
Staying below the Radar: Unraveling a New Family of Ubiquitous “Cryptic” Non-Tailed Temperate Vibriophages and Implications for Their Bacterial Hosts

 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
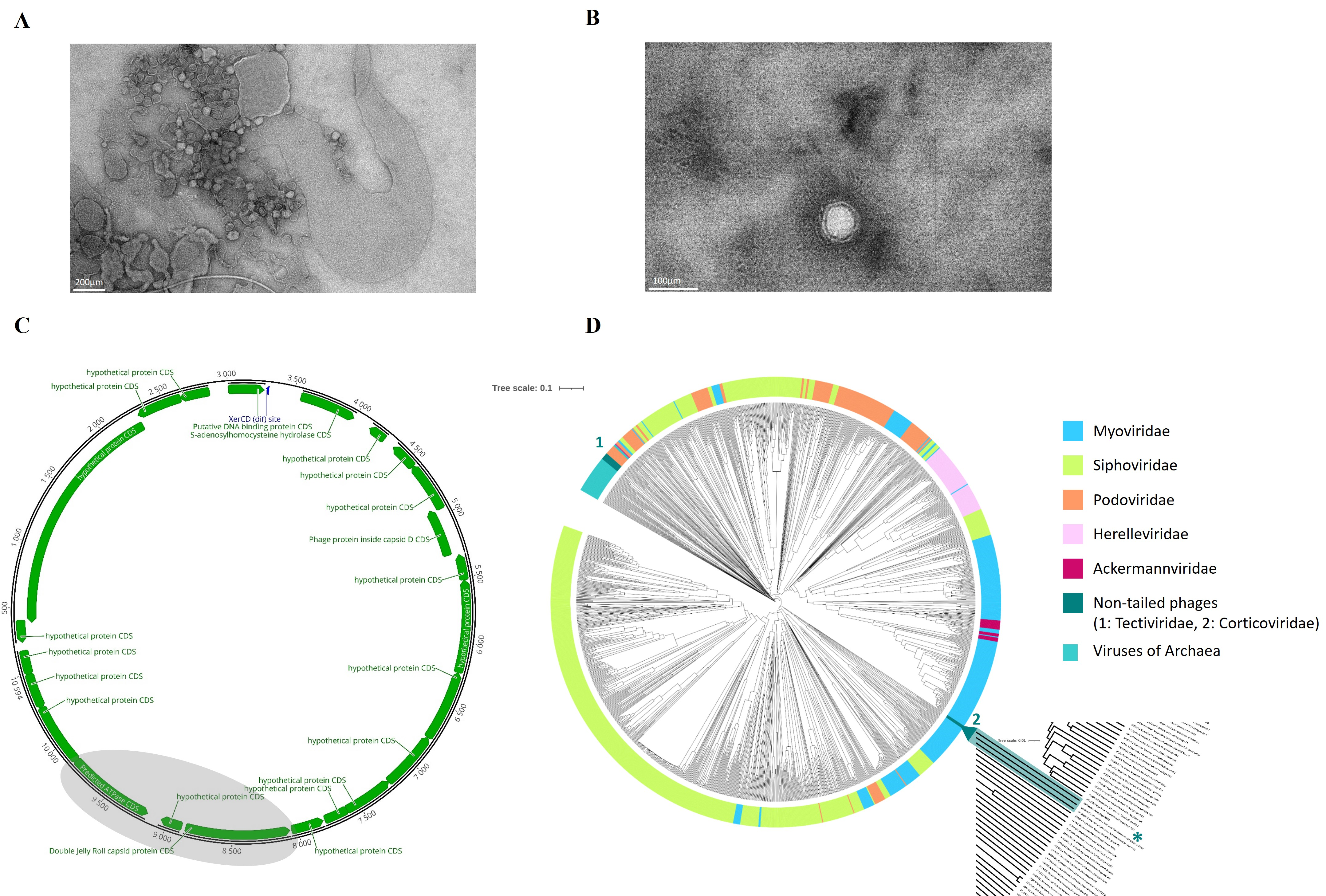
2.1. Characterization of NO16
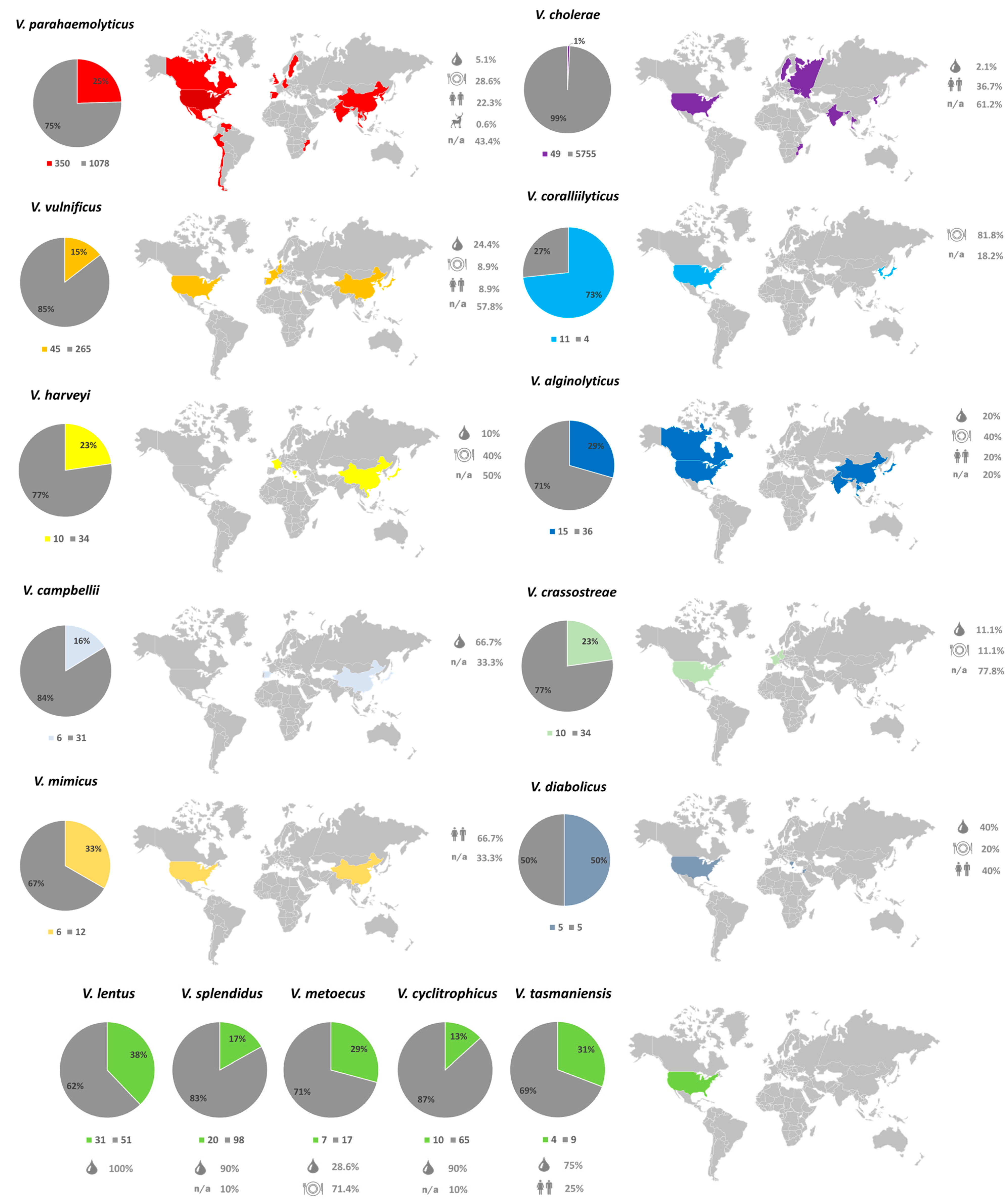
2.2. Genetic Diversity and Distribution of NO16 Viral Elements in Vibrio Genomes
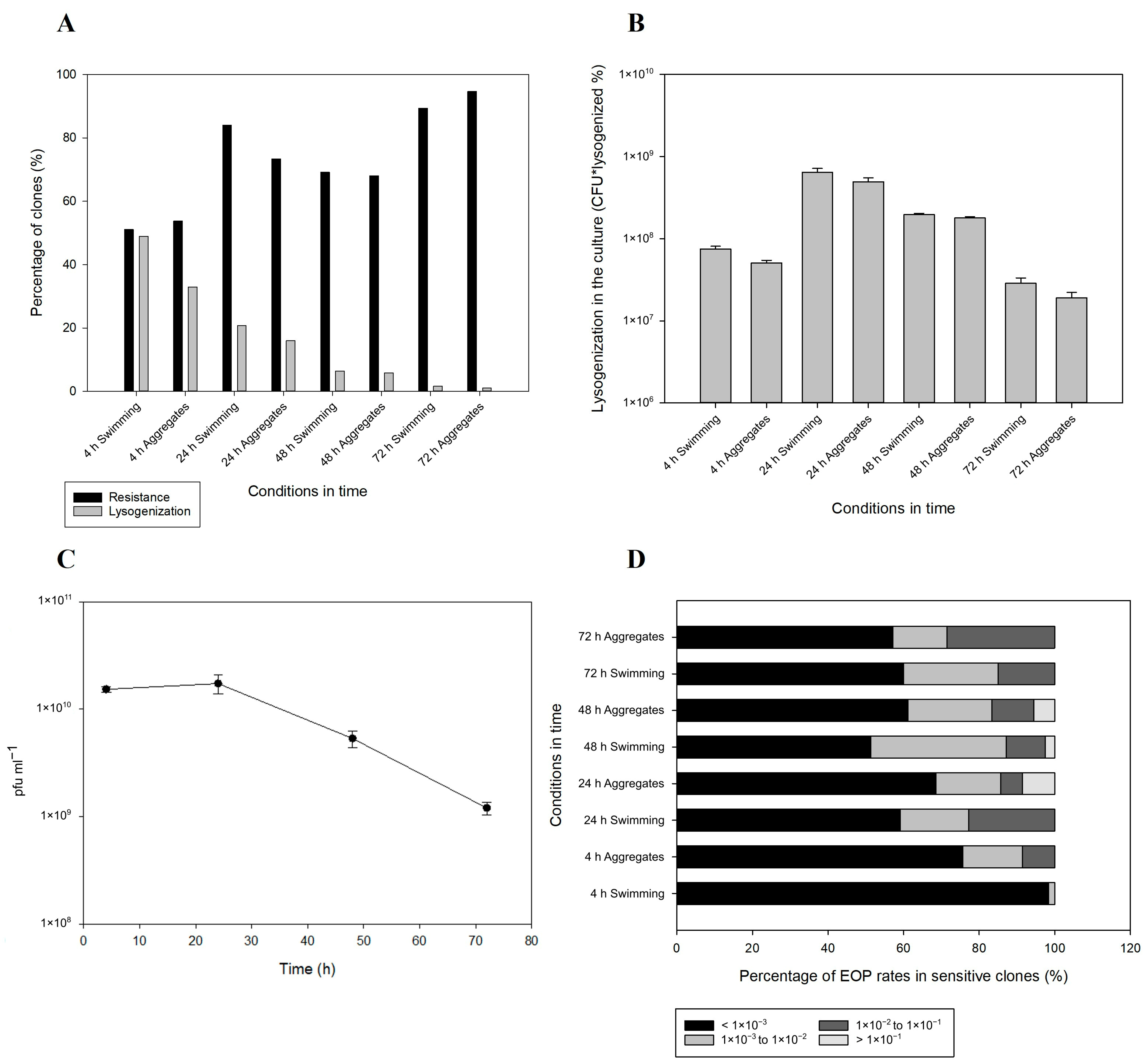
2.3. Lysogenization and Development of Resistance
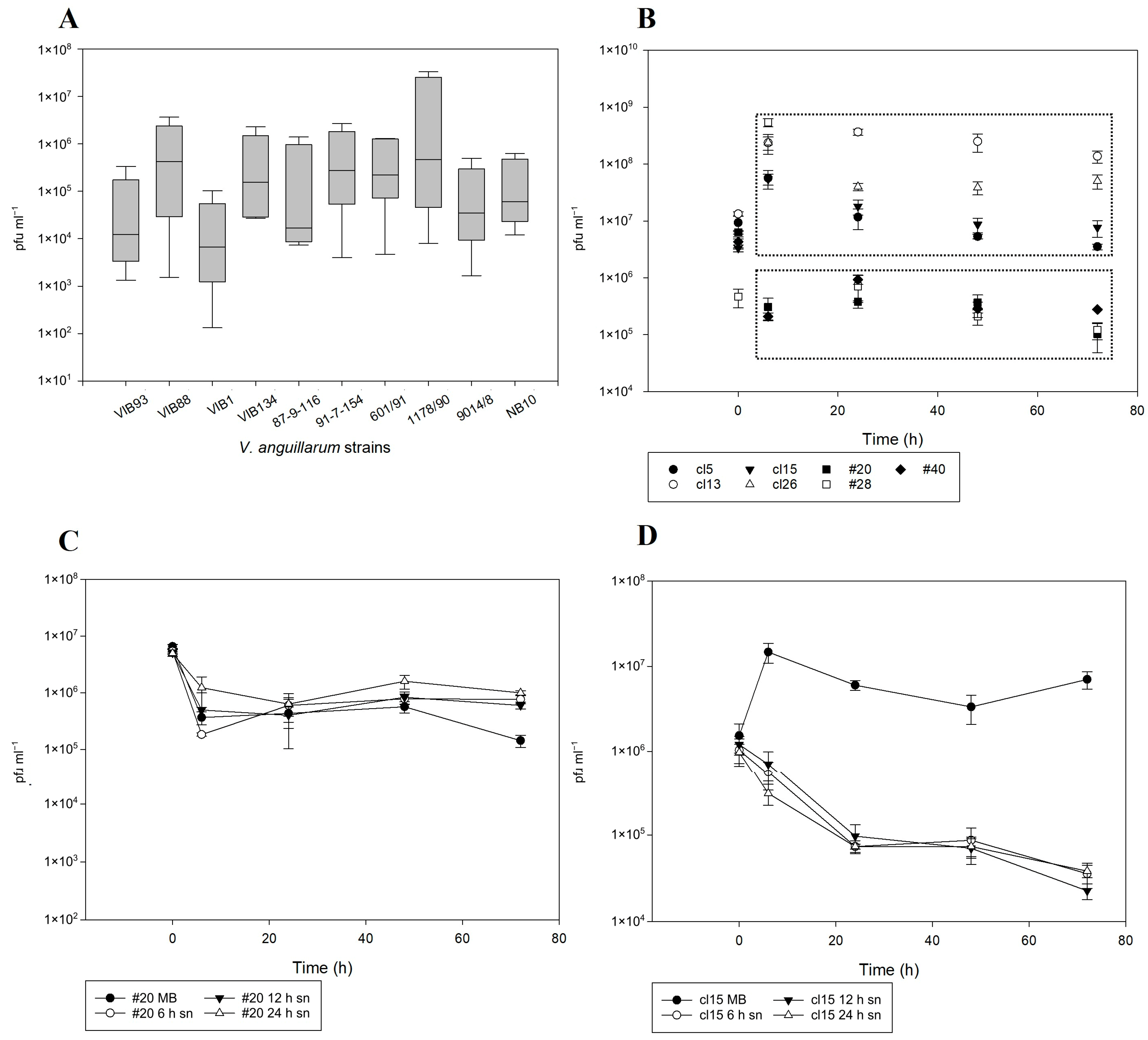
2.4. Prophage Integration and Spontaneous Induction Dynamics
2.5. Genomic Mechanism of Integration and Induction
2.6. Role of NO16 Prophages for Bacterial Functional Properties
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Isolation, Purification and Proliferation of Bacteriophages
4.3. Characterization of Bacteriophages
4.4. Genomic Analysis and HMM Construction
4.5. Resistance Development and Lysogenization Assay
4.6. Bacteriophage Dynamics: Prophage Integration and Spontaneous Induction
4.7. Quantification of Phage and Bacteria Dynamics
4.8. Biofilm Formation in Lysogens and Non-Lysogens
4.9. In Vivo Challenges of Fish Larvae to Assess Lysogenic Conversion
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Middelboe, M.; Brussaard, C.P.D. Marine viruses: Key players in marine ecosystems. Viruses 2017, 9, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalatzis, P.G.; Rørbo, N.; Castillo, D.; Mauritzen, J.J.; Jørgensen, J.; Kokkari, C.; Zhang, F.; Katharios, P.; Middelboe, M. Stumbling across the same phage: Comparative genomics of widespread temperate phages infecting the fish pathogen Vibrio anguillarum. Viruses 2017, 9, 122. [Google Scholar] [CrossRef] [Green Version]
- Sonnenschein, E.C.; Nielsen, K.F.; D’Alvise, P.; Porsby, C.H.; Melchiorsen, J.; Heilmann, J.; Kalatzis, P.G.; López-Pérez, M.; Bunk, B.; Spröer, C.; et al. Global occurrence and heterogeneity of the Roseobacter-clade species Ruegeria mobilis. ISME J. 2017, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Sano, E.; Carlson, S.; Wegley, L.; Rohwer, F. Movement of viruses between biomes. Appl. Environ. Microbiol. 2004, 70, 5842–5846. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Bacteriophage lysis: Mechanism and regulation. Microbiol. Rev. 1992, 56, 430–481. [Google Scholar] [CrossRef]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in Bacteriophage Lambda Development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, P.G.; Castillo, D.; Katharios, P.; Middelboe, M. Bacteriophage interactions with marine pathogenic vibrios: Implications for phage therapy. Antibiotics 2018, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchen-Osmond, C. The universal virus database ICTVdB. Comput. Sci. Eng. 2003, 5, 16–25. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Krupovic, M.; Knezevic, P.; Ackermann, H.W.; Barylski, J.; Brister, J.R.; Clokie, M.R.C.; Duffy, S.; Dutilh, B.E.; Edwards, R.A.; et al. Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2017, 162, 1153–1157. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Martín, C.S.; van Raaij, M.J. The so far farthest reaches of the double jelly roll capsid protein fold. Virol. J. 2018, 15, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamford, D.H. Do viruses form lineages across different domains of life? Res. Microbiol. 2003, 154, 231–236. [Google Scholar] [CrossRef]
- Bamford, D.H.; Grimes, J.M.; Stuart, D.I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Krupovič, M.; Bamford, D.H. Virus evolution: How far does the double β-barrel viral lineage extend? Nat. Rev. Microbiol. 2008, 6, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Laanto, E.; Mäntynen, S.; De Colibus, L.; Marjakangas, J.; Gillum, A.; Stuart, D.I.; Ravantti, J.J.; Huiskonen, J.T.; Sundberg, L.R. Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc. Natl. Acad. Sci. USA 2017, 114, 8378–8383. [Google Scholar] [CrossRef] [Green Version]
- Saren, A.M.; Ravantti, J.J.; Benson, S.D.; Burnett, R.M.; Paulin, L.; Bamford, D.H.; Bamford, J.K.H. A snapshot of viral evolution from genome analysis of the Tectiviridae family. J. Mol. Biol. 2005, 350, 427–440. [Google Scholar] [CrossRef]
- Männistö, R.H.; Kivelä, H.M.; Paulin, L.; Bamford, D.H.; Bamford, J.K.H. The complete genome sequence of PM2, the first lipid-containing bacterial virus to be isolated. Virology 1999, 262, 355–363. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Family-Tectiviridae; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J.B.T.-V.T., Eds.; Elsevier: San Diego, CA, USA, 2012; ISBN 978-0-12-384684-6. [Google Scholar]
- Caruso, S.M.; DeCarvalho, T.N.; Huynh, A.; Morcos, G.; Kuo, N.; Parsa, S.; Erill, I. A novel genus of actinobacterial tectiviridae. Viruses 2019, 11, 1133. [Google Scholar] [CrossRef]
- Kivelä, H.M.; Daugelavicius, R.; Hankkio, R.H.; Bamford, J.K.H.; Bamford, D.H. Penetration of membrane-containing double-stranded-DNA bacteriophage PM2 into Pseudoalteromonas hosts. J. Bacteriol. 2004, 186, 5342–5354. [Google Scholar] [CrossRef] [Green Version]
- Kivelä, H.M.; Madonna, S.; Krupovìč, M.; Tutino, M.L.; Bamford, J.K.H. Genetics for Pseudoalteromonas provides tools to manipulate marine bacterial virus PM2. J. Bacteriol. 2008, 190, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Kivelä, H.M.; Männistö, R.H.; Kalkkinen, N.; Bamford, D.H. Purification and protein composition of PM2, the first lipid-containing bacterial virus to be isolated. Virology 1999, 262, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, B.A.; Breitbart, M.; Oksanen, H.M.; Bamford, D.H.; Dishaw, L.J. Genome Sequence of PM2-Like Phage Cr39582, Induced from a Pseudoalteromonas sp. Isolated from the Gut of Ciona robusta. Genome Announc. 2018, 6, e00368-18. [Google Scholar] [CrossRef] [Green Version]
- Poduval, P.B.; Noronha, J.M.; Bansal, S.K.; Ghadi, S.C. Characterization of a new virulent phage ϕMC1 specific to Microbulbifer strain CMC-5. Virus Res. 2018, 257, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; You, F.; He, Y.; Harn Te, S.; Yew-Hoong Gin, K. Isolation and Characterization of the First Freshwater Cyanophage Infecting Pseudanabaena. J. Virol. 2020, 94, e00682-20. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Hussain, F.A.; Yang, J.; Arevalo, P.; Brown, J.M.; Chang, W.K.; Vaninsberghe, D.; Elsherbini, J.; Sharma, R.S.; Cutler, M.B.; et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nat. Publ. Gr. 2018, 7690, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Krupovič, M.; Bamford, D.H. Putative prophages related to lytic tailless marine dsDNA phage PM2 are widespread in the genomes of aquatic bacteria. BMC Genom. 2007, 8, 236. [Google Scholar] [CrossRef] [Green Version]
- Brum, J.R.; Schenck, R.O.; Sullivan, M.B. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013, 7, 1738–1751. [Google Scholar] [CrossRef] [Green Version]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Richards, G.P.; Chintapenta, L.K.; Watson, M.A.; Abbott, A.G.; Ozbay, G.; Uknalis, J.; Oyelade, A.A.; Parveen, S. Bacteriophages Against Pathogenic Vibrios in Delaware Bay Oysters (Crassostrea virginica) During a Period of High Levels of Pathogenic Vibrio parahaemolyticus. Food Environ. Virol. 2019, 11, 101–112. [Google Scholar] [CrossRef]
- Efrony, R.; Loya, Y.; Bacharach, E.; Rosenberg, E. Phage therapy of coral disease. Coral Reefs 2007, 26, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Ramphul, C.; Casareto, B.E.; Dohra, H.; Suzuki, T.; Yoshimatsu, K.; Yoshinaga, K.; Suzuki, Y. Genome analysis of three novel lytic Vibrio coralliilyticus phages isolated from seawater, Okinawa, Japan. Mar. Genom. 2017, 35, 69–75. [Google Scholar] [CrossRef]
- Pascelli, C.; Laffy, P.W.; Kupresanin, M.; Ravasi, T.; Webster, N.S. Morphological characterization of virus-like particles in coral reef sponges. PeerJ 2018, 6, e5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, N.S.; Roine, E.; Oren, A.; Bamford, D.H.; Oksanen, H.M. Global network of specific virus-host interactions in hypersaline environments. Environ. Microbiol. 2012, 14, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Alexeeva, S.; Liu, Y.; Zhu, J.; Kaczorowska, J.; Kouwen, T.R.H.M.; Abee, T.; Smid, E.J. Genomics of tailless bacteriophages in a complex lactic acid bacteria starter culture. Int. Dairy J. 2021, 114, 104900. [Google Scholar] [CrossRef]
- Alexeeva, S.; Guerra Martínez, J.A.; Spus, M.; Smid, E.J. Spontaneously induced prophages are abundant in a naturally evolved bacterial starter culture and deliver competitive advantage to the host. BMC Microbiol. 2018, 18, 1–16. [Google Scholar] [CrossRef]
- Kalatzis, P.G.; Carstens, A.B.; Katharios, P.; Castillo, D.; Hansen, L.H.; Middelboe, M. Complete Genome Sequence of Vibrio anguillarum Nontailed Bacteriophage NO16. Microbiol. Resour. Announc. 2019, 8, e00020-19. [Google Scholar] [CrossRef] [Green Version]
- Rønneseth, A.; Castillo, D.; D’Alvise, P.; Tønnesen, Ø.; Haugland, G.; Grotkjaer, T.; Engell-Sørensen, K.; Nørremark, L.; Bergh, Ø.; Wergeland, H.I.; et al. Comparative assessment of Vibrio virulence in marine fish larvae. J. Fish Dis. 2017, 10, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Espejo, R.T.; Canelo, E.S. Properties of bacteriophage PM2: A lipid-containing bacterial virus. Virology 1968, 34, 738–747. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.A.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Jacobs-sera, D.; Bustamante, C.A.G.; Garlena, A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.C.; Daniel, A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defense against viral attack and viral counter-defense. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef] [Green Version]
- Castillo, F.; Benmohamed, A.; Szatmari, G. Xer site specific recombination: Double and single recombinase systems. Front. Microbiol. 2017, 8, 453. [Google Scholar] [CrossRef] [Green Version]
- Mauritzen, J.J.; Castillo, D.; Tan, D.; Svenningsen, S.L.; Middelboe, M. Beyond Cholera: Characterization of zot-Encoding Filamentous Phages in the Marine Fish Pathogen Vibrio anguillarum. Viruses 2020, 12, 730. [Google Scholar] [CrossRef]
- Val, M.E.; Bouvier, M.; Campos, J.; Sherratt, D.; Cornet, F.; Mazel, D.; Barre, F.X. The single-stranded genome of phage CTX is the form used for integration into the genome of Vibrio cholerae. Mol. Cell 2005, 19, 559–566. [Google Scholar] [CrossRef]
- Huber, K.E.; Waldor, M.K. Filamentous phage integration requires the host recombinases XerC and XerD. Nature 2002, 417, 656–659. [Google Scholar] [CrossRef]
- Song, W.; Sun, H.X.; Zhang, C.; Cheng, L.; Peng, Y.; Deng, Z.; Wang, D.; Wang, Y.; Hu, M.; Liu, W.; et al. Prophage Hunter: An integrative hunting tool for active prophages. Nucleic Acids Res. 2019, 47, W74–W80. [Google Scholar] [CrossRef]
- Sousa, A.L.d.; Maués, D.; Lobato, A.; Franco, E.F.; Pinheiro, K.; Araújo, F.; Pantoja, Y.; Costa da Silva, A.L.d.; Morais, J.; Ramos, R.T.J. PhageWeb–Web Interface for Rapid Identification and Characterization of Prophages in Bacterial Genomes. Front. Genet. 2018, 9, 644. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, 244–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutin, N.; Bäckström, D.; Ettema, T.J.G.; Krupovic, M.; Koonin, E.V. Vast diversity of prokaryotic virus genomes encoding double jelly-roll major capsid proteins uncovered by genomic and metagenomic sequence analysis. Virol. J. 2018, 15, 67. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Shevchenko, S.; Kapitonov, V.; Krupovic, M.; Koonin, E.V. A novel group of diverse Polinton-like viruses discovered by metagenome analysis. BMC Biol. 2015, 13, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef]
- Tuttle, M.J.; Buchan, A. Lysogeny in the oceans: Lessons from cultivated model systems and a reanalysis of its prevalence. Environ. Microbiol. 2020, 22, 4919–4933. [Google Scholar] [CrossRef]
- Castillo, D.; Kauffman, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 2018, 8, 9973. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Dahl, A.; Middelboe, M. Vibriophages differentially influence biofilm formation by vibrio anguillarum strains. Appl. Environ. Microbiol. 2015, 81, 4489–4497. [Google Scholar] [CrossRef] [Green Version]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Golding, I.; Coleman, S.; Nguyen, T.V.; Yao, T. Decision Making by Temperate Phages. In Reference Module in Life Science; Elsevier: New York, NY, USA, 2019; pp. 1–10. [Google Scholar] [CrossRef]
- Sinha, V.; Goyal, A.; Svenningsen, S.L.; Semsey, S.; Krishna, S. In silico Evolution of Lysis-Lysogeny Strategies Reproduces Observed Lysogeny Propensities in Temperate Bacteriophages. Front. Microbiol. 2017, 8, 1386. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Skinner, S.O.; Sippy, J.; Feiss, M.; Golding, I. Decision Making at a Subcellular Level Determines the Outcome of Bacteriophage Infection. Cell 2009, 4, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigington, C.H.; Sonderegger, D.; Brussaard, C.P.D.; Buchan, A.; Finke, J.F.; Fuhrman, J.A.; Lennon, J.T.; Middelboe, M.; Suttle, C.A.; Stock, C.; et al. Re-examination of the relationship between marine virus and microbial cell abundances. Nat. Microbiol. 2016, 1, 15024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, B.; Silveira, C.B.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobián-Güemes, A.G.; Coutinho, F.H.; Dinsdale, E.A.; Felts, B.; Furby, K.A.; et al. Lytic to temperate switching of viral communities. Nature 2016, 531, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Hansen, M.F.; Nunes, L.; Henriette, D.C.; Røder, L.; Burmølle, M.; Middelboe, M.; Lo, S. High cell densities favor lysogeny: Induction of an H20 prophage is repressed by quorum sensing and enhances bio fi lm formation in Vibrio anguillarum. ISME J. 2020, 7, 1731–1742. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 2019, 176, 268–280.e13. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Roy, K.; Williamson, K.E.; Srinivasiah, S.; Wommack, K.E.; Radosevich, M. Acyl-homoserine lactones can induce virus production in lysogenic bacteria: An alternative paradigm for prophage induction. Appl. Environ. Microbiol. 2009, 75, 7142–7152. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.; Alvise, P.D.; Xu, R.; Zhang, F.; Middelboe, M.; Gram, L. Comparative Genome Analyses of Vibrio anguillarum Strains Reveal a Link with Pathogenicity Traits. mSystems 2017, 2, e00001-17. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Chan, A.M.; Suttle, C.A. Genetic richness of vibriophages isolated in a coastal environment. Environ. Microbiol. 2006, 8, 1164–1176. [Google Scholar] [CrossRef]
- Kutter, E. Phage Host Range and Efficiency of Plating. In Bacteriophages: Methods 448 and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M., Ed.; Springer Science: New York, NY, USA, 2009; pp. 141–149. [Google Scholar]
- Castillo, D.; Andersen, N.; Kalatzis, P.G.; Middelboe, M. Large phenotypic and genetic diversity of prophages induced from the fish pathogen Vibrio anguillarum. Viruses 2019, 11, 983. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, J.; Sato, Y.; Shinozaki, N.; Ye, B.; Tsuboi, A.; Nagasaki, M.; Yamashita, R. Comparison of boiling and robotics automation method in DNA extraction for metagenomic sequencing of human oral microbes. PLoS ONE 2016, 11, e0154389. [Google Scholar] [CrossRef] [Green Version]
- Erez, Z.; Steinberger-levy, I.; Shamir, M.; Doron, S.; Stokar, A. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, fnw015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Rodríguez, A.; García, P. Phage or foe: An insight into the impact of viral predation on microbial communities. ISME J. 2018, 12, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiong DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence with Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 Update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Hair, S.M.; Sexton, J.T.; Landry, B.P.; Olson, E.J.; Igoshin, O.A.; Tabor, J.J. FlowCal: A User-Friendly, Open Source Software Tool for Automatically Converting Flow Cytometry Data from Arbitrary to Calibrated Units. ACS Synth. Biol. 2016, 5, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J.A.; Khodursky, A.B.; Lin, P.H.; Lin-Chao, S.; Cohen, S.N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. USA 2002, 99, 9697–9702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gummesson, B.; Shah, S.A.; Borum, A.S.; Fessler, M. Valine-Induced Isoleucine Starvation in Escherichia coli K-12 Studied by Spike-In Normalized RNA Sequencing. Front. Genet. 2020, 11, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.; Lo Svenningsen, S.; Middelboe, M. Quorum sensing determines the choice of antiphage defense strategy in Vibrio anguillarum. mBio 2015, 6, e00627-15. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-J.; Sun, X.-H.; Xu, X.-Y.; Zhao, Y.; Pan, Y.-J.; Hwang, C.-A.; Wu, V.C.H. Investigation of reference genes in vibrio parahaemolyticus for gene expression analysis using quantitative RT-PCR. PLoS One 2015, 10, e0144362. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalatzis, P.G.; Mauritzen, J.J.; Winther-Have, C.S.; Michniewski, S.; Millard, A.; Tsertou, M.I.; Katharios, P.; Middelboe, M. Staying below the Radar: Unraveling a New Family of Ubiquitous “Cryptic” Non-Tailed Temperate Vibriophages and Implications for Their Bacterial Hosts. Int. J. Mol. Sci. 2023, 24, 3937. https://doi.org/10.3390/ijms24043937
Kalatzis PG, Mauritzen JJ, Winther-Have CS, Michniewski S, Millard A, Tsertou MI, Katharios P, Middelboe M. Staying below the Radar: Unraveling a New Family of Ubiquitous “Cryptic” Non-Tailed Temperate Vibriophages and Implications for Their Bacterial Hosts. International Journal of Molecular Sciences. 2023; 24(4):3937. https://doi.org/10.3390/ijms24043937
Chicago/Turabian StyleKalatzis, Panos G., Jesper Juel Mauritzen, Caroline Sophie Winther-Have, Slawomir Michniewski, Andrew Millard, Maria Ioanna Tsertou, Pantelis Katharios, and Mathias Middelboe. 2023. "Staying below the Radar: Unraveling a New Family of Ubiquitous “Cryptic” Non-Tailed Temperate Vibriophages and Implications for Their Bacterial Hosts" International Journal of Molecular Sciences 24, no. 4: 3937. https://doi.org/10.3390/ijms24043937
APA StyleKalatzis, P. G., Mauritzen, J. J., Winther-Have, C. S., Michniewski, S., Millard, A., Tsertou, M. I., Katharios, P., & Middelboe, M. (2023). Staying below the Radar: Unraveling a New Family of Ubiquitous “Cryptic” Non-Tailed Temperate Vibriophages and Implications for Their Bacterial Hosts. International Journal of Molecular Sciences, 24(4), 3937. https://doi.org/10.3390/ijms24043937