
Evidence for Existence of Multiple Functional Human Small RNAs Derived from Transcripts of Protein-Coding Genes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
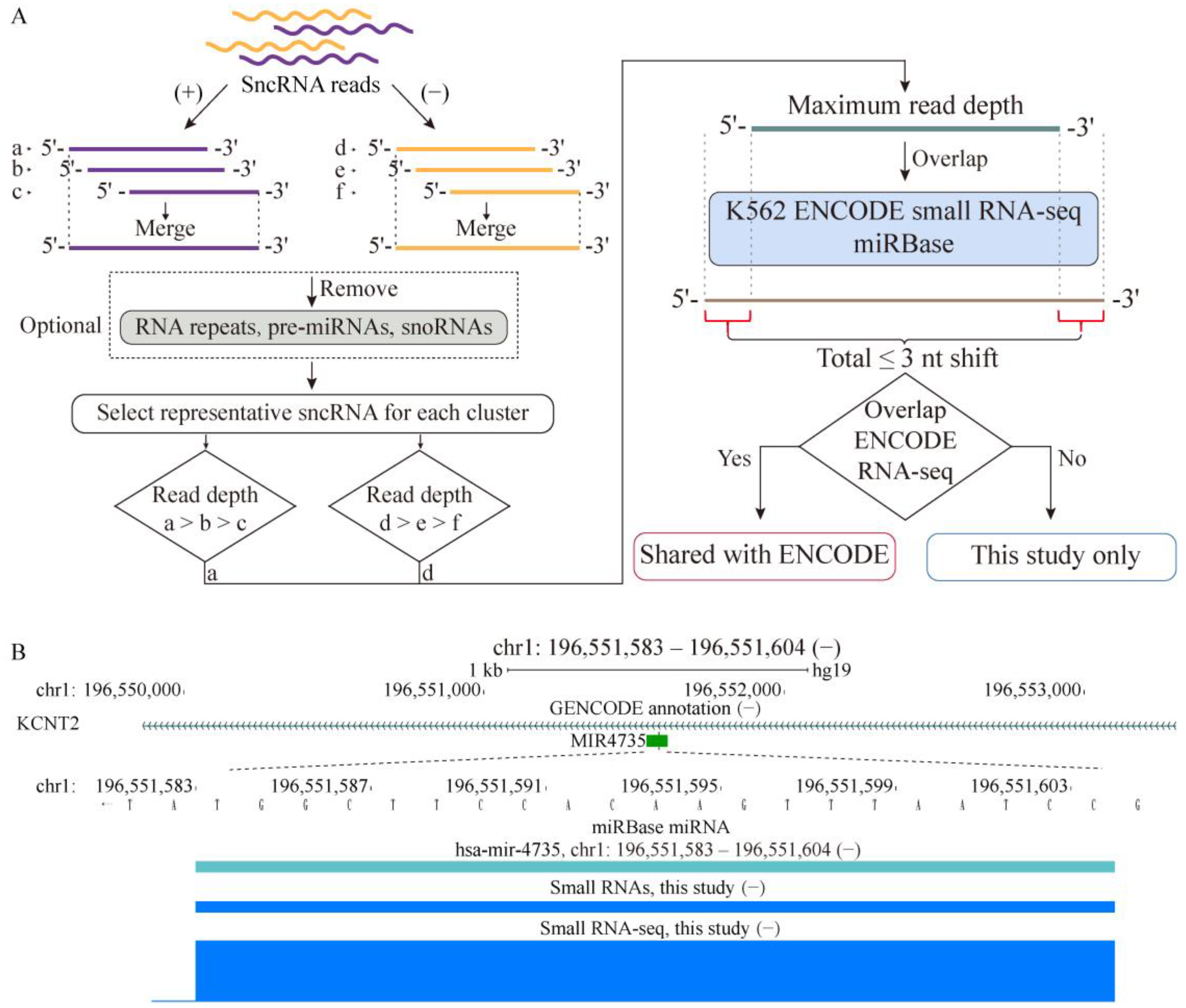
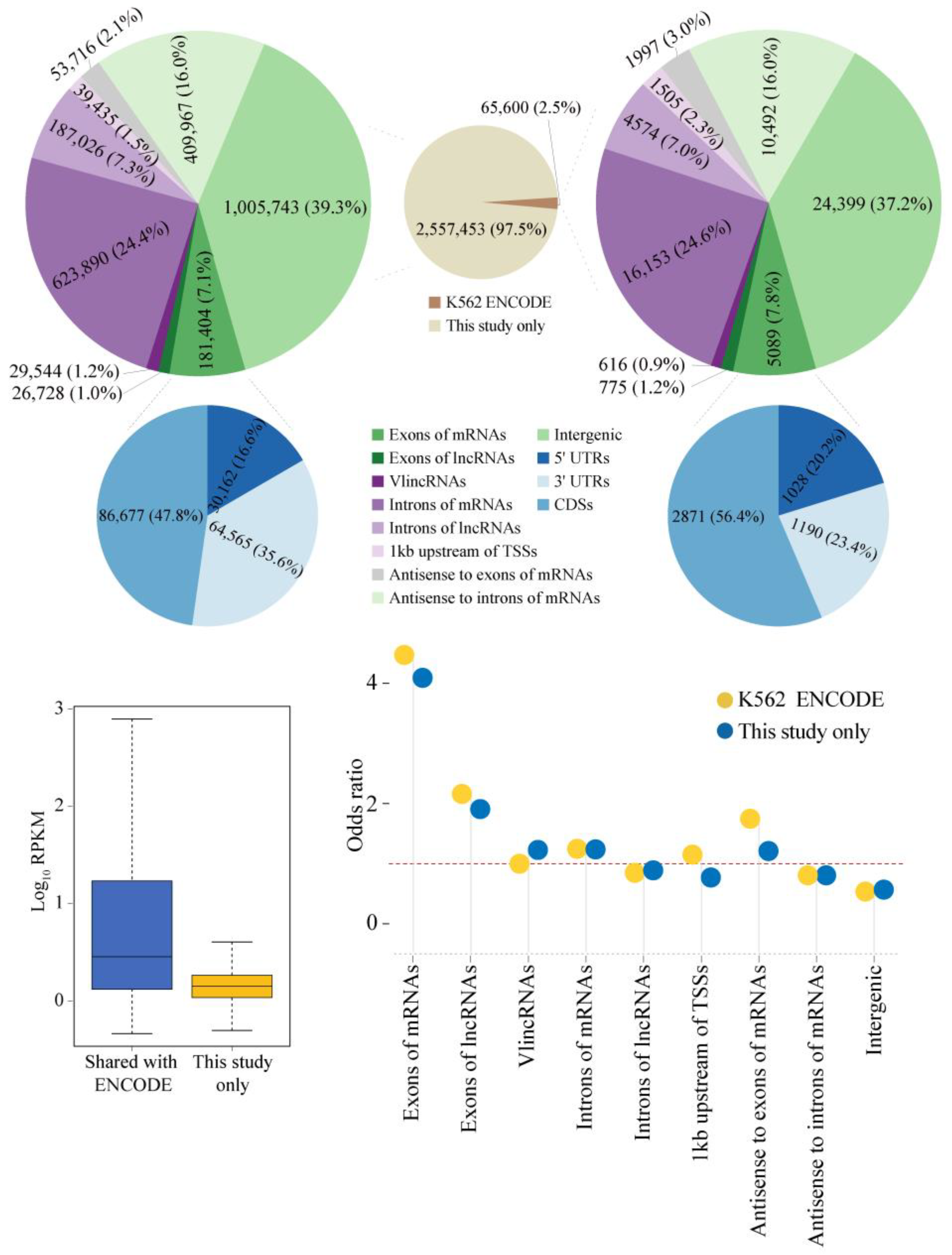
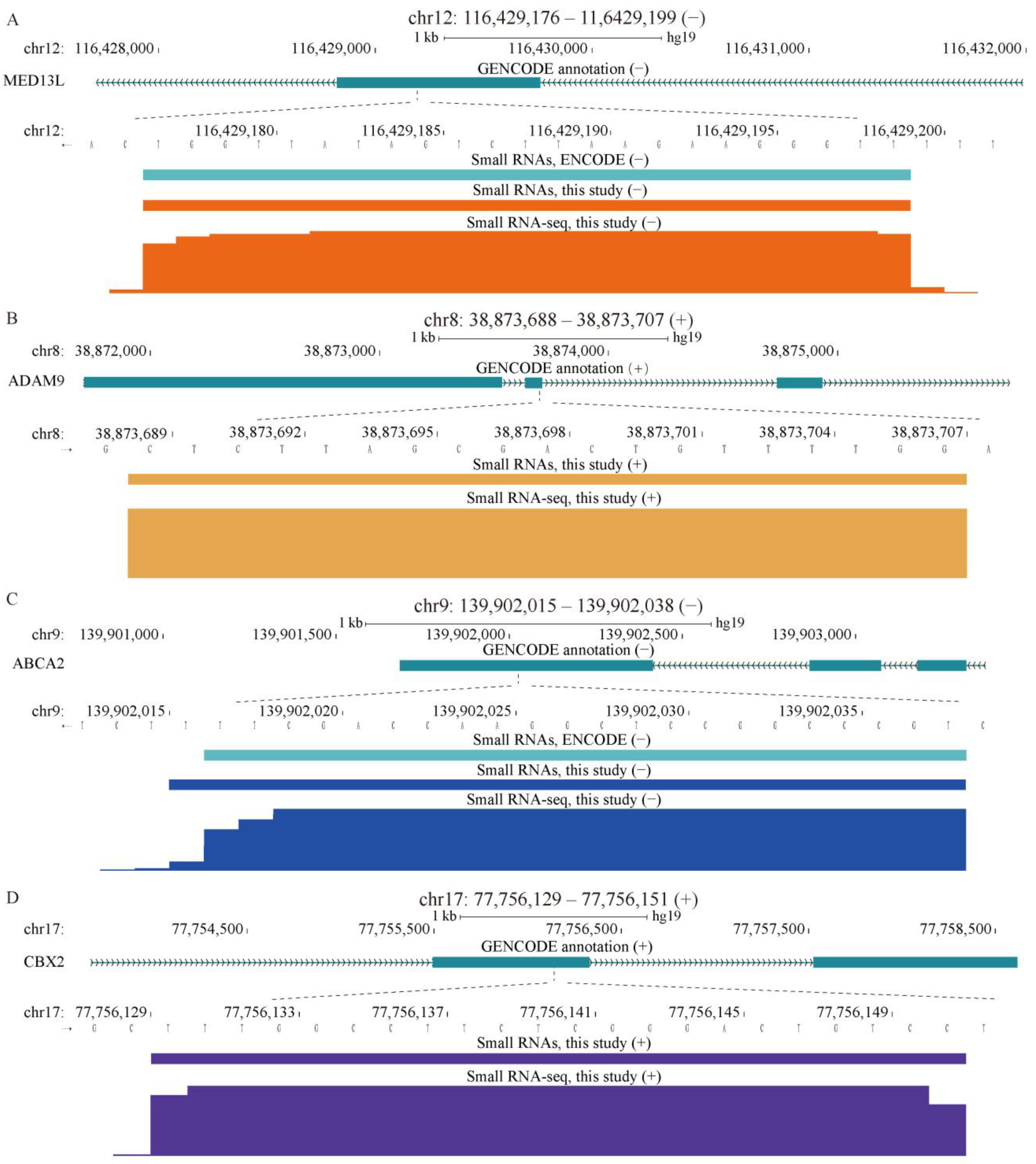
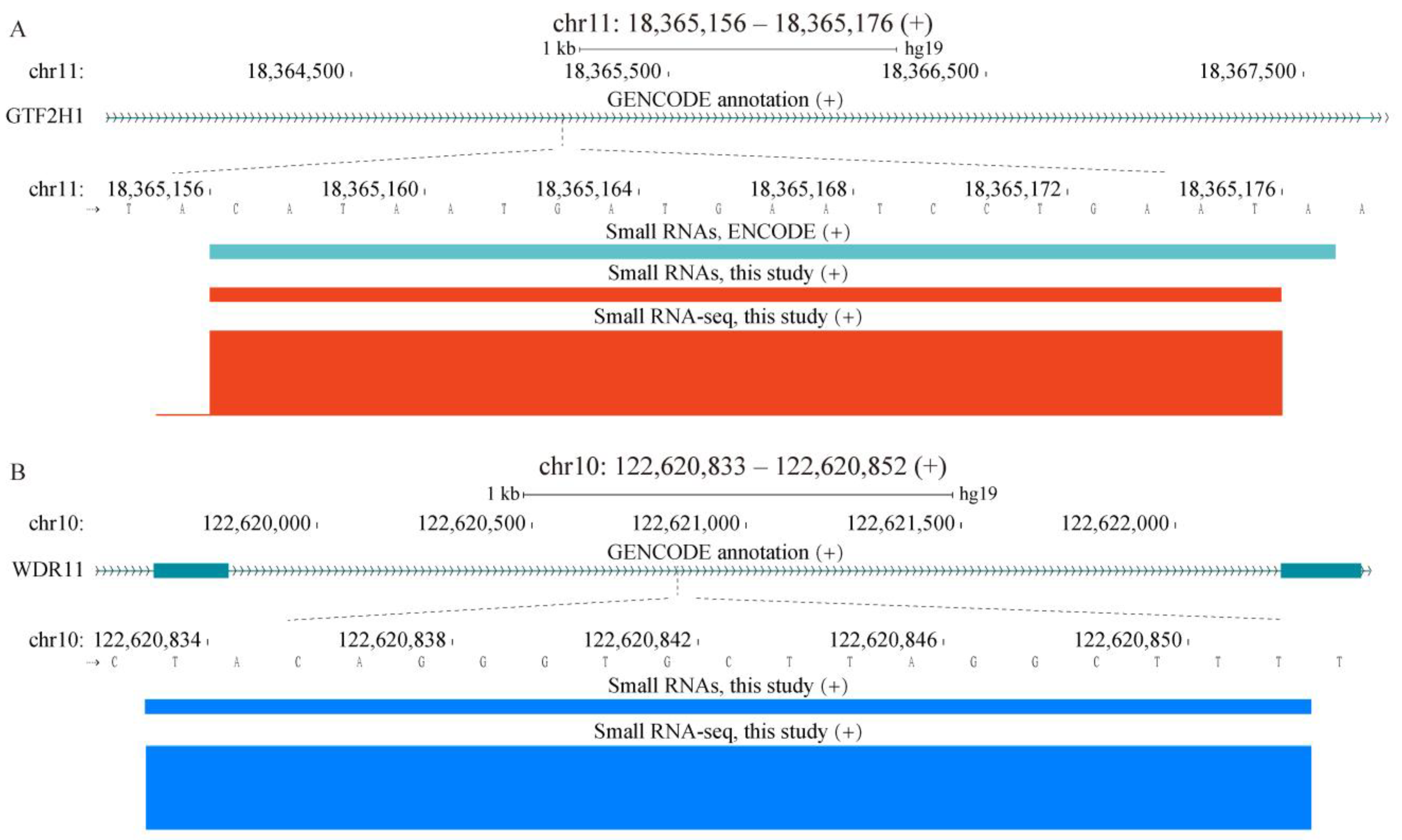
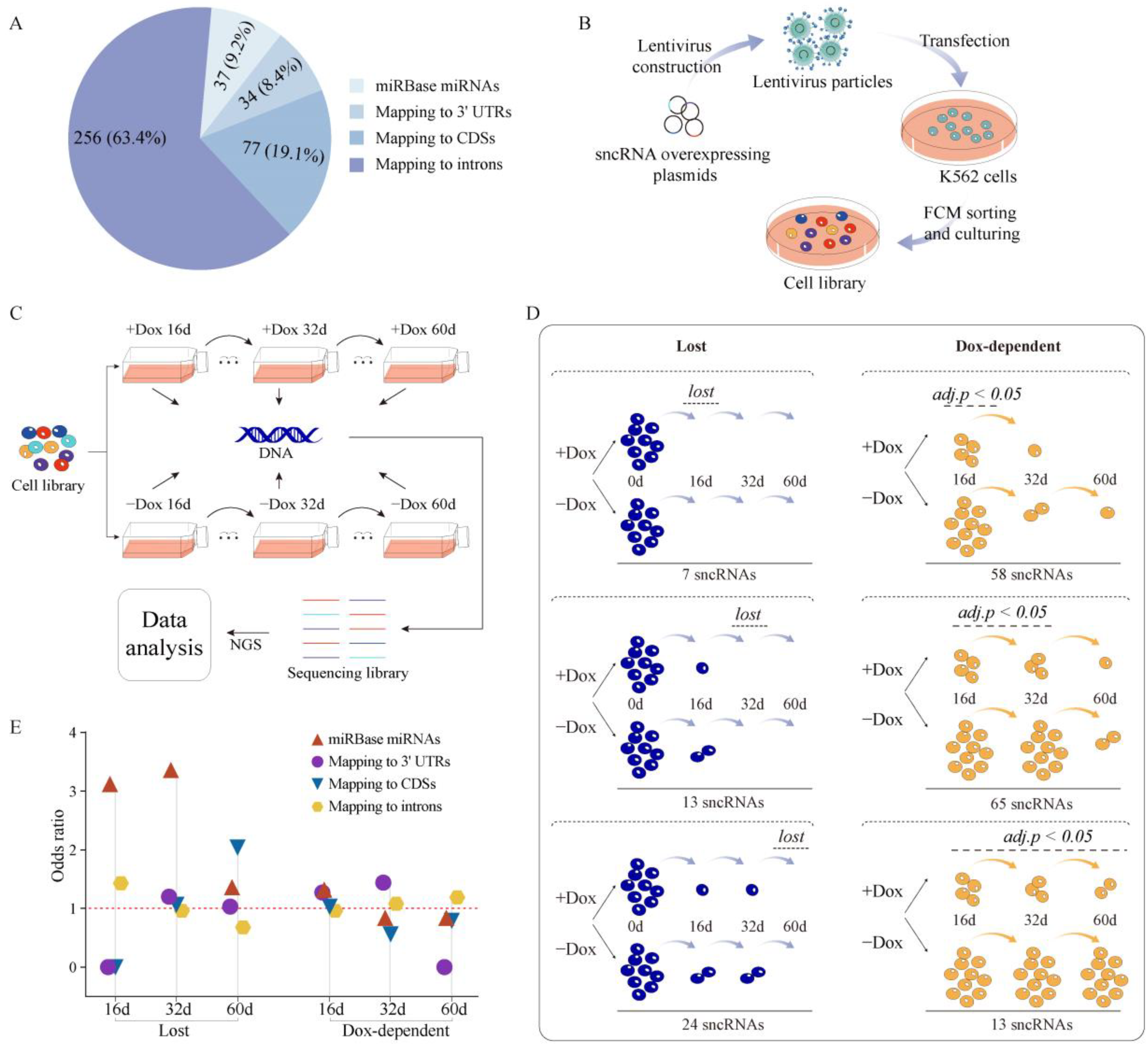
2.1. Properties of sncRNA Transcriptome in a Human Cancer Cell Line
2.2. Establishing Overexpression System for Unannotated sncRNAs
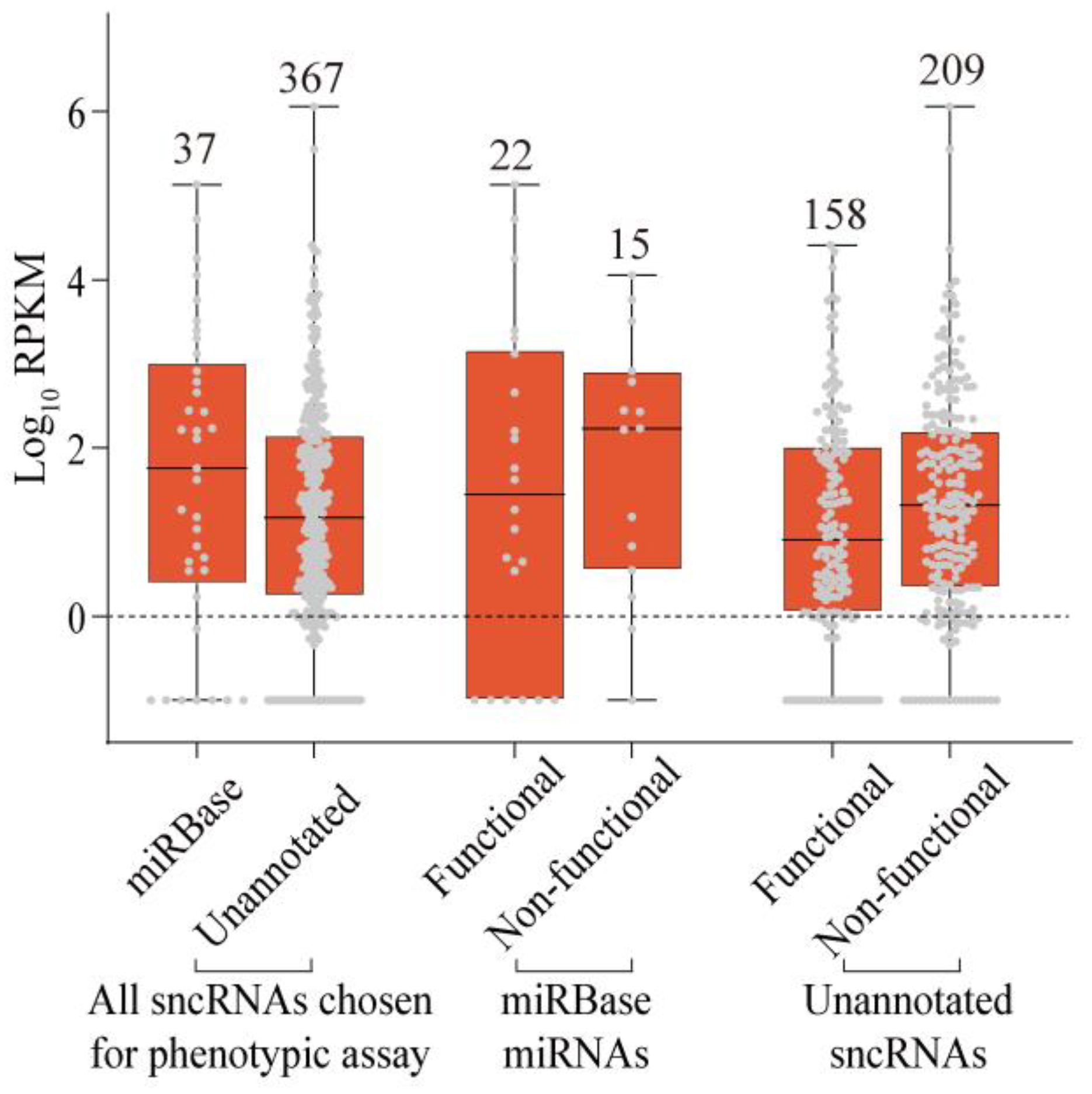
2.3. Phenotypic Analysis of Selected sncRNAs
2.4. Properties of Unannotated sncRNAs Positive in the Phenotypic Screen
3. Discussion
4. Materials and Methods
4.1. Biological Material
4.2. K562 sncRNA Profiling
4.3. Validation of sncRNA Overexpression Using Transient Transfection
4.4. Generation of sncRNA Overexpression Library
4.5. Detection of sncRNAs Affecting Cell Growth and Survival
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapranov, P.; Cawley, S.E.; Drenkow, J.; Bekiranov, S.; Strausberg, R.L.; Fodor, S.P.; Gingeras, T.R. Large-scale transcriptional activity in chromosomes 21 and 22. Science 2002, 296, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, Y.; Furuno, M.; Kasukawa, T.; Adachi, J.; Bono, H.; Kondo, S.; Nikaido, I.; Osato, N.; Saito, R.; Suzuki, H.; et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 2002, 420, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Euskirchen, G.; Bertone, P.; Martone, R.; Luscombe, N.M.; Hartman, S.; Harrison, P.M.; Nelson, F.K.; Miller, P.; Gerstein, M.; et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003, 17, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023. [Google Scholar] [CrossRef]
- Clark, M.B.; Choudhary, A.; Smith, M.A.; Taft, R.J.; Mattick, J.S. The dark matter rises: The expanding world of regulatory RNAs. Essays Biochem. 2013, 54, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; St Laurent, G. Dark Matter RNA: Existence, Function, and Controversy. Front. Genet. 2012, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; St Laurent, G.; Raz, T.; Ozsolak, F.; Reynolds, C.P.; Sorensen, P.H.; Reaman, G.; Milos, P.; Arceci, R.J.; Thompson, J.F.; et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 2010, 8, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S. Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. Bioessays 2003, 25, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. A new paradigm for developmental biology. J. Exp. Biol. 2007, 210, 1526–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Haerty, W. Genome-Wide Analysis of Human Long Noncoding RNAs: A Provocative Review. Annu. Rev. Genom. Hum. Genet. 2022, 23, 153–172. [Google Scholar] [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [Green Version]
- Taft, R.J.; Glazov, E.A.; Cloonan, N.; Simons, C.; Stephen, S.; Faulkner, G.J.; Lassmann, T.; Forrest, A.R.; Grimmond, S.M.; Schroder, K.; et al. Tiny RNAs associated with transcription start sites in animals. Nat. Genet. 2009, 41, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Simons, C.; Nahkuri, S.; Oey, H.; Korbie, D.J.; Mercer, T.R.; Holst, J.; Ritchie, W.; Wong, J.J.; Rasko, J.E.; et al. Nuclear-localized tiny RNAs are associated with transcription initiation and splice sites in metazoans. Nat. Struct. Mol. Biol. 2010, 17, 1030–1034. [Google Scholar] [CrossRef]
- Seila, A.C.; Calabrese, J.M.; Levine, S.S.; Yeo, G.W.; Rahl, P.B.; Flynn, R.A.; Young, R.A.; Sharp, P.A. Divergent transcription from active promoters. Science 2008, 322, 1849–1851. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; Ozsolak, F.; Kim, S.W.; Foissac, S.; Lipson, D.; Hart, C.; Roels, S.; Borel, C.; Antonarakis, S.E.; Monaghan, A.P.; et al. New class of gene-termini-associated human RNAs suggests a novel RNA copying mechanism. Nature 2010, 466, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Laudadio, I.; Formichetti, S.; Gioiosa, S.; Klironomos, F.; Rajewsky, N.; Macino, G.; Carissimi, C.; Fulci, V. Characterization of Transcription Termination-Associated RNAs: New Insights into their Biogenesis, Tailing, and Expression in Primary Tumors. Int. J. Genom. 2018, 2018, 1243858. [Google Scholar] [CrossRef]
- Ma, X.; Han, N.; Shao, C.; Meng, Y. Transcriptome-Wide Discovery of PASRs (Promoter-Associated Small RNAs) and TASRs (Terminus-Associated Small RNAs) in Arabidopsis thaliana. PLoS ONE 2017, 12, e0169212. [Google Scholar] [CrossRef] [Green Version]
- Leng, X.M.; Diao, L.T.; Li, B.; Bi, Y.Z.; Chen, C.J.; Zhou, H.; Qu, L.H. The ribosomal protein rpl26 promoter is required for its 3’ sense terminus ncRNA transcription in Schizosaccharomyces pombe, implicating a new transcriptional mechanism for ncRNAs. Biochem. Biophys. Res. Commun. 2014, 444, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Valen, E.; Preker, P.; Andersen, P.R.; Zhao, X.; Chen, Y.; Ender, C.; Dueck, A.; Meister, G.; Sandelin, A.; Jensen, T.H. Biogenic mechanisms and utilization of small RNAs derived from human protein-coding genes. Nat. Struct. Mol. Biol. 2011, 18, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.; Tomaru, Y.; Suzuki, H.; Hayashizaki, Y.; Daub, C.O. Deep-sequencing of human Argonaute-associated small RNAs provides insight into miRNA sorting and reveals Argonaute association with RNA fragments of diverse origin. RNA Biol. 2011, 8, 158–177. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Batzel, P.; Berezikov, E.; Eilbeck, K.; Eppig, J.T.; McAndrews, M.S.; Singer, A.; Postlethwait, J.H. miRNA Nomenclature: A View Incorporating Genetic Origins, Biosynthetic Pathways, and Sequence Variants. Trends Genet. 2015, 31, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, T. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.Y.; Kuksa, P.P.; Amlie-Wolf, A.; Valladares, O.; Ungar, L.H.; Kannan, S.; Gregory, B.D.; Wang, L.S. DASHR: Database of small human noncoding RNAs. Nucleic Acids Res. 2016, 44, D216–D222. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, P.; Lu, Y.; Li, Y.; Zheng, Y.; Kan, Y.; Chen, R.; He, S. piRBase: A comprehensive database of piRNA sequences. Nucleic Acids Res. 2019, 47, D175–D180. [Google Scholar] [CrossRef] [Green Version]
- Bouchard-Bourelle, P.; Desjardins-Henri, C.; Mathurin-St-Pierre, D.; Deschamps-Francoeur, G.; Fafard-Couture, E.; Garant, J.M.; Elela, S.A.; Scott, M.S. snoDB: An interactive database of human snoRNA sequences, abundance and interactions. Nucleic Acids Res. 2020, 48, D220–D225. [Google Scholar] [CrossRef]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Rendtlew Danielsen, J.M.; Yang, Y.G.; Qi, Y. A role for small RNAs in DNA double-strand break repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Wei, W.; Li, M.M.; Wu, Y.S.; Ba, Z.; Jin, K.X.; Li, M.M.; Liao, Y.Q.; Adhikari, S.; Chong, Z.; et al. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 2014, 24, 532–541. [Google Scholar] [CrossRef]
- Tuck, A.C.; Tollervey, D. RNA in pieces. Trends Genet. 2011, 27, 422–432. [Google Scholar] [CrossRef]
- Pederson, T. Regulatory RNAs derived from transfer RNA? RNA 2010, 16, 1865–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malka, Y.; Alkan, F.; Ju, S.; Korner, P.R.; Pataskar, A.; Shulman, E.; Loayza-Puch, F.; Champagne, J.; Wenzel, C.; Faller, W.J.; et al. Alternative cleavage and polyadenylation generates downstream uncapped RNA isoforms with translation potential. Mol. Cell 2022, 82, 3840–3855.e8. [Google Scholar] [CrossRef] [PubMed]
- Fejes-Toth, K.; Sotirova, V.; Sachidanandam, R.; Assaf, G.; Hannon, G.J.; Kapranov, P.; Foissac, S.; Willingham, A.T.; Duttagupta, R.; Dumais, E.; et al. Post-transcriptional processing generates a diversity of 5’-modified long and short RNAs. Nature 2009, 457, 1028–1032. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Bracken, C.P.; Kolle, G.; Szubert, J.M.; Korbie, D.J.; Askarian-Amiri, M.E.; Gardiner, B.B.; Goodall, G.J.; Grimmond, S.M.; et al. Regulated post-transcriptional RNA cleavage diversifies the eukaryotic transcriptome. Genome Res. 2010, 20, 1639–1650. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Cass, A.A.; Bahn, J.H.; Lee, J.H.; Greer, C.; Lin, X.; Kim, Y.; Hsiao, Y.H.; Xiao, X. Global analyses of endonucleolytic cleavage in mammals reveal expanded repertoires of cleavage-inducing small RNAs and their targets. Nucleic Acids Res. 2016, 44, 3253–3263. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Cai, Y.; Kapranov, P.; Xu, D. Reverse-genetics studies of lncRNAs—What we have learnt and paths forward. Genome Biol. 2020, 21, 93. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA Mimics Should Be Used with Caution. Front. Genet. 2015, 6, 340. [Google Scholar] [CrossRef] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Chiang, H.R.; Schoenfeld, L.W.; Ruby, J.G.; Auyeung, V.C.; Spies, N.; Baek, D.; Johnston, W.K.; Russ, C.; Luo, S.; Babiarz, J.E.; et al. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010, 24, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.H.; Moretti, P.A.; Goodall, G.J.; Pitson, S.M. Attenuation of leakiness in doxycycline-inducible expression via incorporation of 3’ AU-rich mRNA destabilizing elements. Biotechniques 2008, 45, 155–156, 158, 160 passim. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, O.; Chung, M.; Ansai, S.; Takeuchi, H.; Miyaji, M. A modified Tet-ON system minimizing leaky expression for cell-type specific gene induction in medaka fish. Dev. Growth Differ. 2021, 63, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Costello, A.; Lao, N.T.; Gallagher, C.; Capella Roca, B.; Julius, L.A.N.; Suda, S.; Ducree, J.; King, D.; Wagner, R.; Barron, N.; et al. Leaky Expression of the TET-On System Hinders Control of Endogenous miRNA Abundance. Biotechnol. J. 2019, 14, e1800219. [Google Scholar] [CrossRef]
- Nechaev, S.; Fargo, D.C.; dos Santos, G.; Liu, L.; Gao, Y.; Adelman, K. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 2010, 327, 335–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1794. [Google Scholar] [CrossRef] [Green Version]
- Isakova, A.; Neff, N.; Quake, S.R. Single-cell quantification of a broad RNA spectrum reveals unique noncoding patterns associated with cell types and states. Proc. Natl. Acad. Sci. USA 2021, 118, e2113568118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Ouvrard, J.; Muniz, L.; Nicolas, E.; Trouche, D. Small Interfering RNAs Targeting a Chromatin-Associated RNA Induce Its Transcriptional Silencing in Human Cells. Mol. Cell. Biol. 2022, 42, e0027122. [Google Scholar] [CrossRef]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Fazi, F.; Ciaudo, C. Argonaute Proteins: From Structure to Function in Development and Pathological Cell Fate Determination. Front. Cell Dev. Biol. 2019, 7, 360. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Wahlestedt, C.; Kapranov, P. Strategies to Annotate and Characterize Long Noncoding RNAs: Advantages and Pitfalls. Trends Genet. 2018, 34, 704–721. [Google Scholar] [CrossRef]
- Kaneto, C.M.; Nascimento, J.S.; Prado, M.; Mendonca, L.S.O. Circulating miRNAs as biomarkers in cardiovascular diseases. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2234–2243. [Google Scholar] [CrossRef]
- Sohel, M.M.H. Circulating microRNAs as biomarkers in cancer diagnosis. Life Sci. 2020, 248, 117473. [Google Scholar] [CrossRef] [PubMed]
- Sabre, L.; Punga, T.; Punga, A.R. Circulating miRNAs as Potential Biomarkers in Myasthenia Gravis: Tools for Personalized Medicine. Front. Immunol. 2020, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rounge, T.B.; Umu, S.U.; Keller, A.; Meese, E.; Ursin, G.; Tretli, S.; Lyle, R.; Langseth, H. Circulating small non-coding RNAs associated with age, sex, smoking, body mass and physical activity. Sci. Rep. 2018, 8, 17650. [Google Scholar] [CrossRef] [Green Version]
- Hube, F.; Velasco, G.; Rollin, J.; Furling, D.; Francastel, C. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2011, 39, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hube, F.; Ulveling, D.; Sureau, A.; Forveille, S.; Francastel, C. Short intron-derived ncRNAs. Nucleic Acids Res. 2017, 45, 4768–4781. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, F.; Wang, F.; Cao, H.; Chen, Y.; Diao, Y.; Kapranov, P. Evidence for Existence of Multiple Functional Human Small RNAs Derived from Transcripts of Protein-Coding Genes. Int. J. Mol. Sci. 2023, 24, 4163. https://doi.org/10.3390/ijms24044163
Gao F, Wang F, Cao H, Chen Y, Diao Y, Kapranov P. Evidence for Existence of Multiple Functional Human Small RNAs Derived from Transcripts of Protein-Coding Genes. International Journal of Molecular Sciences. 2023; 24(4):4163. https://doi.org/10.3390/ijms24044163
Chicago/Turabian StyleGao, Fan, Fang Wang, Huifen Cao, Yue Chen, Yong Diao, and Philipp Kapranov. 2023. "Evidence for Existence of Multiple Functional Human Small RNAs Derived from Transcripts of Protein-Coding Genes" International Journal of Molecular Sciences 24, no. 4: 4163. https://doi.org/10.3390/ijms24044163
APA StyleGao, F., Wang, F., Cao, H., Chen, Y., Diao, Y., & Kapranov, P. (2023). Evidence for Existence of Multiple Functional Human Small RNAs Derived from Transcripts of Protein-Coding Genes. International Journal of Molecular Sciences, 24(4), 4163. https://doi.org/10.3390/ijms24044163