
NINJ1 Regulates Platelet Activation and PANoptosis in Septic Disseminated Intravascular Coagulation

 ,
,
Abstract
:1. Introduction
2. Results
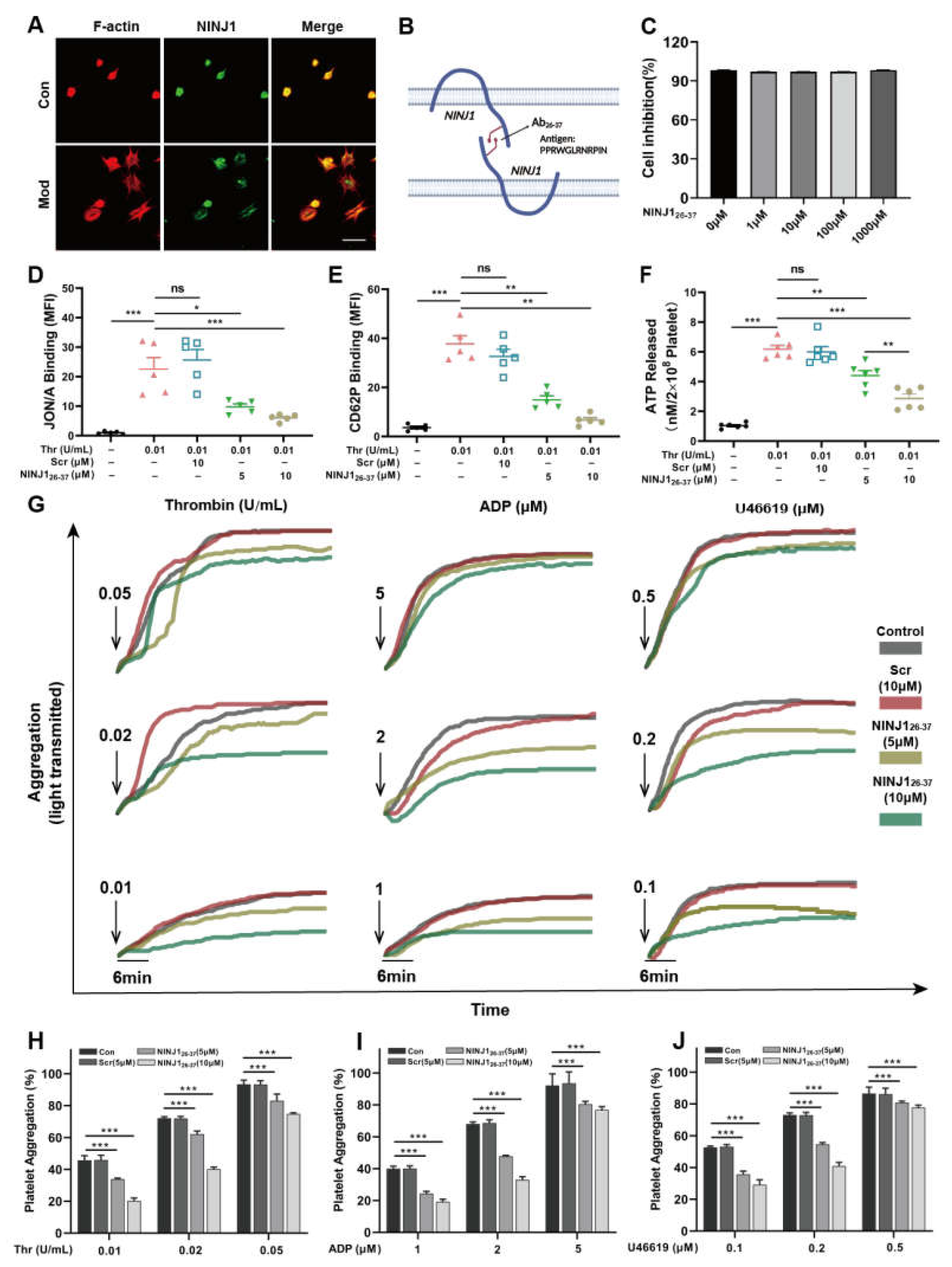
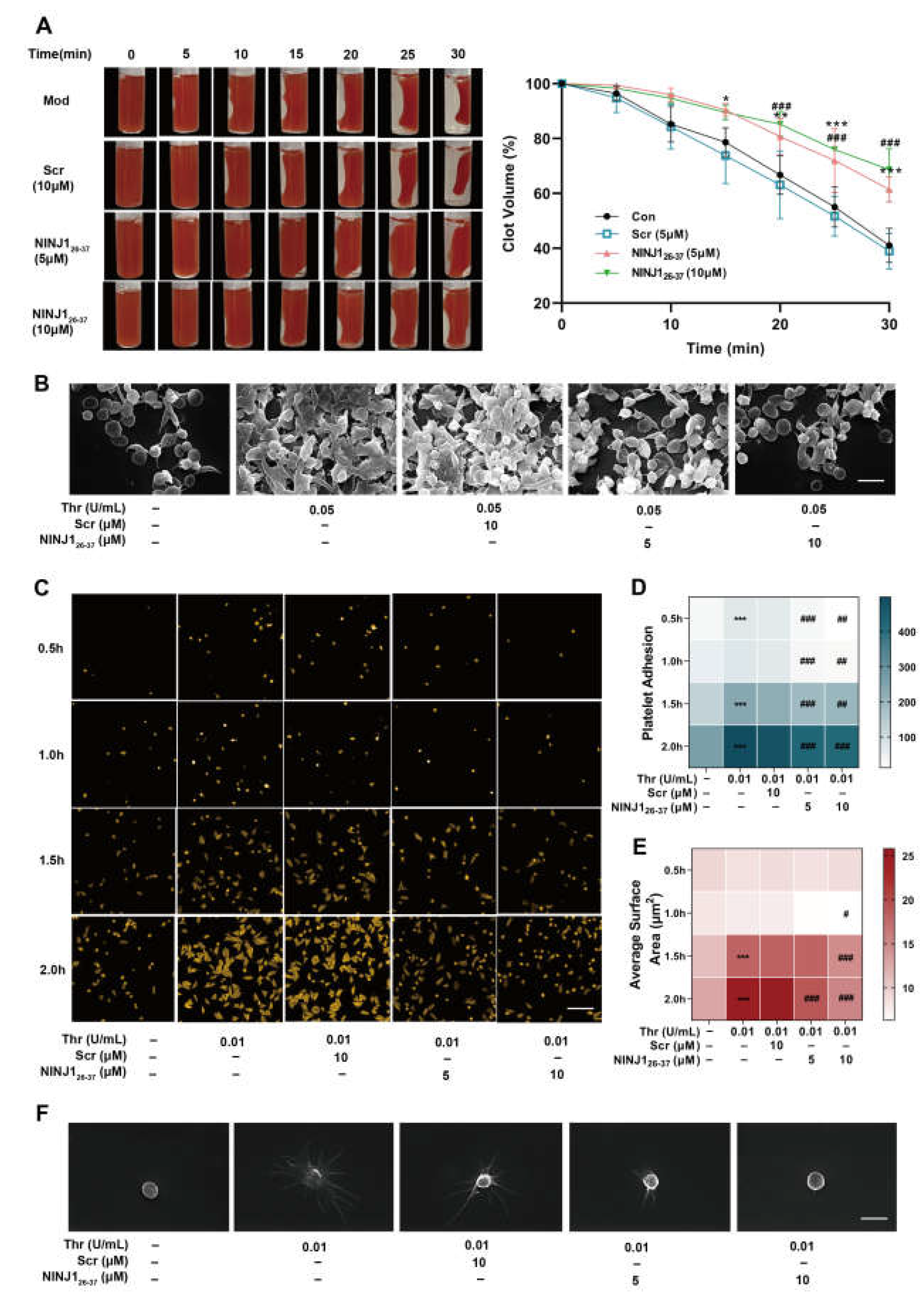
2.1. Inhibition of NINJ1 Reduced the Degree of Agonist-Induced Platelet Activation In Vitro
2.2. Inhibition of NINJ1 Antagonizes the Platelet Integrin Outside-In Signaling In Vitro
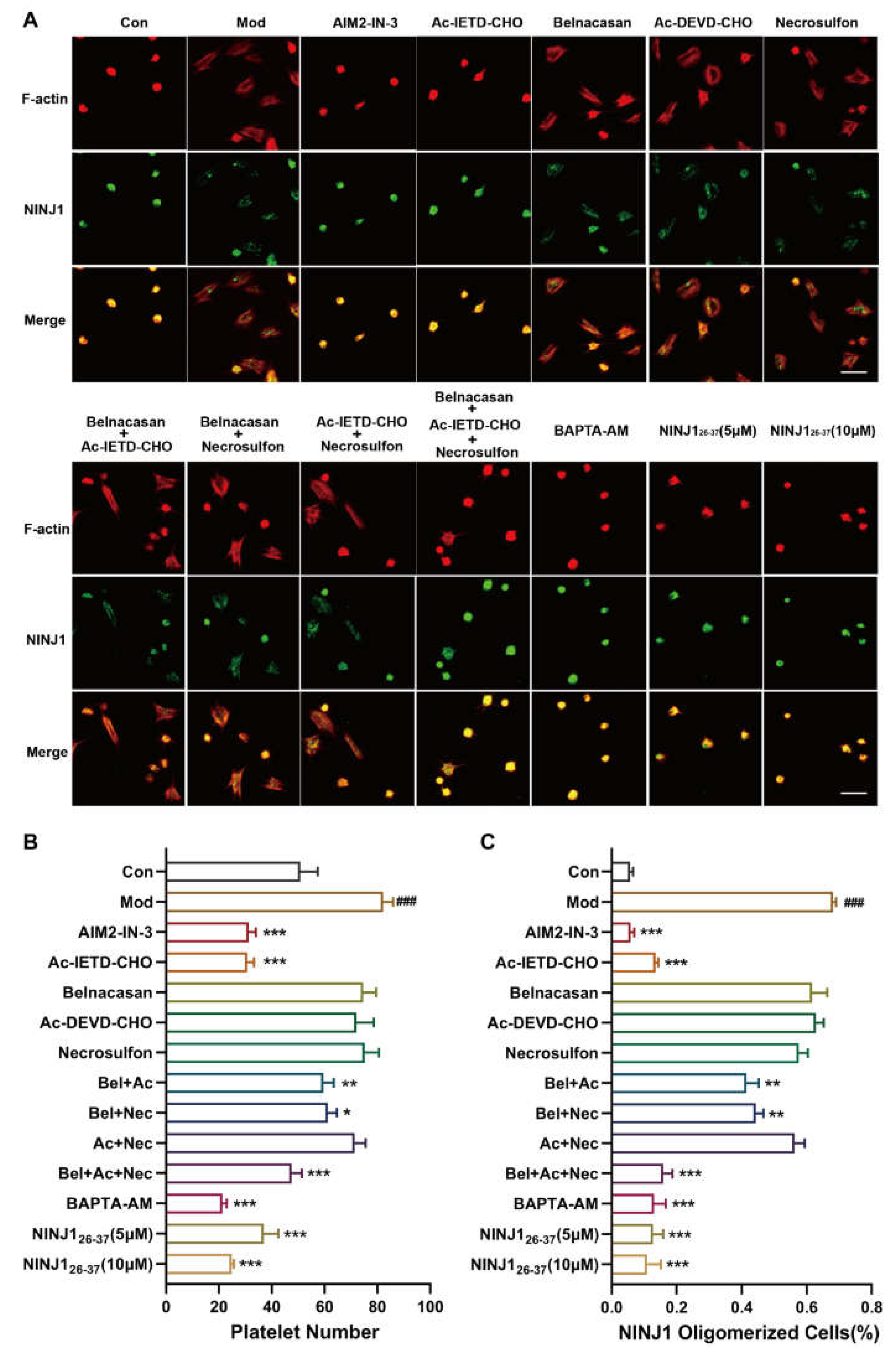
2.3. PANoptosis Pathway Regulates Platelet Plasma Membrane Disruption Related to NINJ1 Oligomerization by Calcium Overload
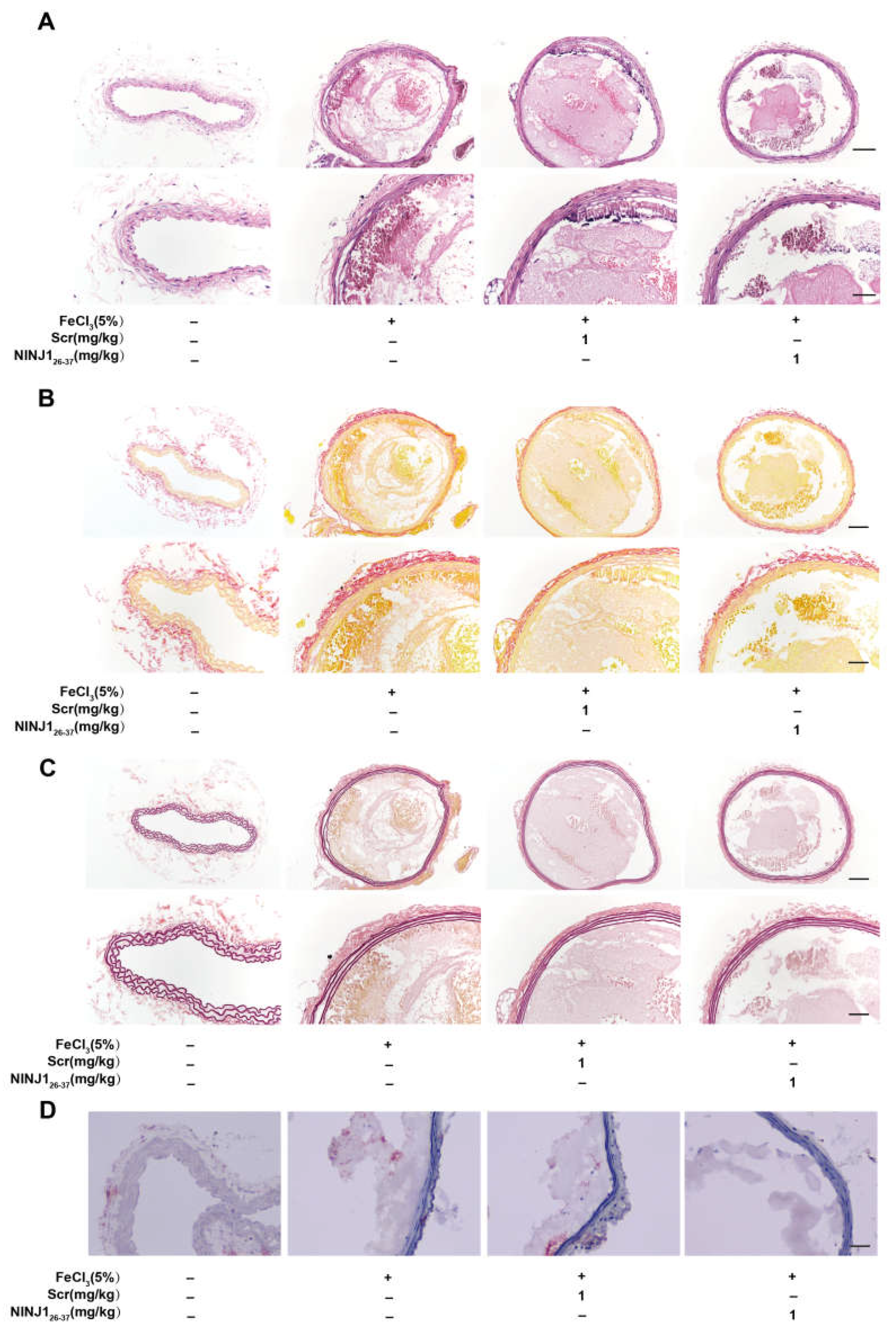
2.4. Inhibition of NINJ1 Reduced Arterial Thrombosis
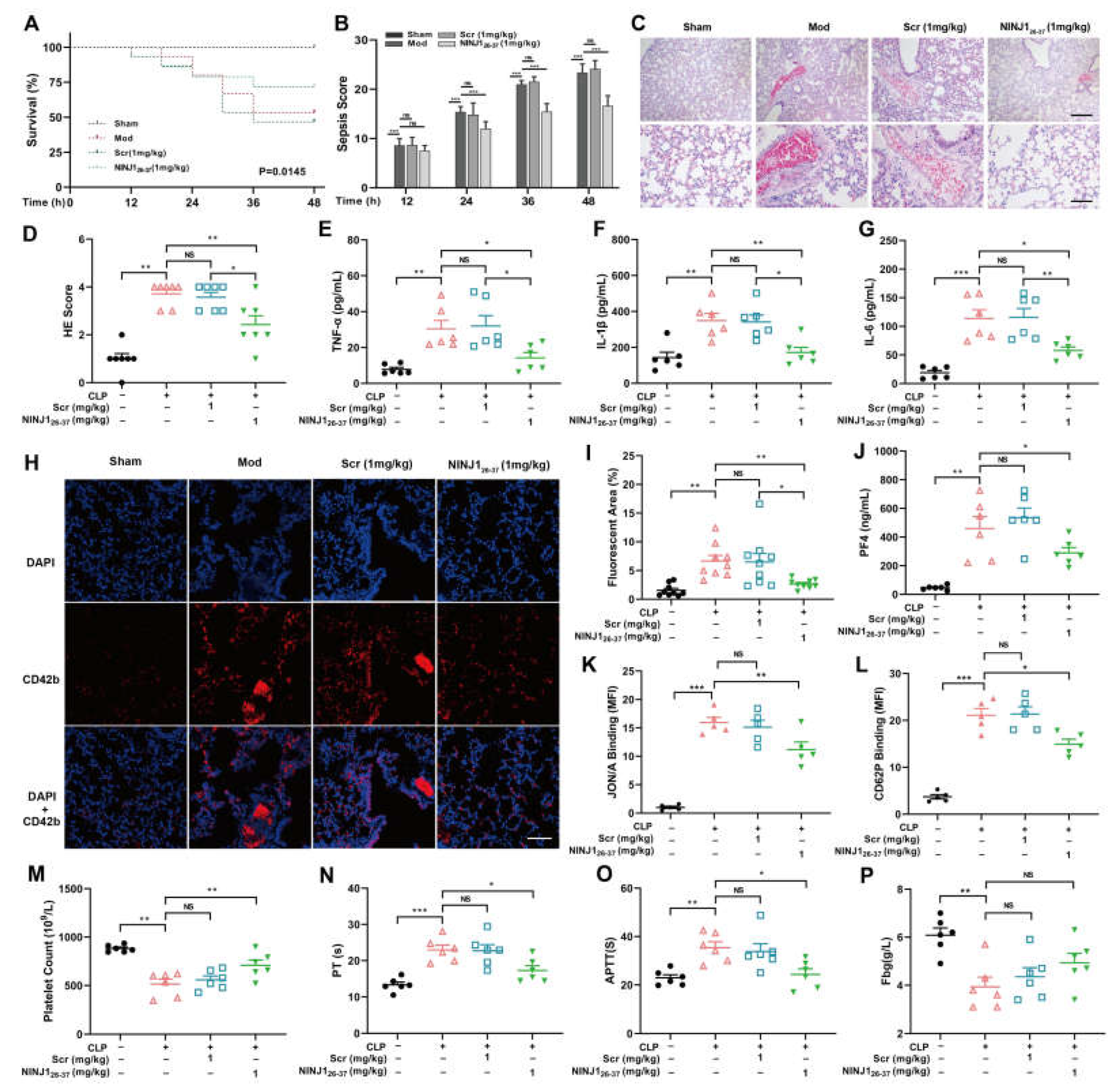
2.5. Inhibition of NINJ1 Attenuates Platelet Activation and Inflammatory Cytokine Release in Sepsis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Platelet Preparation and Count
4.4. Establishment of Experimental Septic Mouse Model
4.5. CCK8
4.6. Flow Cytometry
4.7. Clot Retraction Assay
4.8. Platelet Aggregation
4.9. Platelet Adhesion and Spreading Assays
4.10. Scanning Electron Microscope
4.11. Tail Bleeding Assay
4.12. Vessel Occlusion after FeCl3-Injury of the Carotid Artery
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990–2020: Analysis for the global burden of disease study. Lancet 2021, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef] [PubMed]
- Vardon-Bounes, F.; Ruiz, S.; Gratacap, M.P.; Garcia, C.; Payrastre, B.; Minville, V. Platelets are critical key players in sepsis. Int. J. Mol. Sci. 2019, 20, 3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremmel, T.; Frelinger, A.L.; Michelson, A.D. Platelet physiology. Semin. Thromb. Hemost. 2016, 42, 191–204. [Google Scholar] [PubMed] [Green Version]
- Kim, O.V.; Nevzorova, T.A.; Mordakhanova, E.R.; Ponomareva, A.A.; Andrianova, I.A.; Le Minh, G.; Daminova, A.G.; Peshkova, A.D.; Alber, M.S.; Vagin, O.; et al. Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica 2019, 104, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Liaw, P.C.; Ito, T.; Iba, T.; Thachil, J.; Zeerleder, S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016, 30, 257–261. [Google Scholar] [CrossRef]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef]
- Giustozzi, M.; Ehrlinder, H.; Bongiovanni, D.; Borovac, J.A.; Guerreiro, R.A.; Gąsecka, A.; Papakonstantinou, P.E.; Parker, W.A. Coagulopathy and sepsis: Pathophysiology, clinical manifestations and treatment. Blood Rev. 2021, 50, 100864. [Google Scholar] [CrossRef]
- Araki, T.; Zimonjic, D.B.; Popescu, N.C.; Milbrandt, J. Mechanism of homophilic binding mediated by ninjurin, a novel widely expressed adhesion molecule. J. Biol. Chem. 1997, 272, 21373–21380. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Wang, Y.; Li, H. The role of ninjurin1 and its impact beyond the nervous system. Dev. Neurosci. 2020, 42, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shao, F. NINJ1, rupturing swollen membranes for cataclysmic cell lysis. Mol. Cell 2021, 81, 1370–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Harper, M.T. Auranofin, a thioredoxin reductase inhibitor, causes platelet death through calcium overload. Platelets 2019, 30, 98–104. [Google Scholar] [CrossRef]
- Hu, S.; Wang, Y.; Li, H. The regulation effect of α7nAChRs and M1AChRs on inflammation and immunity in sepsis. Mediat. Inflamm. 2021, 2021, 9059601. [Google Scholar] [CrossRef]
- Wang, Y.; Ouyang, Y.; Liu, B.; Ma, X.; Ding, R. Platelet activation and antiplatelet therapy in sepsis: A narrative review. Thromb Res. 2018, 166, 28–36. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, A.L.; Boyle, J.A.; Di Pietro, S.M. Mechanism of platelet dense granule biogenesis: Study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 2012, 120, 4072–4081. [Google Scholar] [CrossRef] [Green Version]
- Bledzka, K.; Smyth, S.S.; Plow, E.F. Integrin αIIbβ3: From discovery to efficacious therapeutic target. Circ. Res. 2013, 112, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Shattil, S.J.; Newman, P.J. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood 2004, 104, 1606–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin αIIbβ3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Prokopec, J.S.; Zhang, Y.; Sukhoplyasova, M.; Shinglot, H.; Wang, M.-T.; Linkermann, A.; Stewart-Ornstein, J.; Gong, Y.-N. Sensing plasma membrane pore formation induces chemokine production in survivors of regulated necrosis. Dev. Cell 2022, 57, 228–245. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef]
- Hardaway, R.M.; Williams, C.H.; Vasquez, Y. Disseminated intravascular coagulation in sepsis. Semin. Thromb. Hemost. 2001, 27, 577–583. [Google Scholar] [CrossRef]
- Bai, S.; Lan, Y.; Fu, S.; Cheng, H.; Lu, Z.; Liu, G. Connecting Calcium-based nanomaterials and cancer: From diagnosis to therapy. Nano-Micro Lett. 2022, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.-D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Jiang, M.; Qi, L.; Li, L.; Wu, Y.; Song, D.; Li, Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int. J. Cancer 2021, 149, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Kanneganti, T.D. PANoptosis in viral infection: The missing puzzle piece in the cell death field. J. Mol. Biol. 2022, 434, 167249. [Google Scholar] [CrossRef]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, S.R.K.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef]
- Jennewein, C.; Sowa, R.; Faber, A.C.; Dildey, M.; von Knethen, A.; Meybohm, P.; Scheller, B.; Dröse, S.; Zacharowski, K. Contribution of Ninjurin1 to Toll-like receptor 4 signaling and systemic inflammation. Am. J. Respir. Cell Mol. Biol. 2015, 53, 656–663. [Google Scholar] [CrossRef]
- Wang, X.; Qin, J.; Zhang, X.; Peng, Z.; Ye, K.; Wu, X.; Yang, X.; Shi, H.; Zhao, Z.; Guo, X.; et al. Functional blocking of Ninjurin1 as a strategy for protecting endothelial cells in diabetes mellitus. Clin. Sci. 2018, 132, 213–229. [Google Scholar] [CrossRef]
- Kim, M.W.; Kang, J.H.; Jung, H.J.; Park, S.Y.; Hwang, J.-I.; Seong, J.K.; Yoon, Y.S.; Oh, S.H. Deficiency of Ninjurin1 attenuates LPS/D-galactosamine-induced acute liver failure by reducing TNF-α-induced apoptosis in hepatocytes. J. Cell. Mol. Med. 2022, 26, 5122–5134. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, S.; Osuchowski, M. Cecal ligation and puncture. Methods Mol. Biol. 2021, 2321, 1–8. [Google Scholar] [PubMed]
- Shrum, B.; Anantha, R.V.; Xu, S.X.; Donnelly, M.; Haeryfar, S.M.; McCormick, J.K.; Mele, T. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 2014, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Score and description |
|---|---|
| Appearance | 0—Coat is smooth |
| 1—Patches of hair piloerected | |
| 2—Majority of back is piloerected | |
| 3—Piloerection may or may not be present, mouse appears “puffy” | |
| 4—Piloerection may or may not be present, mouse appears emaciated | |
| Level of consciousness | 0—Mouse is active |
| 1—Mouse is active but avoids standing upright | |
| 2—Mouse activity is noticeably slowed. The mouse is still ambulant. | |
| 3—Activity is impaired. Mouse only moves when provoked, movements have a tremor | |
| 4—Activity severely impaired. Mouse remains stationary when provoked, with possible tremor | |
| Activity | 0—Normal amount of activity. Mouse is any of: eating, drinking, climbing, running, fighting |
| 1—Slightly suppressed activity. Mouse is moving around bottom of cage | |
| 2—Suppressed activity. Mouse is stationary with occasional investigative movements | |
| 3—No activity. Mouse is stationary | |
| 4—No activity. Mouse experiencing tremors, particularly in the hind legs | |
| Response to stimulus | 0—Mouse responds immediately to auditory stimulus or touch |
| 1—Slow or no response to auditory stimulus; strong response to touch (moves to escape) | |
| 2—No response to auditory stimulus; moderate response to touch (moves a few steps) | |
| 3—No response to auditory stimulus; mild response to touch (no locomotion) | |
| 4—No response to auditory stimulus. Little or no response to touch. Cannot right itself if pushed over | |
| Eyes | 0—Open |
| 1—Eyes not fully open, possibly with secretions | |
| 2—Eyes at least half closed, possibly with secretions | |
| 3—Eyes half closed or more, possibly with secretions | |
| 4—Eyes closed or milky | |
| Respiration rate | 0—Normal, rapid mouse respiration |
| 1—Slightly decreased respiration (rate not quantifiable by eye) | |
| 2—Moderately reduced respiration (rate at the upper range of quantifying by eye) | |
| 3—Severely reduced respiration (rate easily countable by eye, 0.5 s between breaths) | |
| 4—Extremely reduced respiration (>1 s between breaths) | |
| Respiration quality | 0—Normal |
| 1—Brief periods of labored breathing | |
| 2—Labored, no gasping | |
| 3—Labored with intermittent gasps | |
| 4—Gasping |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Yu, X.; Wan, C.; Li, F.; Wang, Y.; Zhang, K.; Feng, L.; Wen, A.; Deng, J.; Li, S.; et al. NINJ1 Regulates Platelet Activation and PANoptosis in Septic Disseminated Intravascular Coagulation. Int. J. Mol. Sci. 2023, 24, 4168. https://doi.org/10.3390/ijms24044168
Zhou X, Yu X, Wan C, Li F, Wang Y, Zhang K, Feng L, Wen A, Deng J, Li S, et al. NINJ1 Regulates Platelet Activation and PANoptosis in Septic Disseminated Intravascular Coagulation. International Journal of Molecular Sciences. 2023; 24(4):4168. https://doi.org/10.3390/ijms24044168
Chicago/Turabian StyleZhou, Xiaoli, Xiuxian Yu, Chengyu Wan, Fan Li, Yilan Wang, Kun Zhang, Lijuan Feng, Ao Wen, Jiangrong Deng, Shiyi Li, and et al. 2023. "NINJ1 Regulates Platelet Activation and PANoptosis in Septic Disseminated Intravascular Coagulation" International Journal of Molecular Sciences 24, no. 4: 4168. https://doi.org/10.3390/ijms24044168
APA StyleZhou, X., Yu, X., Wan, C., Li, F., Wang, Y., Zhang, K., Feng, L., Wen, A., Deng, J., Li, S., Xin, G., & Huang, W. (2023). NINJ1 Regulates Platelet Activation and PANoptosis in Septic Disseminated Intravascular Coagulation. International Journal of Molecular Sciences, 24(4), 4168. https://doi.org/10.3390/ijms24044168