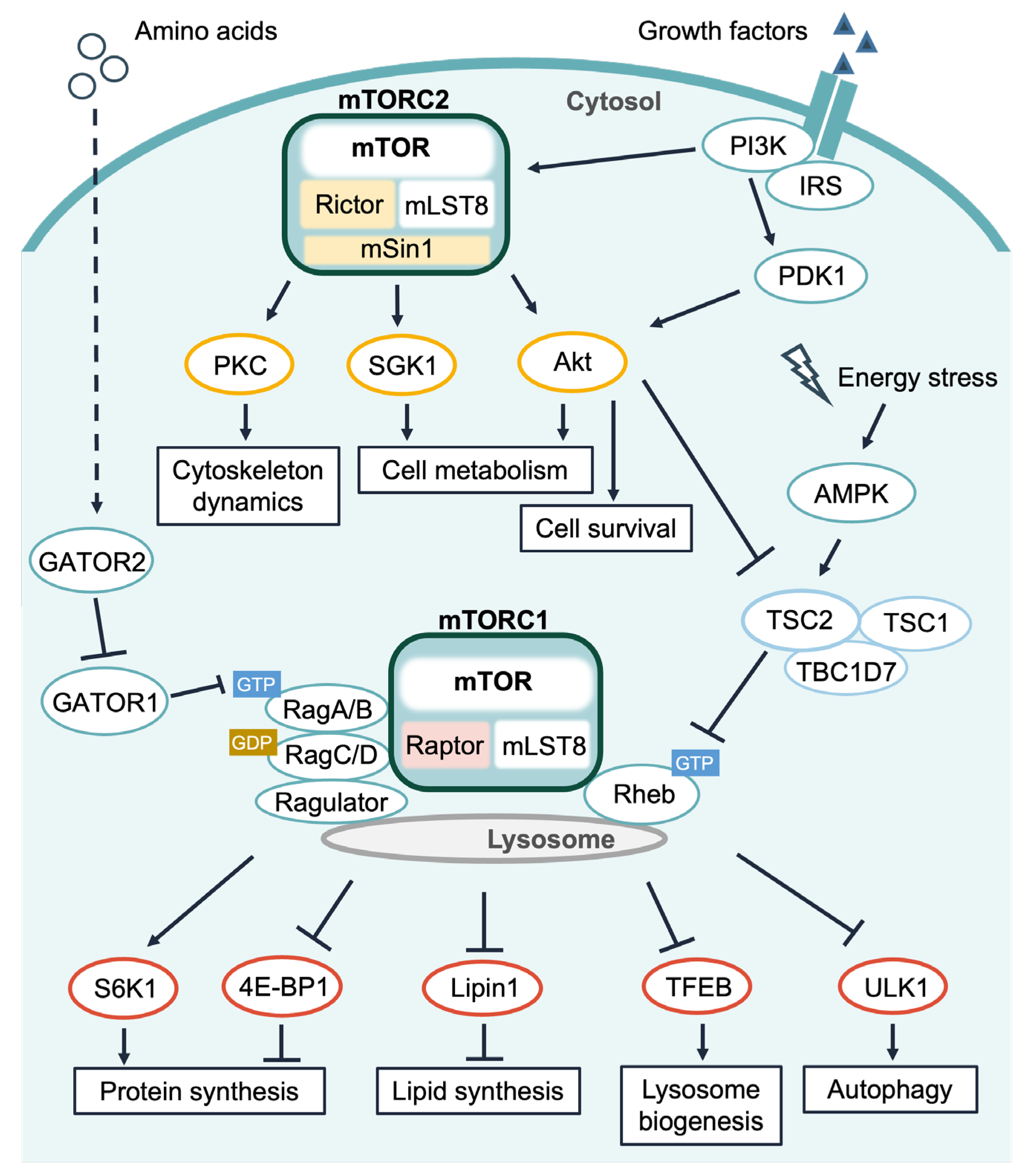
:1. Introduction
1.1. Ca2+ Signals and mTORC1 Signaling
1.2. Calmodulin (CaM) and mTORC1 Signaling
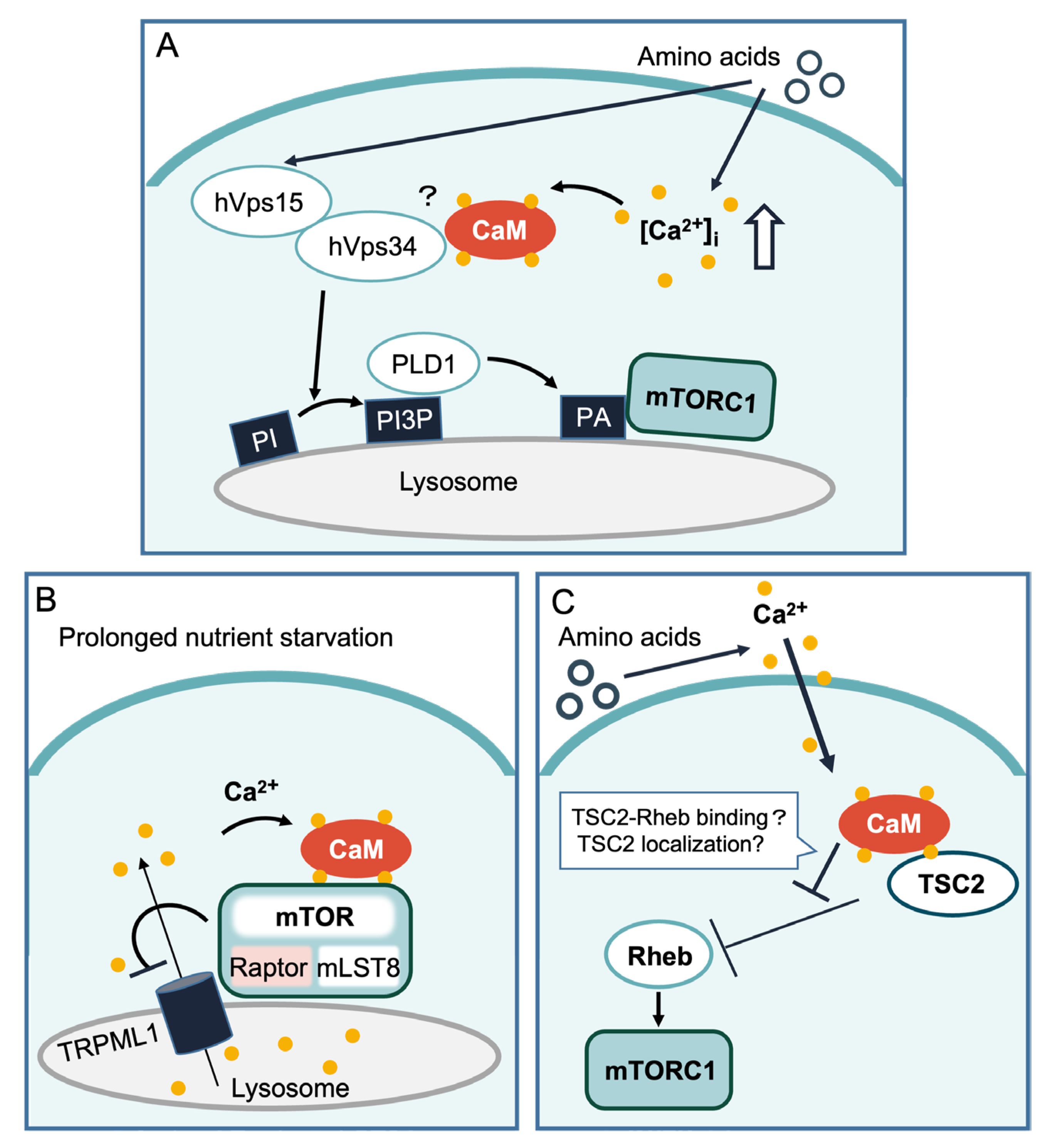
1.2.1. hVps34
1.2.2. TRPML1 and mTOR
1.2.3. Tuberous Sclerosis Complex 2 (TSC2)
2. Ca2+/CaM and mTORC2 Signaling
3. Involvement of Other Ca2+ Sensor Proteins in the mTOR Pathway
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGC | cAMP-dependent, cGMP-dependent, and protein kinase C-type |
| AMPK | AMP-activated protein kinase |
| Atg13 | autophagy-related protein 13 |
| BAPTA-AM | 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis(acetoxymethyl ester) |
| Bax | B cell lymphoma 2-associated X protein |
| BDNF | brain-derived neurotrophic factor |
| Ca2+ | calcium ion |
| CaM | calmodulin |
| CaMK | Ca2+/CaM-dependent protein kinase |
| CaMKK | CaMK kinase |
| CREB | cAMP response element-binding protein |
| ER | endoplasmic reticulum |
| EGF | epidermal growth factor |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| eEF2K | eukaryotic elongation factor 2 kinase |
| eIF4A | eukaryotic initiation factor 4A |
| eIF4B | eukaryotic initiation factor 4B |
| eIF4E | eukaryotic initiation factor 4E |
| 4E-BP1 | eIF4E-binding protein 1 |
| ERK | extracellular signal-regulated kinase |
| GAP | GTPase-activating protein |
| GPCR | G protein-coupled receptor |
| GSK3 | glycogen synthase kinase 3 |
| hVps34 | human vacuolar protein sorting 34 |
| IP3 | inositol-1,4,5-trisphosphate |
| IP3R | inositol-1,4,5-trisphosphate receptor |
| IP4 | inositol 1,3,4, 5-tetrakisphosphate |
| LAM | lymphangioleiomyomatosis |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| mSin1 | mammalian stress-activated protein kinase-interacting protein 1 |
| mTOR | mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| MLCK | myosin light-chain kinase |
| NCS1 | neuronal calcium sensor 1 |
| NF-κB | nuclear factor-kappa B |
| nNOS | neuronal nitric oxide synthase |
| PA | phosphatidic acid |
| PDGF | platelet-derived growth factor |
| PDK1 | 3-phosphoinositide-dependent protein kinase 1 |
| PIP2 | phosphatidylinositol (4,5)-bisphosphate |
| PI3K | phosphatidylinositol-3 kinase |
| PI3P | phosphatidylinositol 3-phosphate |
| PI(3,5)P2 | phosphatidylinositol (3,5)-bisphosphate |
| PI(4,5)P2 | phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | phosphatidylinositol (3,4,5)-trisphosphate |
| PLC | phospholipase C |
| PLD1 | phospholipase D1 |
| PH | pleckstrin homology |
| PKB | protein kinase B |
| PKC | protein kinase C |
| PTEN | phosphatase and tensin homolog deleted on chromosome 10 |
| Rag | Ras-related GTP-binding protein |
| Raptor | the regulatory associated protein of mTOR |
| Rheb | Ras homolog enriched in brain |
| Rictor | the rapamycin-insensitive companion of mTOR |
| S6K1 | p70 S6 kinase 1 |
| SERCA | sarcoplasmic/endoplasmic reticulum Ca2+-ATPase |
| SGK1 | serum- and glucocorticoid-induced kinases 1 |
| SLC38A9 | solute carrier family 38 member 9 |
| SH2 | Src homology 2 |
| SHP-2 | SH2 domain-containing protein tyrosine phosphatase |
| SREBP1 | sterol regulatory element-binding protein 1 |
| STIM1 | stromal interaction molecule 1 |
| T1R1 | taste receptor type1 member1 |
| TBC1D7 | TBC1 domain family member 7 |
| TFEB | transcription factor EB |
| TRPV1 | transient receptor potential cation channel, subfamily V, member 1 |
| TRPML1 | transient receptor potential mucolipin 1 |
| TMBIM6 | Transmembrane B cell lymphoma 2-associated X protein (Bax) inhibitor motif-containing 6 |
| TPC2 | two-pore segment channel 2 |
| TSC2 | tuberous sclerosis complex 2 |
| ULK1 | unc-51-like autophagy-activating kinase 1 |
| v-ATPase | vacuolar-type H+-ATPase |
| VDR | vitamin D receptor |
| VDCCs | voltage-dependent L-type Ca2+ channels |
References
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates Lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Battaglioni, S.; Benjamin, D.; Wälchli, M.; Maier, T.; Hall, M.N. mTOR substrate phosphorylation in growth control. Cell 2022, 185, 1814–1836. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2021, 184, 2255. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and ßTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Wang, X. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. mTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef] [Green Version]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 87. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the Rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit crosstalk in the Rag GTPase heterodimer enables mTORC1 to respond rapidly to amino acid availability. Mol. Cell 2017, 68, 552–565.e8. [Google Scholar] [CrossRef] [PubMed]
- Gollwitzer, P.; Grützmacher, N.; Wilhelm, S.; Kümmel, D.; Demetriades, C. A Rag GTPase dimer code defines the regulation of mTORC1 by amino acids. Nat. Cell Biol. 2022, 24, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+ -ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Ortiz-Vega, S.; Lin, Y.; Avruch, J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J. Biol. Chem. 2005, 280, 23433–23436. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef]
- Liu, X.; Zheng, X.F.S. Endoplasmic reticulum and Golgi localization sequences for mammalian target of rapamycin. Mol. Biol. Cell 2007, 18, 1073–1082. [Google Scholar] [CrossRef] [Green Version]
- Hao, F.; Kondo, K.; Itoh, T.; Ikari, S.; Nada, S.; Okada, M.; Noda, T. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 2017, 131, jcs208017. [Google Scholar] [CrossRef]
- Graves, L.M.; He, Y.; Lambert, J.; Hunter, D.; Li, X.; Earp, H.S. An intracellular calcium signal activates p70 but not p90 ribosomal S6 kinase in liver epithelial cells. J. Biol. Chem. 1997, 272, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Conus, N.M.; Hemmings, B.A.; Pearson, R.B. Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70(S6k). J. Biol. Chem. 1998, 273, 4776–4782. [Google Scholar] [CrossRef] [Green Version]
- Amemiya, Y.; Nakamura, N.; Ikeda, N.; Sugiyama, R.; Ishii, C.; Maki, M.; Shibata, H.; Takahara, T. Amino acid-mediated intracellular Ca2+ rise modulates mTORC1 by regulating the TSC2-Rheb axis through Ca2+/calmodulin. Int. J. Mol. Sci. 2021, 22, 6897. [Google Scholar] [CrossRef]
- Gulati, P.; Gaspers, L.D.; Dann, S.G.; Joaquin, M.; Nobukuni, T.; Natt, F.; Kozma, S.C.; Thomas, A.P.; Thomas, G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008, 7, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Li, R.-J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. Elife 2016, 5, e19360. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.-H.; Zhu, X.; Dong, X.-P. A negative feedback regulation of mTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Sanlialp, A.; Schumacher, D.; Kiper, L.; Varma, E.; Riechert, E.; Ho, T.C.; Hofmann, C.; Kmietczyk, V.; Zimmermann, F.; Dlugosz, S.; et al. Saraf-dependent activation of mTORC1 regulates cardiac growth. J. Mol. Cell. Cardiol. 2020, 141, 30–42. [Google Scholar] [CrossRef]
- Altamirano, F.; Oyarce, C.; Silva, P.; Toyos, M.; Wilson, C.; Lavandero, S.; Uhlén, P.; Estrada, M. Testosterone induces cardiomyocyte hypertrophy through mammalian target of rapamycin complex 1 pathway. J. Endocrinol. 2009, 202, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Hannan, K.M.; Thomas, G.; Pearson, R.B. Activation of S6K1 (p70 ribosomal protein S6 kinase 1) requires an initial calcium-dependent priming event involving formation of a high-molecular-mass signalling complex. Biochem. J. 2003, 370, 469–477. [Google Scholar] [CrossRef]
- Mercan, F.; Lee, H.; Kolli, S.; Bennett, A.M. Novel role for SHP-2 in nutrient-responsive control of S6 kinase 1 signaling. Mol. Cell. Biol. 2013, 33, 293–306. [Google Scholar] [CrossRef]
- Wauson, E.M.; Zaganjor, E.; Lee, A.-Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G protein-coupled taste receptor T1R1/T1R3 regulates mTORC1 and autophagy. Mol. Cell 2012, 47, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Ren, J.; Song, T.; Peng, J.; Wei, H. Methionine regulates mTORC1 via the T1R1/T1R3-PLCβ-Ca2+-ERK1/2 signal transduction process in C2C12 cells. Int. J. Mol. Sci. 2016, 17, 1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahara, T.; Inoue, K.; Arai, Y.; Kuwata, K.; Shibata, H.; Maki, M. The calcium-binding protein ALG-2 regulates protein secretion and trafficking via interactions with MISSL and MAP1B proteins. J. Biol. Chem. 2017, 292, 17057–17072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, S.; Kakizaki, M.; Hirose, Y.; Ishikawa, Y.; Funato, H.; Yanagisawa, M.; Yu, Y.; Liu, Q. Orexin/hypocretin activates mTOR complex 1 (mTORC1) via an Erk/Akt-independent and calcium-stimulated lysosome v-ATPase pathway. J. Biol. Chem. 2014, 289, 31950–31959. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef] [Green Version]
- Chemelli, R.M.; Willie, J.T.; Sinton, C.M.; Elmquist, J.K.; Scammell, T.; Lee, C.; Richardson, J.A.; Williams, S.C.; Xiong, Y.; Kisanuki, Y.; et al. Narcolepsy in orexin Knockout Mice. Cell 1999, 98, 437–451. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Faraco, J.; Li, R.; Kadotani, H.; Rogers, W.; Lin, X.; Qiu, X.; de Jong, P.J.; Nishino, S.; Mignot, E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 1999, 98, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Ito, N.; Ruegg, U.T.; Kudo, A.; Miyagoe-Suzuki, Y.; Takeda, S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat. Med. 2013, 19, 101–106. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2015, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Ito, N.; Ruegg, U.T.; Kudo, A.; Miyagoe-Suzuki, Y.; Takeda, S. Capsaicin mimics mechanical load-induced intracellular signaling events. Channels 2013, 7, 221–224. [Google Scholar] [CrossRef]
- Garg, R.; Borbora, S.M.; Bansia, H.; Rao, S.; Singh, P.; Verma, R.; Balaji, K.N.; Nagaraja, V. Mycobacterium tuberculosis calcium pump CtpF modulates the autophagosome in an mTOR-dependent manner. Front. Cell. Infect. Microbiol. 2020, 10, 461. [Google Scholar] [CrossRef]
- Ogawa, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X.-J. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am. J. Physiol. Physiol. 2012, 302, C405–C411. [Google Scholar] [CrossRef] [Green Version]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Hisatsune, C.; Shimada, T.; Miyamoto, A.; Lee, A.; Yamagata, K. Tuberous Sclerosis Complex (TSC) inactivation increases neuronal network activity by enhancing Ca2+ influx via L-type Ca2+ channels. J. Neurosci. 2021, 41, 8134–8149. [Google Scholar] [CrossRef]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal two-pore channel subtype 2 (TPC2) regulates skeletal muscle autophagic signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef] [Green Version]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. mTORC1 controls lysosomal Ca2+ release through the two-pore channel TPC2. Sci. Signal. 2018, 11, eaao5775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Mol. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef]
- Léger, C.; Pitard, I.; Sadi, M.; Carvalho, N.; Brier, S.; Mechaly, A.; Raoux-Barbot, D.; Davi, M.; Hoos, S.; Weber, P.; et al. Dynamics and structural changes of calmodulin upon interaction with the antagonist calmidazolium. BMC Biol. 2022, 20, 176. [Google Scholar] [CrossRef]
- Klee, C.B.; Crouch, T.H.; Richman, P.G. Calmodulin. Annu. Rev. Biochem. 1980, 49, 489–515. [Google Scholar] [CrossRef]
- Rasmussen, C.D.; Means, A.R. Calmodulin is required for cell-cycle progression during G1 and mitosis. EMBO J. 1989, 8, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, C.D.; Means, A.R. Calmodulin is involved in regulation of cell proliferation. EMBO J. 1987, 6, 3961–3968. [Google Scholar] [CrossRef] [PubMed]
- Tebar, F.; Villalonga, P.; Sorkina, T.; Agell, N.; Sorkin, A.; Enrich, C. Calmodulin regulates intracellular trafficking of epidermal growth factor receptor and the MAPK signaling pathway. Mol. Biol. Cell 2002, 13, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, M.P.; Mrwa, U.; Hofmann, F.; Rüegg, J.C. Calmodulin is essential for smooth muscle contraction. FEBS Lett. 1981, 125, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Lenz, G.; Avruch, J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J. Biol. Chem. 2005, 280, 38121–38124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Daniel, E.A.; Metcalf, J.; Dai, Y.; Reif, G.A.; Wallace, D.P. CaMK4 overexpression in polycystic kidney disease promotes mTOR-mediated cell proliferation. J. Mol. Cell Biol. 2022, 12, 4683–4698. [Google Scholar] [CrossRef]
- Clodfelder-Miller, B.; De Sarno, P.; Zmijewska, A.A.; Song, L.; Jope, R.S. Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J. Biol. Chem. 2005, 280, 39723–39731. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef] [Green Version]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 2005, 280, 33076–33082. [Google Scholar] [CrossRef]
- Yoon, M.-S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid–sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.-S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.; Chen, J. Leucyl-tRNA synthetase activates Vps34 in amino acid-sensing mTORC1 signaling. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Flinn, R.J.; Wu, H.; Schnur, R.S.; Backer, J.M. hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem. J. 2009, 417, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the center of cell signaling: Interplay between Endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef] [Green Version]
- Gan, N.; Han, Y.; Zeng, W.; Wang, Y.; Xue, J.; Jiang, Y. Structural mechanism of allosteric activation of TRPML1 by PI(3,5)P 2 and rapamycin. Proc. Natl. Acad. Sci. USA 2022, 119, e2120404119. [Google Scholar] [CrossRef]
- Fine, M.; Schmiege, P.; Li, X. Structural basis for PtdInsP2-mediated human TRPML1 regulation. Nat. Commun. 2018, 9, 4192. [Google Scholar] [CrossRef] [Green Version]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Díaz, M.C.H.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuammar, H.; Bhattacharjee, A.; Simon-Vecsei, Z.; Blastyák, A.; Csordás, G.; Páli, T.; Juhász, G. Ion channels and pumps in autophagy: A reciprocal relationship. Cells 2021, 10, 3537. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating TSC2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.K.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. Elife 2016, 5, e11058. [Google Scholar] [CrossRef]
- Roccio, M.; Bos, J.L.; Zwartkruis, F.J.T. Regulation of the small GTPase Rheb by amino acids. Oncogene 2006, 25, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, Y.; Arrazola, P.; Hino, O.; Kobayashi, T.; Yeung, R.S.; Ru, B.; Pan, D. Tsc tumour suppressor proteins antagonize amino-acid–TOR signalling. Nat. Cell Biol. 2002, 4, 699–704. [Google Scholar] [CrossRef]
- Fawal, M.-A.; Brandt, M.; Djouder, N. MCRS1 binds and couples Rheb to amino acid-dependent mTORC1 activation. Dev. Cell 2015, 33, 67–81. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, Y.; Ting, C.-Y.; Bettedi, L.; Kim, K.; Ghaniam, E.; Lilly, M.A. The Rag GTPase regulates the dynamic behavior of TSC downstream of both amino acid and growth factor restriction. Dev. Cell 2020, 55, 272–288.e5. [Google Scholar] [CrossRef]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef] [Green Version]
- Noonan, D.J.; Lou, D.; Griffith, N.; Vanaman, T.C. A calmodulin binding site in the tuberous sclerosis 2 gene product is essential for regulation of transcription events and is altered by mutations linked to tuberous sclerosis and lymphangioleiomyomatosis. Arch. Biochem. Biophys. 2002, 398, 132–140. [Google Scholar] [CrossRef]
- Yang, H.; Yu, Z.; Chen, X.; Li, J.; Li, N.; Cheng, J.; Gao, N.; Yuan, H.-X.; Ye, D.; Guan, K.-L.; et al. Structural insights into TSC complex assembly and GAP activity on Rheb. Nat. Commun. 2021, 12, 339. [Google Scholar] [CrossRef]
- York, B.; Lou, D.; Noonan, D.J. Tuberin nuclear localization can be regulated by phosphorylation of its carboxyl terminus. Mol. Cancer Res. 2006, 4, 885–897. [Google Scholar] [CrossRef] [Green Version]
- Kazami, M.; Sakamoto, T.; Suzuki, T.; Inoue, H.; Kato, H.; Kobayashi, K.-I.; Tadokoro, T.; Yamamoto, Y. Ca2+/Calmodulin induces translocation of membrane-associated TSC2 to the nucleus where it suppresses CYP24A1 expression. Biosci. Biotechnol. Biochem. 2022, 87, 45–53. [Google Scholar] [CrossRef]
- Franz, D.N.; Capal, J.K. mTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders. Orphanet J. Rare Dis. 2017, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, M.; Ohsawa, M.; Kashii, H.; Sato, A. Brain symptoms of tuberous sclerosis complex: Pathogenesis and treatment. Int. J. Mol. Sci. 2021, 22, 6677. [Google Scholar] [CrossRef]
- Palavra, F.; Robalo, C.; Reis, F. Recent advances and challenges of mTOR inhibitors use in the treatment of patients with tuberous sclerosis complex. Oxid. Med. Cell. Longev. 2017, 2017, 9820181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneike, M.; Nishida, K.; Omiya, S.; Zarrinpashneh, E.; Misaka, T.; Kitazume-Taneike, R.; Austin, R.; Takaoka, M.; Yamaguchi, O.; Gambello, M.J.; et al. mTOR hyperactivation by ablation of tuberous sclerosis complex 2 in the mouse heart induces cardiac dysfunction with the increased number of small mitochondria mediated through the down-regulation of autophagy. PLoS ONE 2016, 11, e0152628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassberg, M.K.; Elliot, S.J.; Fritz, J.; Catanuto, P.; Potier, M.; Donahue, R.; Stetler-Stevenson, W.; Karl, M. Activation of the estrogen receptor contributes to the progression of pulmonary lymphangioleiomyomatosis via matrix metalloproteinase-induced cell invasiveness. J. Clin. Endocrinol. Metab. 2008, 93, 1625–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhang, E.; Lao, T.; Pereira, A.M.; Li, C.; Xiong, L.; Morrison, T.; Haley, K.J.; Zhou, X.; Yu, J.J. Progesterone and estradiol synergistically promote the lung metastasis of tuberin-deficient cells in a preclinical model of lymphangioleiomyomatosis. Horm. Cancer 2014, 5, 284–298. [Google Scholar] [CrossRef] [PubMed]
- Prizant, H.; Hammes, S.R. Minireview: Lymphangioleiomyomatosis (LAM): The “other” steroid-sensitive cancer. Endocrinology 2016, 157, 3374–3383. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Joyal, J.L.; Sacks, D.B. Calmodulin enhances the stability of the estrogen receptor. J. Biol. Chem. 2001, 276, 17354–17360. [Google Scholar] [CrossRef] [Green Version]
- York, B.; Lou, D.; Panettieri, R.A.; Krymskaya, V.P.; Vanaman, T.C.; Noonan, D.J. Cross-talk between tuberin, calmodulin, and estrogen signaling pathways. FASEB J. 2005, 19, 1202–1204. [Google Scholar] [CrossRef]
- Joyal, J.L.; Burks, D.J.; Pons, S.; Matter, W.F.; Vlahos, C.J.; White, M.F.; Sacks, D.B. Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 1997, 272, 28183–28186. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, Z.; Wang, G.; Jang, H.; Sacks, D.B.; Zhang, J.; Gaponenko, V.; Nussinov, R. Calmodulin (CaM) activates PI3Kα by targeting the “soft” CaM-binding motifs in both the nSH2 and cSH2 domains of p85α. J. Phys. Chem. B 2018, 122, 11137–11146. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Gaponenko, V.; Nussinov, R. Phosphorylated calmodulin promotes PI3K activation by binding to the SH2 domains. Biophys. J. 2017, 113, 1956–1967. [Google Scholar] [CrossRef]
- Corti, C.; LeClerc L’Hostis, E.; Quadroni, M.; Schmid, H.; Durussel, I.; Cox, J.; Dainese Hatt, P.; James, P.; Carafoli, E. Tyrosine phosphorylation modulates the interaction of calmodulin with its target proteins. Eur. J. Biochem. 1999, 262, 790–802. [Google Scholar] [CrossRef] [Green Version]
- Deb, T.B.; Coticchia, C.M.; Dickson, R.B. Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 2004, 279, 38903–38911. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.; Valencia, C.A.; Liu, R. Ca2+/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J. Biol. Chem. 2007, 282, 25131–25140. [Google Scholar] [CrossRef] [Green Version]
- Coticchia, C.M.; Revankar, C.M.; Deb, T.B.; Dickson, R.B.; Johnson, M.D. Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res. Treat. 2009, 115, 545–560. [Google Scholar] [CrossRef] [Green Version]
- Agamasu, C.; Ghanam, R.H.; Xu, F.; Sun, Y.; Chen, Y.; Saad, J.S. The interplay between calmodulin and membrane interactions with the pleckstrin homology domain of Akt. J. Biol. Chem. 2017, 292, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Agamasu, C.; Ghanam, R.H.; Saad, J.S. Structural and biophysical characterization of the interactions between calmodulin and the pleckstrin homology domain of Akt. J. Biol. Chem. 2015, 290, 27403–27413. [Google Scholar] [CrossRef] [Green Version]
- Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J. Biol. Chem. 2011, 286, 6128–6142. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Ikegami, S.; Villa, G.R.; Yang, H.; Yong, W.H.; Cloughesy, T.F.; Yamagata, K.; Arai, N.; Cavenee, W.K.; et al. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc. Natl. Acad. Sci. USA 2015, 112, 9406–9411. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Shibata, N.; Cavenee, W.K.; Mischel, P.S. mTORC2 activity in brain cancer: Extracellular nutrients are required to maintain oncogenic signaling. BioEssays 2016, 38, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Merhi, A.; Delrée, P.; Marini, A.M. The metabolic waste ammonium regulates mTORC2 and mTORC1 signaling. Sci. Rep. 2017, 7, 44602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, C.; Kresse, W.; Kettenmann, H. Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes, an effect of pH. J. Biol. Chem. 2005, 280, 20937–20944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-K.; Bhattarai, K.R.; Junjappa, R.P.; Ahn, J.H.; Pagire, S.H.; Yoo, H.J.; Han, J.; Lee, D.; Kim, K.-W.; Kim, H.-R.; et al. TMBIM6/BI-1 contributes to cancer progression through assembly with mTORC2 and AKT activation. Nat. Commun. 2020, 11, 4012. [Google Scholar] [CrossRef] [PubMed]
- Bultynck, G.; Kiviluoto, S.; Henke, N.; Ivanova, H.; Schneider, L.; Rybalchenko, V.; Luyten, T.; Nuyts, K.; De Borggraeve, W.; Bezprozvanny, I.; et al. The C terminus of bax inhibitor-1 forms a Ca2+-permeable channel pore. J. Biol. Chem. 2012, 287, 2544–2557. [Google Scholar] [CrossRef] [Green Version]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lin, C.; Tao, Q.; Zhao, S.; Liu, H.; Li, L. Evaluation of calcium-binding protein A11 promotes the carcinogenesis of hypopharygeal squamous cell carcinoma via the PI3K/AKT signaling pathway. Am. J. Transl. Res. 2019, 11, 3472–3480. [Google Scholar]
- Teng, F.; Jiang, J.; Zhang, J.; Yuan, Y.; Li, K.; Zhou, B.; Zhou, X.; Liu, W.; Zhang, P.; Liu, D.; et al. The S100 calcium-binding protein A11 promotes hepatic steatosis through RAGE-mediated AKT-mTOR signaling. Metabolism 2021, 117, 154725. [Google Scholar] [CrossRef]
- Seguella, L.; Capuano, R.; Pesce, M.; Annunziata, G.; Pesce, M.; de Conno, B.; Sarnelli, G.; Aurino, L.; Esposito, G. S100B protein stimulates proliferation and angiogenic mediators release through RAGE/pAkt/mTOR pathway in human colon adenocarcinoma Caco-2 cells. Int. J. Mol. Sci. 2019, 20, 3240. [Google Scholar] [CrossRef]
- Zhong, X.; Xie, F.; Chen, L.; Liu, Z.; Wang, Q. S100A8 and S100A9 promote endothelial cell activation through the RAGE-mediated mammalian target of rapamycin complex 2 pathway. Mol. Med. Rep. 2020, 22, 5293–5303. [Google Scholar] [CrossRef]
- Apasu, J.E.; Schuette, D.; Laranger, R.; Steinle, J.A.; Nguyen, L.D.; Grosshans, H.K.; Zhang, M.; Cai, W.L.; Yan, Q.; Robert, M.E.; et al. Neuronal calcium sensor 1 (NCS1) promotes motility and metastatic spread of breast cancer cells in vitro and in vivo. FASEB J. 2019, 33, 4802–4813. [Google Scholar] [CrossRef]
- Schuette, D.; Moore, L.M.; Robert, M.E.; Taddei, T.H.; Ehrlich, B.E. Hepatocellular carcinoma outcome is predicted by expression of Neuronal Calcium Sensor 1. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Grosshans, H.K.; Fischer, T.T.; Steinle, J.A.; Brill, A.L.; Ehrlich, B.E. Neuronal Calcium Sensor 1 is up-regulated in response to stress to promote cell survival and motility in cancer cells. Mol. Oncol. 2020, 14, 1134–1151. [Google Scholar] [CrossRef]
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain research for drug discovery: Challenges and potential. Nat. Rev. Drug Discov. 2016, 15, 854–876. [Google Scholar] [CrossRef]
- Rao, S.S.; Mu, Q.; Zeng, Y.; Cai, P.C.; Liu, F.; Yang, J.; Xia, Y.; Zhang, Q.; Song, L.J.; Zhou, L.L.; et al. Calpain-activated mTORC2/Akt pathway mediates airway smooth muscle remodelling in asthma. Clin. Exp. Allergy 2017, 47, 176–189. [Google Scholar] [CrossRef]
- Briz, V.; Hsu, Y.T.; Li, Y.; Lee, E.; Bi, X.; Baudry, M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J. Neurosci. 2013, 33, 4317–4328. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amemiya, Y.; Maki, M.; Shibata, H.; Takahara, T. New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins. Int. J. Mol. Sci. 2023, 24, 3923. https://doi.org/10.3390/ijms24043923
Amemiya Y, Maki M, Shibata H, Takahara T. New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins. International Journal of Molecular Sciences. 2023; 24(4):3923. https://doi.org/10.3390/ijms24043923
Chicago/Turabian StyleAmemiya, Yuna, Masatoshi Maki, Hideki Shibata, and Terunao Takahara. 2023. "New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins" International Journal of Molecular Sciences 24, no. 4: 3923. https://doi.org/10.3390/ijms24043923
APA StyleAmemiya, Y., Maki, M., Shibata, H., & Takahara, T. (2023). New Insights into the Regulation of mTOR Signaling via Ca2+-Binding Proteins. International Journal of Molecular Sciences, 24(4), 3923. https://doi.org/10.3390/ijms24043923