
Genome-Wide Gene-Set Analysis Identifies Molecular Mechanisms Associated with ALS
 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Gene-Level Meta-Analysis
2.2. Gene-Set Analysis
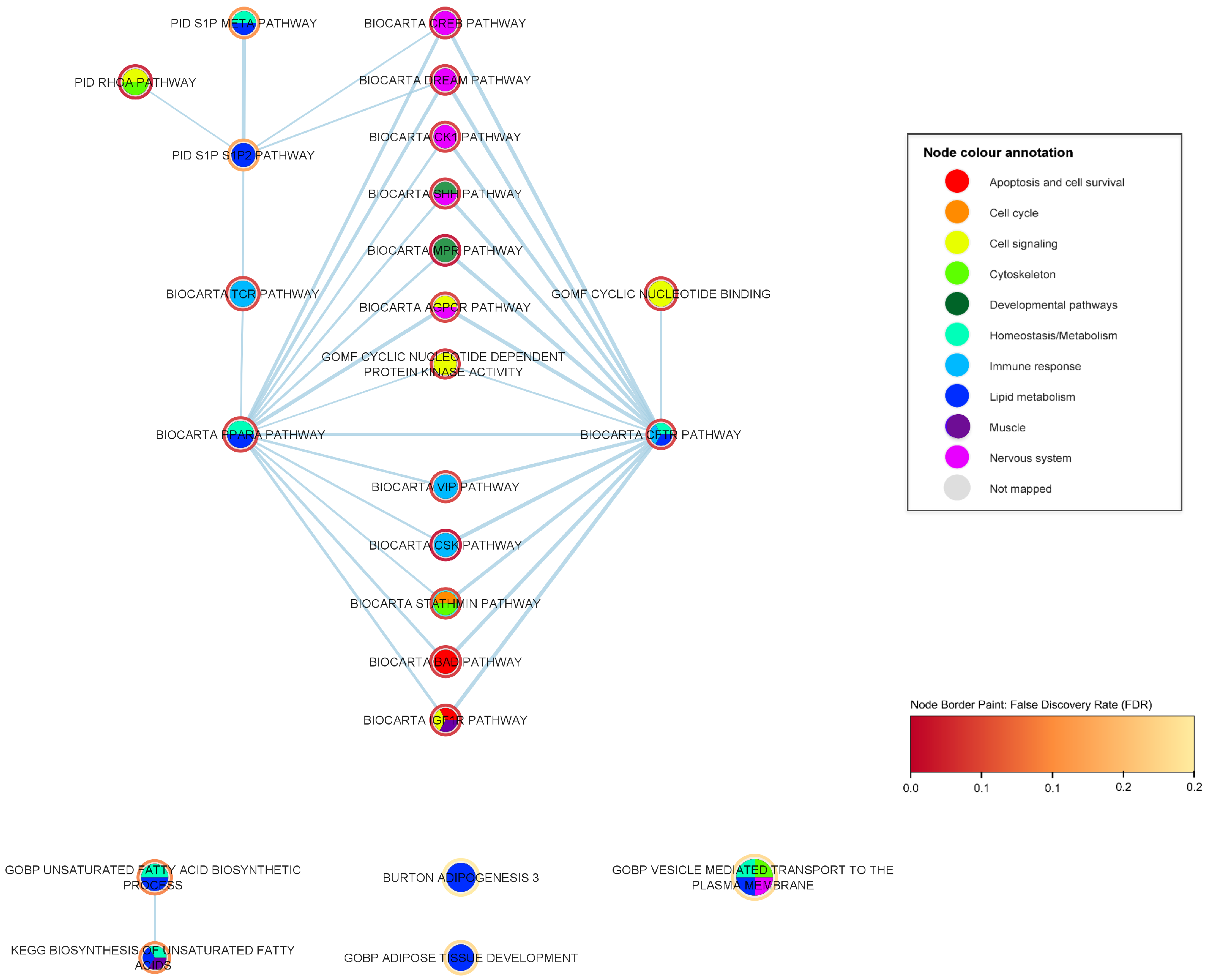
2.3. Mechanistic Relationships of ALS-Associated Gene Sets
2.3.1. Immune-Response Pathways
2.3.2. Developmental Pathways
2.3.3. Nervous System Pathways
2.3.4. Muscle Pathways
2.3.5. Lipid Metabolism Pathways
2.4. Interaction Analysis
3. Discussion
3.1. Gene Level Confirmation
3.2. Gene Set Association
3.3. Gene Set Interaction Analysis
3.4. Limitations
4. Materials and Methods
4.1. Datasets
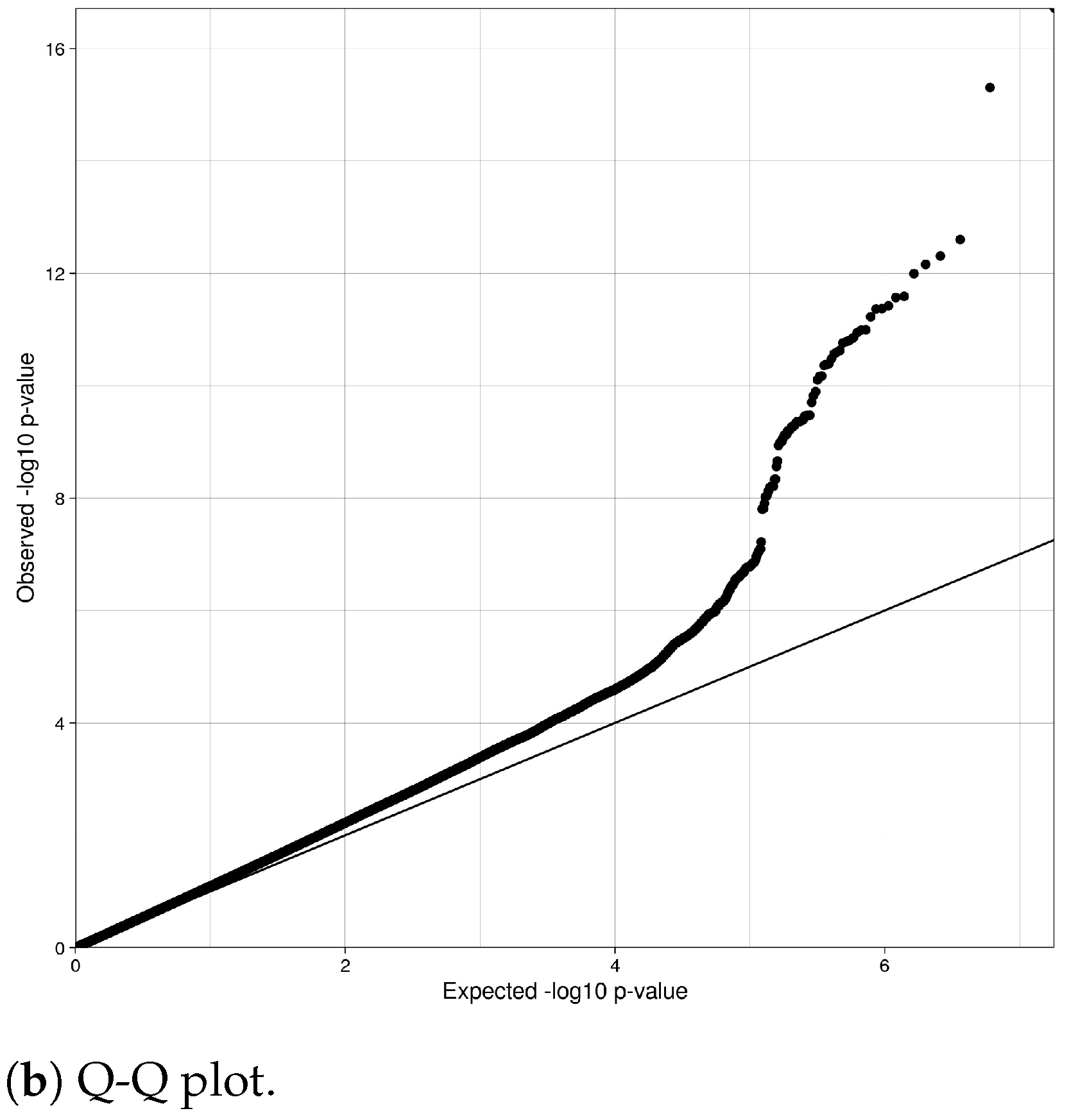
4.2. Genomic Quality Control Analysis
4.3. Imputation
4.4. Genome-Wide Association Analysis
4.5. Annotation and Gene Analysis
4.6. Gene Meta-Analysis
4.7. Gene-Set Analysis
4.8. Interaction Analysis
4.9. Enrichment Networks
- Jaccard coefficient = [size of (A intersect B)]/[size of (A union B)].
- Overlap coefficient = [size of (A intersect B)]/[size of (minimum(A, B))].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Traynor, B.; Collins, J.; Simeone, J.; Goldstein, L.; White, L. Global Epidemiology of Amyotrophic Lateral Sclerosis: A Systematic Review of the Published Literature. Neuroepidemiology 2013, 41, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.; Traynor, B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Vijayakumar, U.G.; Milla, V.; Stafford, M.Y.C.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.R. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef]
- Turner, M.R.; Al-Chalabi, A.; Chio, A.; Hardiman, O.; Kiernan, M.C.; Rohrer, J.D.; Rowe, J.; Seeley, W.; Talbot, K. Genetic screening in sporadic ALS and FTD. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1042–1044. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef]
- Smukowski, S.N.; Maioli, H.; Latimer, C.S.; Bird, T.D.; Jayadev, S.; Valdmanis, P.N. Progress in Amyotrophic Lateral Sclerosis Gene Discovery. Neurol. Genet. 2022, 8, e669. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Connolly, O.; Le Gall, L.; McCluskey, G.; Donaghy, C.G.; Duddy, W.J.; Duguez, S. A Systematic Review of Genotype–Phenotype Correlation across Cohorts Having Causal Mutations of Different Genes in ALS. J. Pers. Med. 2020, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, L.R.; Vajda, A.; Hardiman, O. Heritability of amyotrophic lateral sclerosis insights from disparate numbers. JAMA Neurol. 2015, 72, 857–858. [Google Scholar] [CrossRef]
- Vasilopoulou, C.; Morris, A.P.; Giannakopoulos, G.; Duguez, S.; Duddy, W. What Can Machine Learning Approaches in Genomics Tell Us about the Molecular Basis of Amyotrophic Lateral Sclerosis? J. Pers. Med. 2020, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Gall, L.L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duguez, S. Molecular and cellular mechanisms affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wen, Y.; Guo, X.; Hao, J.; Wang, W.; He, A.; Fan, Q.; Li, P.; Liu, L.; Liang, X.; et al. A Genome-wide Expression Association Analysis Identifies Genes and Pathways Associated with Amyotrophic Lateral Sclerosis. Cell. Mol. Neurobiol. 2018, 38, 635–639. [Google Scholar] [CrossRef]
- De Leeuw, C.A.; Neale, B.M.; Heskes, T.; Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 2016, 17, 353–364. [Google Scholar] [CrossRef]
- Vasilopoulou, C.; Duguez, S.; Duddy, W. Genome-Wide Gene-Set Analysis Approaches in Amyotrophic Lateral Sclerosis. J. Pers. Med. 2022, 12, 1932. [Google Scholar] [CrossRef]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C.A.; Stringer, S.; Dekkers, I.A.; Heskes, T.; Posthuma, D. Conditional and interaction gene-set analysis reveals novel functional pathways for blood pressure. Nat. Commun. 2018, 9, 3768. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mailman, M.D.; Feolo, M.; Jin, Y.; Kimura, M.; Tryka, K.; Bagoutdinov, R.; Hao, L.; Kiang, A.; Paschall, J.; Phan, L.; et al. The NCBI dbGaP database of genotypes and phenotypes. Nucleic Acids Res. 2007, 42, D975–D979. [Google Scholar] [CrossRef]
- Anderson, C.A.; Pettersson, F.H.; Clarke, G.M.; Cardon, L.R.; Morris, P.; Zondervan, K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2011, 5, 1564–1573. [Google Scholar] [CrossRef]
- Laurie, C.C.; Doheny, K.F.; Mirel, D.B.; Pugh, E.W.; Bierut, J.L.; Bhangale, T.; Boehm, F.; Caporaso, N.E.; Cornelis, M.C.; Edenberg, H.J.; et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet. Epidemiol. 2011, 34, 591–602. [Google Scholar] [CrossRef]
- Li, C.Y.; Yang, T.M.; Ou, R.W.; Wei, Q.Q.; Shang, H.F. Genome-wide genetic links between amyotrophic lateral sclerosis and autoimmune diseases. BMC Med. 2021, 19, 27. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257. [Google Scholar] [CrossRef]
- Van Es, M.A.; Veldink, J.H.; Saris, C.G.; Blauw, H.M.; Van Vught, P.W.; Birve, A.; Lemmens, R.; Schelhaas, H.J.; Groen, E.J.; Huisman, M.H.; et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 2009, 41, 1083–1087. [Google Scholar] [CrossRef]
- Vang, T.; Torgersen, K.M.; Sundvold, V.; Saxena, M.; Levy, F.O.; Skålhegg, B.S.; Hansson, V.; Mustelin, T.; Taskén, K. Activation of the Cooh-Terminal Src Kinase (Csk) by Camp-Dependent Protein Kinase Inhibits Signaling through the T Cell Receptor. J. Exp. Med. 2001, 193, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.I.; Broome, S.T.; Castorina, A. Exploring the Pro-Phagocytic and Anti-Inflammatory Functions of PACAP and VIP in Microglia: Implications for Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 4788. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Murga-Zamalloa, C.; Wilcox, R.A. GATA-3 in T-cell lymphoproliferative disorders. IUBMB Life 2020, 72, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dakemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, Y.; Chen, C.; Yang, D.; Ding, T.; Zhu, L.; Deng, J.; Xu, Z. The Triangle Relationship Between Long Noncoding RNA, RIG-I-like Receptor Signaling Pathway, and Glycolysis. Front. Microbiol. 2021, 12, 807737. [Google Scholar] [CrossRef]
- Misra, U.K.; Gawdi, G.; Akabani, G.; Pizzo, S.V. Cadmium-induced DNA synthesis and cell proliferation in macrophages: The role of intracellular calcium and signal transduction mechanisms. Cell. Signal. 2002, 14, 327–340. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Gargalovic, P.S.; Imura, M.; Zhang, B.; Gharavi, N.M.; Clark, M.J.; Pagnon, J.; Yang, W.P.; He, A.; Truong, A.; Patel, S.; et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc. Natl. Acad. Sci. USA 2006, 103, 12741. [Google Scholar] [CrossRef]
- Freigang, S. The regulation of inflammation by oxidized phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 family of cytokines in health and disease. Immunity 2019, 50, 892. [Google Scholar] [CrossRef] [PubMed]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Paoletti, G.; Puggioni, F.; Racca, F.; Pelaia, G.; Canonica, G.W.; Heffler, E. Interleukin-5 in the Pathophysiology of Severe Asthma. Front. Physiol. 2019, 10, 1514. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 2021, 89, 1619. [Google Scholar] [CrossRef]
- Holesh, J.E.; Bass, A.N.; Lord, M. Physiology, Ovulation; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Fair, T.; Lonergan, P. The role of progesterone in oocyte acquisition of developmental competence. Reprod. Domest. Anim. Zuchthyg. 2012, 47, 142–147. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y. Membrane Progesterone Receptors (mPRs): Evidence for Neuroprotective, Neurosteroid Signaling and Neuroendocrine Functions in Neuronal Cells. Neuroendocrinology 2012, 96, 162. [Google Scholar] [CrossRef]
- Choudhry, Z.; Rikani, A.A.; Choudhry, A.M.; Tariq, S.; Zakaria, F.; Asghar, M.W.; Sarfraz, M.K.; Haider, K.; Shafiq, A.A.; Mobassarah, N.J. Sonic hedgehog signalling pathway: A complex network. Ann. Neurosci. 2014, 21, 28. [Google Scholar] [CrossRef]
- Jha, N.K.; Chen, W.C.; Kumar, S.; Dubey, R.; Tsai, L.W.; Kar, R.; Jha, S.K.; Gupta, P.K.; Sharma, A.; Gundamaraju, R.; et al. Molecular mechanisms of developmental pathways in neurological disorders: A pharmacological and therapeutic review. Open Biol. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Maronde, E. Cyclic Nucleotide (cNMP) Analogues: Past, Present and Future. Int. J. Mol. Sci. 2021, 22, 12879. [Google Scholar] [CrossRef]
- Linder, J.U.; Schultz, J.E. Use of Chimeric Adenylyl Cyclases to Study Cyclic Nucleotide Signaling. In Handbook of Cell Signaling, 2nd ed.; Academic Press: San Diego, CA, USA, 2010; pp. 1537–1542. [Google Scholar] [CrossRef]
- Atwood, B.K.; Lopez, J.; Wager-Miller, J.; Mackie, K.; Straiker, A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genom. 2011, 12, 14. [Google Scholar] [CrossRef]
- Cheng, H.Y.M.; Pitcher, G.M.; Laviolette, S.R.; Whishaw, I.Q.; Tong, K.I.; Kockeritz, L.K.; Wada, T.; Joza, N.A.; Crackower, M.; Goncalves, J.; et al. DREAM is a critical transcriptional repressor for pain modulation. Cell 2002, 108, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Seliger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049. [Google Scholar] [CrossRef]
- Jiang, J. CK1 in Developmental signaling: Hedgehog and Wnt. Curr. Top. Dev. Biol. 2017, 123, 303. [Google Scholar] [CrossRef]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52. [Google Scholar] [CrossRef]
- Brusés, J.L. N-cadherin signaling in synapse formation and neuronal physiology. Mol. Neurobiol. 2006, 33, 237–252. [Google Scholar] [CrossRef]
- Lelièvre, E.C.; Plestant, C.; Boscher, C.; Wolff, E.; Mège, R.M.; Birbes, H. N-cadherin mediates neuronal cell survival through Bim down-regulation. PLoS ONE 2012, 7, 939. [Google Scholar] [CrossRef]
- Redies, C.; Treubert-Zimmermann, U.; Luo, J. Cadherins as regulators for the emergence of neural nets from embryonic divisions. J. Physiol. Paris 2003, 97, 5–15. [Google Scholar] [CrossRef]
- Takeichi, M.; Matsunami, H.; Inoue, T.; Kimura, Y.; Suzuki, S.; Tanaka, T. Roles of cadherins in patterning of the developing brain. Dev. Neurosci. 1997, 19, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Parrish, A.R.; Hill, M.A.; Meininger, G.A. N-cadherin, a vascular smooth muscle cell-cell adhesion molecule: Function and signaling for vasomotor control. Microcirculation 2014, 21, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Rosen, C.J.; Naski, M.C. N-cadherin adherens junctions mediate osteogenesis through PI3K signaling. Bone 2012, 50, 54–62. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, H.S.; Park, J.H.; Kim, M.J.; Lee, H.G.; Petersen, R.B.; Kim, Y.S.; Park, J.B.; Choi, E.K. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis. 2017, 8, e2668. [Google Scholar] [CrossRef] [PubMed]
- Marrs, G.S.; Theisen, C.S.; Brusés, J.L. N-cadherin modulates voltage activated calcium influx via RhoA, p120-catenin, and myosin-actin interaction. Mol. Cell. Neurosci. 2009, 40, 390–400. [Google Scholar] [CrossRef]
- Muhr, J.; Ackerman, K.M. Embryology, Gastrulation; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Zhang, Y.; Ulvmar, M.H.; Stanczuk, L.; Martinez-Corral, I.; Frye, M.; Alitalo, K.; Mäkinen, T. Heterogeneity in VEGFR3 levels drives lymphatic vessel hyperplasia through cell-autonomous and non-cell-autonomous mechanisms. Nat. Commun. 2018, 9, 1296. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef] [PubMed]
- Kopp-Scheinpflug, C.; Forsythe, I.D. Nitric Oxide Signaling in the Auditory Pathway. Front. Neural Circuits 2021, 15, 12. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Combs, C.K. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells). J. Neurosci. 2010, 30, 9641–9646. [Google Scholar] [CrossRef]
- Yoshida, S.; Hasegawa, T. Deciphering the prion-like behavior of pathogenic protein aggregates in neurodegenerative diseases. Neurochem. Int. 2022, 155, 105307. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Cleveland, D.W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS. Cell 2011, 147, 498. [Google Scholar] [CrossRef] [PubMed]
- Gau, D.; Roy, P. SRF’ing and SAP’ing—The role of MRTF proteins in cell migration. J. Cell Sci. 2018, 131, jcs218222. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Selvaraj, A.; Prywes, R. Myocardin/MKL family of SRF coactivators: Key regulators of immediate early and muscle specific gene expression. J. Cell. Biochem. 2004, 93, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Signaling, I.; Hakuno, F.; Takahashi, S.I.; Hakuno, F.; Takahashi, S.I. 40 YEARS OF IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef]
- Krauss, R.S.; Joseph, G.A.; Goel, A.J. Keep Your Friends Close: Cell–Cell Contact and Skeletal Myogenesis. Cold Spring Harb. Perspect. Biol. 2017, 9, a029298. [Google Scholar] [CrossRef]
- Lehka, L.; Rȩdowicz, M.J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92. [Google Scholar] [CrossRef]
- De Mello, W.C.; Danser, A.H. Angiotensin II and the Heart. Hypertension 2000, 35, 1183–1188. [Google Scholar] [CrossRef]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.D.; Huang, P. Redox Regulation of Cell Survival. Antioxidants Redox Signal. 2008, 10, 1343. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Q.; Long, X.Y.; Xie, Y.; Zhao, Z.H.; Fang, L.Z.; Liu, L.; Fu, W.P.; Shu, J.K.; Wu, J.H.; Dai, L.M. Relationship between PPARα mRNA expression and mitochondrial respiratory function and ultrastructure of the skeletal muscle of patients with COPD. Bioengineered 2017, 8, 723. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef]
- Sun, Y.; Curle, A.J.; Haider, A.M.; Balmus, G. The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem. 2020, 64, 847–861. [Google Scholar] [CrossRef]
- Herzog, J.J.; Xu, W.; Deshpande, M.; Rahman, R.; Suib, H.; Rodal, A.A.; Rosbash, M.; Paradis, S. TDP-43 dysfunction restricts dendritic complexity by inhibiting CREB activation and altering gene expression. Proc. Natl. Acad. Sci. USA 2020, 117, 11760–11769. [Google Scholar] [CrossRef]
- Herzog, J.J.; Deshpande, M.; Shapiro, L.; Rodal, A.A.; Paradis, S. TDP-43 misexpression causes defects in dendritic growth. Sci. Rep. 2017, 7, 15656. [Google Scholar] [CrossRef] [PubMed]
- Kweon, J.H.; Kim, S.; Lee, S.B. The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep. 2017, 50, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.J.; Spruston, N. Dendritic integration: 60 years of progress. Nat. Neurosci. 2015, 18, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Catanese, A.; Rajkumar, S.; Sommer, D.; Freisem, D.; Wirth, A.; Aly, A.; Massa-L Opez, D.; Olivieri, A.; Torelli, F.; Ioannidis, V.; et al. Synaptic disruption and CREB-regulated transcription are restored by K+ channel blockers in ALS. EMBO Mol. Med. 2021, 13, e13131. [Google Scholar] [CrossRef]
- Larrodé, P.; Calvo, A.C.; Moreno-Martínez, L.; De La Torre, M.; Moreno-García, L.; Molina, N.; Castiella, T.; Iñiguez, C.; Pascual, L.F.; Mena, F.J.M.; et al. DREAM-dependent activation of astrocytes in amyotrophic lateral sclerosis. Mol. Neurobiol. 2018, 55, 1–12. [Google Scholar] [CrossRef]
- Kametani, F.; Nonaka, T.; Suzuki, T.; Arai, T.; Dohmae, N.; Akiyama, H.; Hasegawa, M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [Google Scholar] [CrossRef]
- Xie, Y.; Luo, X.; He, H.; Tang, M. Novel Insight Into the Role of Immune Dysregulation in Amyotrophic Lateral Sclerosis Based on Bioinformatic Analysis. Front. Neurosci. 2021, 15, 657465. [Google Scholar] [CrossRef]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Neuroinflammation and ALS: Transcriptomic Insights into Molecular Disease Mechanisms and Therapeutic Targets. Mediat. Inflamm. 2017, 2017, 7070469. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715. [Google Scholar] [CrossRef] [PubMed]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Yong, V.W. Oxidized phospholipids as novel mediators of neurodegeneration. Trends Neurosci. 2022, 45, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Catalá, A. Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem. Phys. Lipids 2009, 157, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Tomita, Y.; Allen, W.; Tsubota, K.; Negishi, K.; Kurihara, T. PPARα Modulation-Based Therapy in Central Nervous System Diseases. Life 2021, 11, 1168. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Yadav, S.; Gupta, R.K.; Waggoner, G.R.; Deloach, A.; Calingasan, N.Y.; Flint Beal, M.; Kiaei, M. Preferential PPAR-α activation reduces neuroinflammation, and blocks neurodegeneration in vivo. Hum. Mol. Genet. 2016, 25, 317–327. [Google Scholar] [CrossRef]
- Spitaler, M.; Cantrell, D.A. Protein kinase C and beyond. Nat. Immunol. 2004, 5, 785–790. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, F.; Zhong, S.; Zhou, Y.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, M.; Zhao, C. Molecular identification of protein kinase C beta in Alzheimer’s disease. Aging 2020, 12, 21798–21808. [Google Scholar] [CrossRef]
- Lo, C.; Cooper-Knock, J.; Garrard, K.; Martindale, J.; Williams, T.; Shaw, P. Concurrent amyotrophic lateral sclerosis and cystic fibrosis supports common pathways of pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 473–475. [Google Scholar] [CrossRef]
- De Nicola, A.F.; Meyer, M.; Garay, L.; Kruse, M.S.; Schumacher, M.; Guennoun, R.; Gonzalez Deniselle, M.C. Progesterone and Allopregnanolone Neuroprotective Effects in the Wobbler Mouse Model of Amyotrophic Lateral Sclerosis. Cell. Mol. Neurobiol. 2021, 42, 23–40. [Google Scholar] [CrossRef]
- Drannik, A.; Martin, J.; Peterson, R.; Ma, X.; Jiang, F.; Turnbull, J. Cerebrospinal fluid from patients with amyotrophic lateral sclerosis inhibits sonic hedgehog function. PLoS ONE 2017, 12, e0171668. [Google Scholar] [CrossRef]
- Peterson, R.; Turnbull, J. Sonic hedgehog is cytoprotective against oxidative challenge in a cellular model of amyotrophic lateral sclerosis. J. Mol. Neurosci. 2012, 47, 31–41. [Google Scholar] [CrossRef]
- Liu, Y.J.; Ju, T.C.; Chen, H.M.; Jang, Y.S.; Lee, L.M.; Lai, H.L.; Tai, H.C.; Fang, J.M.; Lin, Y.L.; Tu, P.H.; et al. Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2015, 24, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Zhang, H.; Wagey, R.; Krieger, C.; Pelech, S.L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [Google Scholar] [CrossRef]
- Vasilopoulou, C.; Duddy, W.; Wingfield, B.; Morris, A.P. snpQT: Flexible, reproducible, and comprehensive quality control and imputation of genomic data. F1000Research 2021, 10, 567. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zuvich, R.L.; Armstrong, L.L.; Bielinski, S.J.; Bradford, Y.; Carlson, C.S.; Crawford, D.C.; Crenshaw, A.T.; de Andrade, M.; Doheny, K.F.; Haines, J.L.; et al. Pitfalls of Merging GWAS Data: Lessons Learned in the eMERGE Network and Quality Control Procedures to Maintain High Data Quality. Genet. Epidemiol. 2011, 35, 887. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Durbin, R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014, 30, 1266–1272. [Google Scholar] [CrossRef]
- Tsunoda, T.; Lathrop, G.M.; Sekine, A.; Yamada, R.; Takahashi, A.; Ohnishi, Y.; Tanaka, T.; Nakamura, Y. Variation of gene-based SNPs and linkage disequilibrium patterns in the human genome. Hum. Mol. Genet. 2004, 13, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Springer: Singapore, 2021; pp. 27–56. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Lomax, J.; Mungall, C.; Hitz, B.; Balakrishnan, R.; et al. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Taylor, T.; Awad, A.; Baker, D.; Giovanonni, G.; Noyce, A.J.; Dobson, R. Summary-data-based Mendelian randomization prioritizes potential druggable targets for multiple sclerosis. Brain Commun. 2020, 2, fcaa119. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome | No. SNPs | p-Value | FDR |
|---|---|---|---|---|
| MOB3B | 9 | 979 | 9.20 | 1.77 |
| IFNK | 9 | 206 | 4.14 | 3.98 |
| C9ORF72 | 9 | 289 | 3.90 | 2.50 |
| UNC13A | 19 | 487 | 2.37 | 1.14 |
| ADARB1 | 21 | 726 | 3.13 | 1.20 |
| KIF5A | 12 | 142 | 2.44 | 0.78 |
| Gene sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_MPR_PATHWAY | 6.27 | 9.15 | 38 | 21 |
| BIOCARTA_CSK_PATHWAY | 5.02 | 9.15 | 29 | 22 |
| PID_RHOA_PATHWAY | 1.16 | 2.28 | 11 | 45 |
| GARGALOVIC_RESPONSE_TO_ | ||||
| OXIDIZED_PHOSPHOLIPIDS_BLACK_UP | 9.54 | 3.22 | 1 | 35 |
| GOMF_CYCLIC_NUCLEOTIDE_ | ||||
| DEPENDENT_PROTEIN_KINASE_ACTIVITY | 6.70 | 3.88 | 26 | 10 |
| GOMF_CYCLIC_NUCLEOTIDE_BINDING | 6.59 | 3.88 | 21 | 38 |
| GOMF_DIOXYGENASE_ACTIVITY | 6.11 | 3.88 | 5 | 94 |
| BIOCARTA_CREB_PATHWAY | 4.05 | 3.94 | 50 | 22 |
| MIR12119 | 1.68 | 4.00 | 1 | 185 |
| BIOCARTA_IGF1R_PATHWAY | 9.71 | 4.03 | 45 | 23 |
| BIOCARTA_BAD_PATHWAY | 8.07 | 4.03 | 37 | 25 |
| BIOCARTA_DREAM_PATHWAY | 1.10 | 4.03 | 34 | 13 |
| BIOCARTA_SHH_PATHWAY | 6.60 | 4.03 | 28 | 16 |
| BIOCARTA_MONOCYTE_PATHWAY | 9.34 | 4.03 | 11 | 11 |
| MIR4286 | 3.76 | 4.47 | 0 | 92 |
| BIOCARTA_PPARA_PATHWAY | 1.71 | 4.63 | 42 | 52 |
| BIOCARTA_CK1_PATHWAY | 1.71 | 4.63 | 26 | 16 |
| BIOCARTA_CFTR_PATHWAY | 1.81 | 4.63 | 25 | 11 |
| BIOCARTA_LYMPHOCYTE_PATHWAY | 1.91 | 4.63 | 11 | 9 |
| BIOCARTA_TCR_PATHWAY | 2.49 | 4.85 | 39 | 44 |
| BIOCARTA_AGPCR_PATHWAY | 2.56 | 4.85 | 36 | 11 |
| BIOCARTA_STATHMIN_PATHWAY | 2.30 | 4.85 | 31 | 20 |
| BIOCARTA_GHRELIN_PATHWAY | 2.66 | 4.85 | 1 | 13 |
| BIOCARTA_VIP_PATHWAY | 2.87 | 4.93 | 33 | 26 |
| Gene Sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_CSK_PATHWAY | 5.02 | 9.15 | 20 | 22 |
| GARGALOVIC_RESPONSE_TO | ||||
| _OXIDIZED_PHOSPHOLIPIDS_BLACK_UP | 9.54 | 3.22 | 0 | 35 |
| BIOCARTA_MONOCYTE_PATHWAY | 9.34 | 4.03 | 4 | 11 |
| BIOCARTA_CFTR_PATHWAY | 1.81 | 4.63 | 16 | 11 |
| BIOCARTA_LYMPHOCYTE_PATHWAY | 1.90 | 4.63 | 4 | 9 |
| BIOCARTA_TCR_PATHWAY | 2.49 | 4.85 | 16 | 44 |
| BIOCARTA_VIP_PATHWAY | 2.87 | 4.93 | 20 | 26 |
| BIOCARTA_CTLA4_PATHWAY | 3.53 | 5.42 | 7 | 22 |
| BIOCARTA_TCRA_PATHWAY | 4.17 | 5.53 | 5 | 14 |
| GSE29615_CTRL_VS_DAY3 | ||||
| _LAIV_IFLU_VACCINE_PBMC_DN | 1.26 | 6.12 | 0 | 194 |
| BIOCARTA_NFAT_PATHWAY | 5.41 | 6.87 | 18 | 51 |
| GSE1112_OT1_CD8AB_VS_HY | ||||
| _CD8AA_THYMOCYTE_RTOC_CULTURE_DN | 3.54 | 8.00 | 0 | 194 |
| GSE12963_UNINF_VS_ENV_AND_NEF | ||||
| _DEFICIENT_HIV1_INF_CD4_TCELL_DN | 4.93 | 8.00 | 0 | 146 |
| BIOCARTA_GATA3_PATHWAY | 9.52 | 9.93 | 16 | 14 |
| KEGG_RIG_I_LIKE_RECEPTOR | ||||
| _SIGNALING_PATHWAY | 0.22 | 1.15 | 3 | 71 |
| BIOCARTA_LYM_PATHWAY | 2.23 | 1.58 | 5 | 14 |
| BIOCARTA_CDMAC_PATHWAY | 2.40 | 1.79 | 12 | 16 |
| GARGALOVIC_RESPONSE_TO | ||||
| _OXIDIZED_PHOSPHOLIPIDS_CYAN_UP | 2.38 | 1.87 | 0 | 17 |
| BIOCARTA_NKCELLS_PATHWAY | 2.84 | 1.90 | 6 | 20 |
| BIOCARTA_MSP_PATHWAY | 3.20 | 2.01 | 2 | 6 |
| BIOCARTA_GRANULOCYTES_PATHWAY | 4.03 | 2.26 | 6 | 15 |
| BIOCARTA_NEUTROPHIL_PATHWAY | 4.02 | 2.26 | 4 | 8 |
| BIOCARTA_IL17_PATHWAY | 4.31 | 2.29 | 7 | 15 |
| BIOCARTA_RNA_PATHWAY | 4.28 | 2.29 | 5 | 10 |
| PID_AVB3_INTEGRIN_PATHWAY | 1.20 | 2.42 | 4 | 74 |
| GOBP_NEGATIVE_REGULATION_OF | ||||
| _INTERLEUKIN_5_PRODUCTION | 6.38 | 2.42 | 0 | 8 |
| Gene Sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_MPR_PATHWAY | 6.27 | 9.15 | 18 | 21 |
| BIOCARTA_SHH_PATHWAY | 6.60 | 4.03 | 18 | 16 |
| PID_NCADHERIN_PATHWAY | 5.77 | 5.66 | 4 | 33 |
| BIOCARTA_AGR_PATHWAY | 7.82 | 8.78 | 3 | 33 |
| PID_LYMPH_ANGIOGENESIS_PATHWAY | 1.45 | 9.46 | 14 | 25 |
| HP_ABNORMAL_RIB_CAGE_MORPHOLOGY | 6.03 | 1.02 | 9 | 354 |
| HP_THORACIC_HYPOPLASIA | 5.01 | 1.02 | 3 | 138 |
| HP_ONYCHOLYSIS | 5.21 | 1.02 | 2 | 16 |
| BIOCARTA_PTC1_PATHWAY | 1.17 | 1.09 | 3 | 11 |
| GOBP_MESODERM_DEVELOPMENT | 7.52 | 1.11 | 6 | 132 |
| GOBP_MESODERM_MORPHOGENESIS | 1.65 | 1.11 | 5 | 75 |
| GOBP_ANTEROGRADE_DENDRITIC_TRANSPORT | ||||
| _OF_NEUROTRANSMITTER_RECEPTOR_COMPLEX | 5.60 | 1.11 | 1 | 5 |
| KEGG_HEMATOPOIETIC_CELL_LINEAGE | 3.68 | 1.15 | 4 | 87 |
| HP_THORACIC_DYSPLASIA | 9.63 | 1.22 | 3 | 6 |
| NKX2_3_TARGET_GENES | 2.65 | 1.36 | 0 | 544 |
| GOBP_CARDIAC_MUSCLE_CELL_FATE | ||||
| _COMMITMENT | 1.99 | 1.90 | 4 | 11 |
| GOBP_GASTRULATION | 5.13 | 2.18 | 10 | 190 |
| GOBP_FORMATION_OF_PRIMARY_GERM_LAYER | 4.68 | 2.18 | 10 | 123 |
| HP_ABNORMALITY_OF_THE_RIBS | 2.65 | 2.24 | 4 | 292 |
| PID_AVB3_INTEGRIN_PATHWAY | 1.20 | 2.36 | 7 | 74 |
| GOBP_CHORIO_ALLANTOIC_FUSION | 7.28 | 2.42 | 4 | 7 |
| BIOCARTA_KERATINOCYTE_PATHWAY | 4.95 | 2.46 | 10 | 46 |
| Overlapping Neighbors | p-Value | FDR |
|---|---|---|
| BIOCARTA_MPR_PATHWAY | 6.27 | 9.15 |
| BIOCARTA_CSK_PATHWAY | 5.02 | 9.15 |
| GOMF_CYCLIC_NUCLEOTIDE_DEPENDENT | ||
| _PROTEIN_KINASE_ACTIVITY | 6.70 | 3.88 |
| GOMF_CYCLIC_NUCLEOTIDE_BINDING | 6.59 | 3.88 |
| BIOCARTA_CREB_PATHWAY | 4.05 | 3.94 |
| BIOCARTA_DREAM_PATHWAY | 1.10 | 4.03 |
| BIOCARTA_SHH_PATHWAY | 6.60 | 4.03 |
| BIOCARTA_BAD_PATHWAY | 8.07 | 4.03 |
| BIOCARTA_IGF1R_PATHWAY | 9.71 | 4.03 |
| BIOCARTA_CFTR_PATHWAY | 1.81 | 4.63 |
| BIOCARTA_CK1_PATHWAY | 1.71 | 4.63 |
| BIOCARTA_PPARA_PATHWAY | 1.71 | 4.63 |
| BIOCARTA_AGPCR_PATHWAY | 2.56 | 4.85 |
| BIOCARTA_STATHMIN_PATHWAY | 2.30 | 4.85 |
| BIOCARTA_VIP_PATHWAY | 2.87 | 4.93 |
| HP_ABNORMAL_RIB_CAGE_MORPHOLOGY | 6.03 | 1.02 |
| BIOCARTA_PTC1_PATHWAY | 1.17 | 1.09 |
| Gene Sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_CREB_PATHWAY | 4.05 | 3.94 | 21 | 22 |
| BIOCARTA_SHH_PATHWAY | 6.60 | 4.03 | 16 | 16 |
| BIOCARTA_DREAM_PATHWAY | 1.10 | 4.03 | 19 | 13 |
| BIOCARTA_CK1_PATHWAY | 1.70 | 4.63 | 16 | 16 |
| BIOCARTA_AGPCR_PATHWAY | 2.56 | 4.85 | 18 | 11 |
| BIOCARTA_PRION_PATHWAY | 3.36 | 5.42 | 1 | 12 |
| BIOCARTA_NOS1_PATHWAY | 3.95 | 5.53 | 18 | 21 |
| PID_NCADHERIN_PATHWAY | 5.77 | 5.66 | 7 | 33 |
| BIOCARTA_NFAT_PATHWAY | 5.41 | 6.87 | 20 | 51 |
| BIOCARTA_AGR_PATHWAY | 7.82 | 8.78 | 4 | 33 |
| GOBP_ANTEROGRADE_DENDRITIC | ||||
| _TRANSPORT_OF_NEUROTRANSMITTER | ||||
| _RECEPTOR_COMPLEX | 5.60 | 1.11 | 1 | 5 |
| BIOCARTA_TRKA_PATHWAY | 1.92 | 1.57 | 11 | 14 |
| BIOCARTA_CB1R_PATHWAY | 2.19 | 1.69 | 1 | 7 |
| BIOCARTA_PDZS_PATHWAY | 2.53 | 1.80 | 0 | 18 |
| DIERICK_SEROTONIN_FUNCTION_GENES | 2.77 | 1.87 | 0 | 7 |
| GOBP_VESICLE_MEDIATED_TRANSPORT | ||||
| _TO_THE_PLASMA_MEMBRANE | 3.83 | 2.18 | 0 | 140 |
| BIOCARTA_ERK5_PATHWAY | 3.65 | 2.19 | 12 | 14 |
| BIOCARTA_MAL_PATHWAY | 4.39 | 2.29 | 10 | 19 |
| BIOCARTA_NGF_PATHWAY | 5.07 | 2.47 | 12 | 20 |
| Gene Sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_IGF1R_PATHWAY | 9.71 | 4.03 | 19 | 23 |
| PID_NCADHERIN_PATHWAY | 5.77 | 5.66 | 3 | 33 |
| BIOCARTA_NFAT_PATHWAY | 5.41 | 6.87 | 18 | 51 |
| BIOCARTA_AT1R_PATHWAY | 6.55 | 7.65 | 13 | 27 |
| BIOCARTA_AGR_PATHWAY | 7.82 | 8.78 | 4 | 33 |
| BIOCARTA_IGF1MTOR_PATHWAY | 1.21 | 1.09 | 4 | 19 |
| KEGG_BIOSYNTHESIS_OF | ||||
| _UNSATURATED_FATTY_ACIDS | 2.59 | 1.15 | 0 | 22 |
| HP_TORSADE_DE_POINTES | 1.57 | 1.59 | 0 | 24 |
| GOBP_CARDIAC_MUSCLE_CELL | ||||
| _FATE_COMMITMENT | 1.99 | 1.90 | 0 | 11 |
| GOBP_VASCULAR_ASSOCIATED_SMOOTH | ||||
| _MUSCLE_CELL_MIGRATION | 3.42 | 2.18 | 2 | 45 |
| GOBP_MUSCLE_CELL_MIGRATION | 4.75 | 2.18 | 3 | 104 |
| BIOCARTA_MAL_PATHWAY | 4.39 | 2.29 | 9 | 19 |
| BIOCARTA_MYOSIN_PATHWAY | 4.81 | 2.46 | 8 | 11 |
| Gene Sets | p-Value | FDR | Degree | No. Genes |
|---|---|---|---|---|
| BIOCARTA_CFTR_PATHWAY | 1.81 | 4.63 | 14 | 11 |
| BIOCARTA_PPARA_PATHWAY | 1.71 | 4.63 | 14 | 52 |
| GOBP_UNSATURATED_FATTY | ||||
| _ACID_BIOSYNTHETIC_PROCESS | 4.90 | 1.11 | 1 | 52 |
| KEGG_BIOSYNTHESIS_OF | ||||
| _UNSATURATED_FATTY_ACIDS | 2.59 | 1.15 | 1 | 22 |
| PID_S1P_META_PATHWAY | 2.55 | 1.25 | 1 | 21 |
| PID_S1P_S1P2_PATHWAY | 3.92 | 1.49 | 5 | 24 |
| GOBP_VESICLE_MEDIATED_TRANSPORT | ||||
| _TO_THE_PLASMA_MEMBRANE | 3.83 | 2.18 | 0 | 140 |
| GOBP_ADIPOSE_TISSUE_DEVELOPMENT | 4.76 | 2.18 | 0 | 46 |
| BURTON_ADIPOGENESIS_2 | 6.41 | 2.41 | 0 | 71 |
| Gene Set A | Gene Set B | Overlapping Genes | p-Value |
|---|---|---|---|
| BIOCARTA_PPARA_PATHWAY | BIOCARTA_GPCR_PATHWAY | 10 | 1.36 |
| KEGG_HEMATOPOIETIC_CELL_LINEAGE | KEGG_ECM_RECEPTOR_INTERACTION | 14 | 1.95 |
| BIOCARTA_PPARA_PATHWAY | BIOCARTA_CREB_PATHWAY | 12 | 3.15 |
| KEGG_GAP_JUNCTION | BIOCARTA_GPCR_PATHWAY | 10 | 4.67 |
| Main Category | Sub-Categories | No. Gene Sets |
|---|---|---|
| C2: curated gene sets | CGP: chemical and genetic perturbations | 3383 |
| Canonical Pathways: BioCarta | 292 | |
| Canonical Pathways: KEGG | 186 | |
| Canonical Pathways: PID | 196 | |
| Canonical Pathways: REACTOME | 1615 | |
| Canonical Pathways: WikiPathways | 664 | |
| C3: regulatory target gene sets | miRDB subset of MIR | 2377 |
| MIR_Legacy subset of MIR | 221 | |
| GTRD subset of TFT | 518 | |
| TFT_Legacy subset of TFT | 610 | |
| C5: ontology gene sets | Gene Ontology: Biological Process | 7658 |
| Gene Ontology: Cellular Component | 1006 | |
| Gene Ontology: Molecular Function | 1738 | |
| HPO: Human Phenotype Ontology | 5071 | |
| C7: immunologic signature gene sets | ImmuneSigDB subset of C7 | 4872 |
| VAX: vaccine response gene sets | 347 | |
| C8: cell type signature gene sets | - | 700 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilopoulou, C.; McDaid-McCloskey, S.L.; McCluskey, G.; Duguez, S.; Morris, A.P.; Duddy, W. Genome-Wide Gene-Set Analysis Identifies Molecular Mechanisms Associated with ALS. Int. J. Mol. Sci. 2023, 24, 4021. https://doi.org/10.3390/ijms24044021
Vasilopoulou C, McDaid-McCloskey SL, McCluskey G, Duguez S, Morris AP, Duddy W. Genome-Wide Gene-Set Analysis Identifies Molecular Mechanisms Associated with ALS. International Journal of Molecular Sciences. 2023; 24(4):4021. https://doi.org/10.3390/ijms24044021
Chicago/Turabian StyleVasilopoulou, Christina, Sarah L. McDaid-McCloskey, Gavin McCluskey, Stephanie Duguez, Andrew P. Morris, and William Duddy. 2023. "Genome-Wide Gene-Set Analysis Identifies Molecular Mechanisms Associated with ALS" International Journal of Molecular Sciences 24, no. 4: 4021. https://doi.org/10.3390/ijms24044021
APA StyleVasilopoulou, C., McDaid-McCloskey, S. L., McCluskey, G., Duguez, S., Morris, A. P., & Duddy, W. (2023). Genome-Wide Gene-Set Analysis Identifies Molecular Mechanisms Associated with ALS. International Journal of Molecular Sciences, 24(4), 4021. https://doi.org/10.3390/ijms24044021