
Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model
 , ,
, ,
Abstract
:1. Significance Statement
2. Introduction
3. Results
3.1. Chronic Nicotine Exposure Attenuates PD-Associated Locomotor Deficits and Increases Nurr1 Expression in the SNr
3.2. Chronic Nicotine Exposure Induces De Novo TH Expression in Non-DAergic Cells
3.3. Nicotine-Induced Neurotransmitter Plasticity Occurs via Translational Induction of Nurr1 and Transcriptional TH Regulation in Non-DAergic Neurons
3.4. The Pool of Neurons Recruitable for Nicotine-Induced TH Plasticity Is GABAergic
3.5. A Fraction of SNr GABAergic Nurr1+ Neurons Project to the Striatum
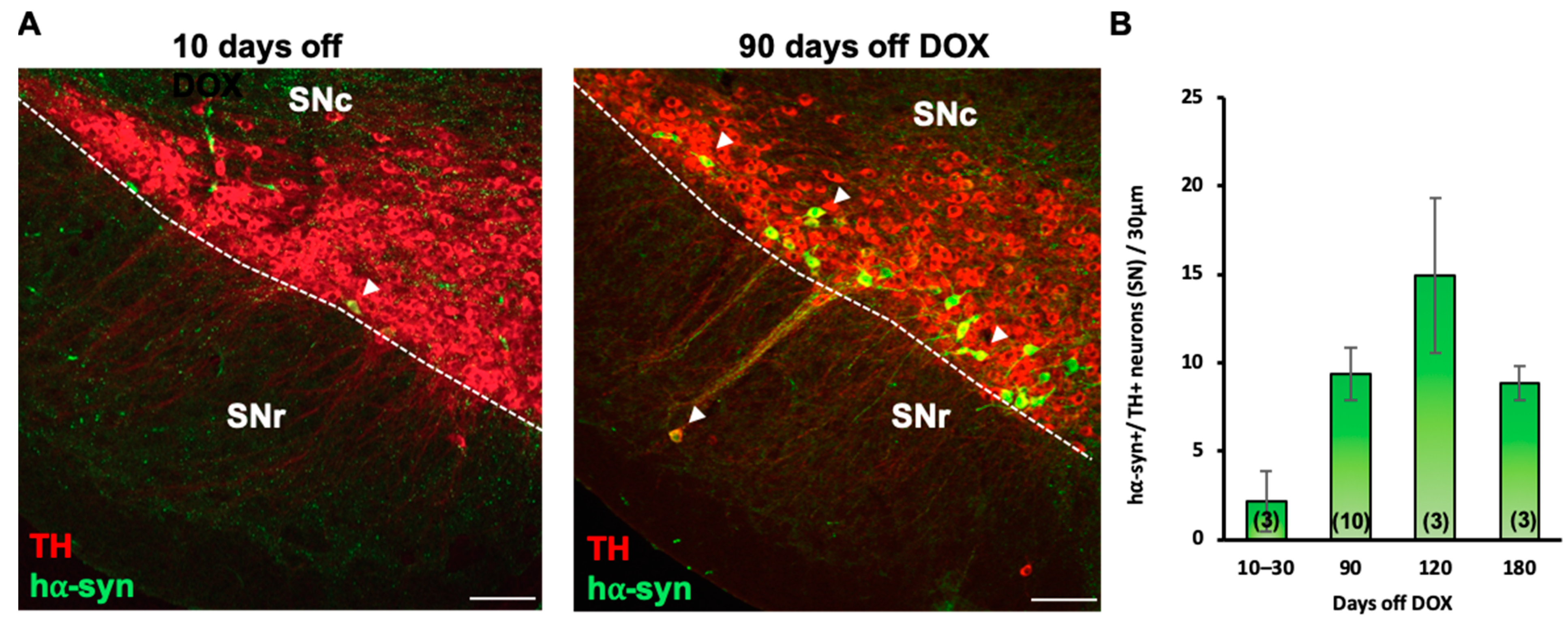
3.6. Nurr1 Upregulation Is Sufficient to Ameliorate PD-Related Locomotor Deficits and Decrease the Number of hα-syn+ Neurons in the SN
3.7. Selective Nurr1 Upregulation in GABAergic Cells Is Not Sufficient to Induce a TH Phenotype
3.8. Chemogenetic Activation of SN GABAergic Neurons Is Sufficient to Induce the Acquisition of Nurr1 but Not TH Phenotype
3.9. Concomitant Chemogenetic Activation of SN GABAergic Neurons and Nurr1 Upregulation Recapitulate Nicotine-Mediated Acquisition of the TH Phenotype
4. Discussion
5. Methods and Materials
5.1. Mice
5.2. Stereotactic Injections
5.3. Drug Treatment and Blood Collection
5.4. Immunohistochemistry and In Situ Hybridization
5.5. Behavioral Testing
5.6. Experimental Design and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, S.; Jahangeer, M.; Maknoon Razia, D.; Ashiq, M.; Ghaffar, A.; Akram, M.; El Allam, A.; Bouyahya, A.; Garipova, L.; Ali Shariati, M.; et al. Dopamine in Parkinson’s disease. Clin. Chim. Acta 2021, 522, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729. [Google Scholar] [CrossRef]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.; Sato, F.; Wu, Y.; Gauthier, J.; Lévesque, M.; Parent, M. Organization of the basal ganglia: The importance of axonal collateralization. Trends Neurosci. 2000, 23, S20–S27. [Google Scholar] [CrossRef]
- Benabid, A.L.; Krack, P.P.; Benazzouz, A.; Limousin, P.; Koudsie, A.; Pollak, P. Deep brain stimulation of the subthalamic nucleus for Parkinson’s disease: Methodologic aspects and clinical criteria. Neurology 2000, 55, S40–S44. [Google Scholar]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Perez de la Mora, M.; Hernandez-Mondragon, C.; Crespo-Ramirez, M.; Rejon-Orantes, J.; Borroto-Escuela, D.O.; Fuxe, K. Conventional and Novel Pharmacological Approaches to Treat Dopamine-Related Disorders: Focus on Parkinson’s Disease and Schizophrenia. Neuroscience 2020, 439, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Neuroprotection in Parkinson’s disease. Parkinsonism Relat Disord 2009, 15 (Suppl. S4), S41–S43. [Google Scholar] [CrossRef] [PubMed]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s disease therapeutics: New developments and challenges since the introduction of levodopa. Neuropsychopharmacology 2012, 37, 213–246. [Google Scholar] [CrossRef]
- Fratiglioni, L.; Wang, H.X. Smoking and Parkinson’s and Alzheimer’s disease: Review of the epidemiological studies. Behav. Brain Res. 2000, 113, 117–120. [Google Scholar] [CrossRef]
- Ishikawa, A.; Miyatake, T. Effects of smoking in patients with early-onset Parkinson’s disease. J. Neurol. Sci. 1993, 117, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Bordia, T.; Zhang, D.; Perez, X.A. Nicotine and Nicotinic Receptor Drugs: Potential for Parkinson’s Disease and Drug-Induced Movement Disorders. Int. Rev. Neurobiol. 2015, 124, 247–271. [Google Scholar] [CrossRef]
- Ritz, B.; Ascherio, A.; Checkoway, H.; Marder, K.S.; Nelson, L.M.; Rocca, W.A.; Ross, G.W.; Strickland, D.; Van Den Eeden, S.K.; Gorell, J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch. Neurol. 2007, 64, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Z.; Parameswaran, N.; Bordia, T.; Michael McIntosh, J.; Quik, M. Nicotine is neuroprotective when administered before but not after nigrostriatal damage in rats and monkeys. J. Neurochem. 2009, 109, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Quik, M.; Chen, L.; Parameswaran, N.; Xie, X.; Langston, J.W.; McCallum, S.E. Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J. Neurosci. 2006, 26, 4681–4689. [Google Scholar] [CrossRef]
- Ji, D.; Lape, R.; Dani, J.A. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron 2001, 31, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Nashmi, R.; McKinney, S.; Cai, H.; McIntosh, J.M.; Lester, H.A. Chronic nicotine selectively enhances alpha4beta2* nicotinic acetylcholine receptors in the nigrostriatal dopamine pathway. J. Neurosci. 2009, 29, 12428–12439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajas-Bailador, F.A.; Mogg, A.J.; Wonnacott, S. Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: Contribution of voltage-operated Ca2+ channels and Ca2+ stores. J. Neurochem. 2002, 81, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Rathouz, M.M.; Berg, D.K. Synaptic-type acetylcholine receptors raise intracellular calcium levels in neurons by two mechanisms. J. Neurosci. 1994, 14, 6935–6945. [Google Scholar] [CrossRef]
- Shen, J.X.; Yakel, J.L. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Klink, R.; de Kerchove d’Exaerde, A.; Zoli, M.; Changeux, J.P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 2001, 21, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, P.D.; Wonnacott, S. Nicotinic acetylcholine receptors and the ascending dopamine pathways. Biochem. Pharmacol. 2009, 78, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Nashmi, R.; Xiao, C.; Deshpande, P.; McKinney, S.; Grady, S.R.; Whiteaker, P.; Huang, Q.; McClure-Begley, T.; Lindstrom, J.M.; Labarca, C.; et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: Basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J. Neurosci. 2007, 27, 8202–8218. [Google Scholar] [CrossRef] [Green Version]
- Bordia, T.; McGregor, M.; Papke, R.L.; Decker, M.W.; McIntosh, J.M.; Quik, M. The α7 nicotinic receptor agonist ABT-107 protects against nigrostriatal damage in rats with unilateral 6-hydroxydopamine lesions. Exp. Neurol. 2015, 263, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.E.; Ross, S.A.; Drago, J.; Loiacono, R.E. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br. J. Pharmacol. 2001, 132, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Kadkhodaei, B.; Ito, T.; Joodmardi, E.; Mattsson, B.; Rouillard, C.; Carta, M.; Muramatsu, S.; Sumi-Ichinose, C.; Nomura, T.; Metzger, D.; et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 2009, 29, 15923–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuoka, H.; Hatanaka, T.; Metzger, D.; Ichinose, H. Nurr1 expression is regulated by voltage-dependent calcium channels and calcineurin in cultured hippocampal neurons. Neurosci. Lett. 2014, 559, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Preux, F.; Bores, L.R.; Tulloch, I.; Ladenheim, B.; Kim, R.; Thanos, P.K.; Volkow, N.D.; Cadet, J.L. Chronic co-administration of nicotine and methamphetamine causes differential expression of immediate early genes in the dorsal striatum and nucleus accumbens of rats. Neuroscience 2013, 243, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Barneda-Zahonero, B.; Servitja, J.M.; Badiola, N.; Miñano-Molina, A.J.; Fadó, R.; Saura, C.A.; Rodríguez-Alvarez, J. Nurr1 protein is required for N-methyl-D-aspartic acid (NMDA) receptor-mediated neuronal survival. J. Biol. Chem. 2012, 287, 11351–11362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volakakis, N.; Kadkhodaei, B.; Joodmardi, E.; Wallis, K.; Panman, L.; Silvaggi, J.; Spiegelman, B.M.; Perlmann, T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. USA 2010, 107, 12317–12322. [Google Scholar] [CrossRef] [Green Version]
- Decressac, M.; Volakakis, N.; Björklund, A.; Perlmann, T. NURR1 in Parkinson disease--from pathogenesis to therapeutic potential. Nat. Rev. Neurol. 2013, 9, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Chen, S.; Le, W.D. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog. Neurobiol. 2005, 77, 128–138. [Google Scholar] [CrossRef]
- Kim, C.-H.; Han, B.-S.; Moon, J.; Kim, D.-J.; Shin, J.; Rajan, S.; Quoc Toan, N.; Sohn, M.; Kim, W.-G.; Han, M.; et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, 8756–8761. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Li, S.; Mo, J.L.; Cai, H.B.; Le, W.D. Nurr1-Based Therapies for Parkinson’s Disease. CNS Neurosci. Ther. 2016, 22, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.M.; Chang, M.Y.; Song, J.J.; Rhee, Y.H.; Joe, E.H.; Lee, H.S.; Yi, S.H.; Lee, S.H. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson’s disease. EMBO Mol. Med. 2015, 7, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Parisiadou, L.; Sgobio, C.; Liu, G.; Yu, J.; Sun, L.; Shim, H.; Gu, X.L.; Luo, J.; Long, C.X.; et al. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 2012, 32, 9248–9264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooltorton, J.R.; Pidoplichko, V.I.; Broide, R.S.; Dani, J.A. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 2003, 23, 3176–3185. [Google Scholar] [CrossRef]
- Lai, I.C.; Dulcis, D. Nicotine-induced dopamine plasticity: A gateway to neurotransmitter replacement? Neural. Regen. Res. 2020, 15, 73–74. [Google Scholar] [CrossRef]
- Dulcis, D.; Spitzer, N.C. Reserve pool neuron transmitter respecification: Novel neuroplasticity. Dev. Neurobiol. 2012, 72, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, T.; Rodriguez, M.; Patt, S.; Gertz, H.J.; Gerhard, L.; Cervos-Navarro, J.; Grofova, I.; Deniau, J.M.; Kitai, S.T. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J. Comp. Neurol. 2000, 421, 107–135. [Google Scholar] [CrossRef]
- Pan, W.X.; Mao, T.; Dudman, J.T. Inputs to the dorsal striatum of the mouse reflect the parallel circuit architecture of the forebrain. Front. Neuroanat. 2010, 4, 147. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, M.; González-Hernández, T. Electrophysiological and morphological evidence for a GABAergic nigrostriatal pathway. J. Neurosci. 1999, 19, 4682–4694. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.W.; Jin, Y.; Matta, S.G.; Xu, M.; Zhou, F.M. An ultra-short dopamine pathway regulates basal ganglia output. J. Neurosci. 2009, 29, 10424–10435. [Google Scholar] [CrossRef] [Green Version]
- Borodinsky, L.N.; Root, C.M.; Cronin, J.A.; Sann, S.B.; Gu, X.N.; Spitzer, N.C. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 2004, 429, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Dulcis, D.; Spitzer, N.C. Illumination controls differentiation of dopamine neurons regulating behaviour. Nature 2008, 456, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Dulcis, D.; Jamshidi, P.; Leutgeb, S.; Spitzer, N.C. Neurotransmitter Switching in the Adult Brain Regulates Behavior. Science 2013, 340, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Romoli, B.; Lozada, A.F.; Sandoval, I.M.; Manfredsson, F.P.; Hnasko, T.S.; Berg, D.K.; Dulcis, D. Neonatal Nicotine Exposure Primes Midbrain Neurons to a Dopaminergic Phenotype and Increases Adult Drug Consumption. Biological. Psychiatry 2019, 86, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, J.A. Cigarette smoking and Parkinson’s disease. Neurology 1986, 36, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Janson, A.M.; Jansson, A.; Andersson, K.; Eneroth, P.; Agnati, L.F. Chronic nicotine treatment increases dopamine levels and reduces dopamine utilization in substantia nigra and in surviving forebrain dopamine nerve terminal systems after a partial di-mesencephalic hemitransection. Naunyn. Schmiedebergs Arch. Pharm. 1990, 341, 171–181. [Google Scholar] [CrossRef]
- Quik, M.; Huang, L.Z.; Parameswaran, N.; Bordia, T.; Campos, C.; Perez, X.A. Multiple roles for nicotine in Parkinson’s disease. Biochem. Pharmacol. 2009, 78, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Aumann, T.D.; Egan, K.; Lim, J.; Boon, W.C.; Bye, C.R.; Chua, H.K.; Baban, N.; Parish, C.L.; Bobrovskaya, L.; Dickson, P.; et al. Neuronal activity regulates expression of tyrosine hydroxylase in adult mouse substantia nigra pars compacta neurons. J. Neurochem. 2011, 116, 646–658. [Google Scholar] [CrossRef]
- Gu, X.; Spitzer, N.C. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+ transients. Nature 1995, 375, 784–787. [Google Scholar] [CrossRef]
- Gutierrez, R.; Romo-Parra, H.; Maqueda, J.; Vivar, C.; Ramirez, M.; Morales, M.A.; Lamas, M. Plasticity of the GABAergic phenotype of the “glutamatergic” granule cells of the rat dentate gyrus. J. Neurosci. 2003, 23, 5594–5598. [Google Scholar] [CrossRef] [Green Version]
- Dulcis, D.; Lippi, G.; Stark, C.J.; Do, L.H.; Berg, D.K.; Spitzer, N.C. Neurotransmitter Switching Regulated by miRNAs Controls Changes in Social Preference. Neuron 2017, 95, 1319–1333.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Ulloa, N.; Spitzer, N.; Dulcis, D. Activity-dependent specification of dopaminergic neurons in the embryonic nervous system. Int. J. Dev. Neurosci. 2008, 26, 887. [Google Scholar] [CrossRef]
- Prakash, N.; Stark, C.J.; Keisler, M.N.; Luo, L.; Der-Avakian, A.; Dulcis, D. Serotonergic Plasticity in the Dorsal Raphe Nucleus Characterizes Susceptibility and Resilience to Anhedonia. J. Neurosci. 2020, 40, 569–584. [Google Scholar] [CrossRef]
- Tandé, D.; Höglinger, G.; Debeir, T.; Freundlieb, N.; Hirsch, E.C.; François, C. New striatal dopamine neurons in MPTP-treated macaques result from a phenotypic shift and not neurogenesis. Brain 2006, 129, 1194–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quik, M.; Parameswaran, N.; McCallum, S.E.; Bordia, T.; Bao, S.; McCormack, A.; Kim, A.; Tyndale, R.F.; Langston, J.W.; Di Monte, D.A. Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J. Neurochem. 2006, 98, 1866–1875. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellnow, R.C.; Steece-Collier, K.; Kanaan, N.M.; Sortwell, C.E.; Collier, T.J.; Cole-Strauss, A.; Lipton, J.W.; Manfredsson, F.P. rAAV-Mediated Regulation of Striatal Nurr1 Expression Alters Development and Severity of Levodopa-Induced Dyskinesias in the 6-OHDA Rat Model of Parkinson’s Disease. Mol. Ther. 2015, 23, S282–S283. [Google Scholar] [CrossRef] [Green Version]
- Willmore, L.; Cameron, C.; Yang, J.; Witten, I.B.; Falkner, A.L. Behavioural and dopaminergic signatures of resilience. Nature 2022, 611, 124–132. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer Lot # | Dilution Ratio |
|---|---|---|
| Chicken anti GFP | Invitrogen, A10262 | 1:500 |
| Chicken anti GFAP | Invitrogen, AB5541 | 1:1000 |
| Goat anti Foxa2 | Boster, A01032 | 1:250 |
| Guinea Pig anti NeuN | Millipore, AB2251 | 1:1500 |
| Mouse anti TH | Millipore, MAB318 | 1:1000 |
| Mouse anti α-syn | Santa Cruz, sc-12767 | 1:500 |
| Rabbit anti Nurr1 | Santa Cruz, sc-990 | 1:300 |
| Sheep anti TH | Novus, NB300-110 | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, J.I.; Porcu, A.; Romoli, B.; Keisler, M.; Manfredsson, F.P.; Powell, S.B.; Dulcis, D. Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model. Int. J. Mol. Sci. 2023, 24, 4204. https://doi.org/10.3390/ijms24044204
Lai JI, Porcu A, Romoli B, Keisler M, Manfredsson FP, Powell SB, Dulcis D. Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model. International Journal of Molecular Sciences. 2023; 24(4):4204. https://doi.org/10.3390/ijms24044204
Chicago/Turabian StyleLai, Jessica IChi, Alessandra Porcu, Benedetto Romoli, Maria Keisler, Fredric P. Manfredsson, Susan B. Powell, and Davide Dulcis. 2023. "Nicotine-Mediated Recruitment of GABAergic Neurons to a Dopaminergic Phenotype Attenuates Motor Deficits in an Alpha-Synuclein Parkinson’s Model" International Journal of Molecular Sciences 24, no. 4: 4204. https://doi.org/10.3390/ijms24044204