

MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism

Abstract
:
1. Introduction
2. An Overview of DNA Methylation
2.1. History
2.2. DNA Methylation in the Central Nervous System (CNS)
2.3. Different Factors Involved in DNA Methylation as an Epigenetic Modification
2.3.1. Writers of DNA Methylation
2.3.2. Erasers/Modifiers of DNA Methylation
2.3.3. Readers of DNA Methylation
2.4. MeCP2, a Well-Studied Epigenetic Reader of DNA Methylation
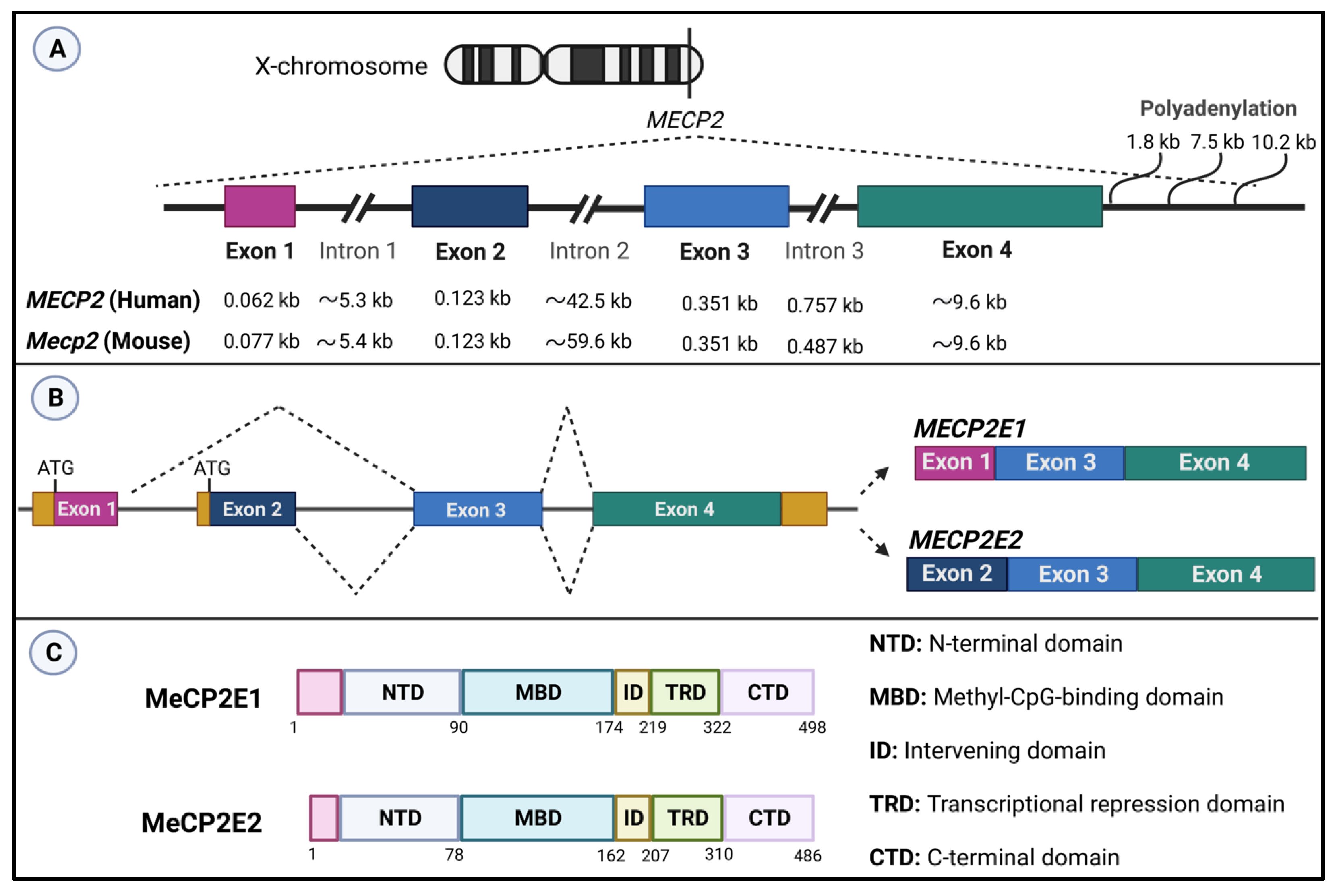
2.4.1. MeCP2—Expression, Key Functional Domains, and DNA Binding Activities
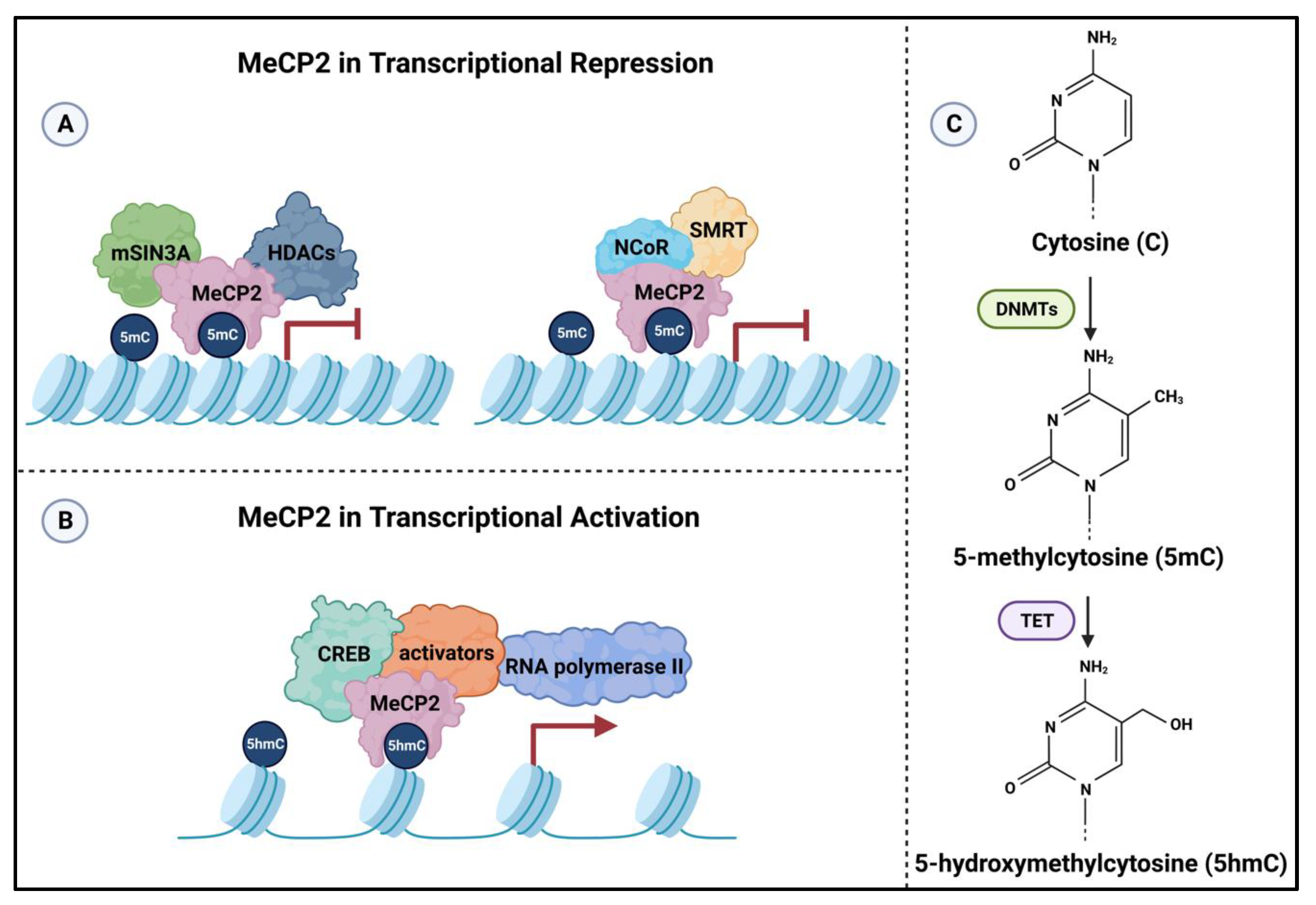
2.4.2. Mechanism of Action of MeCP2 in Transcriptional Regulation
2.4.3. MeCP2 and Regulation of Gene Expression in the Central Nervous System
2.4.4. Impaired Metabolism in Mutated MeCP2-Linked Neurodevelopmental Disorders
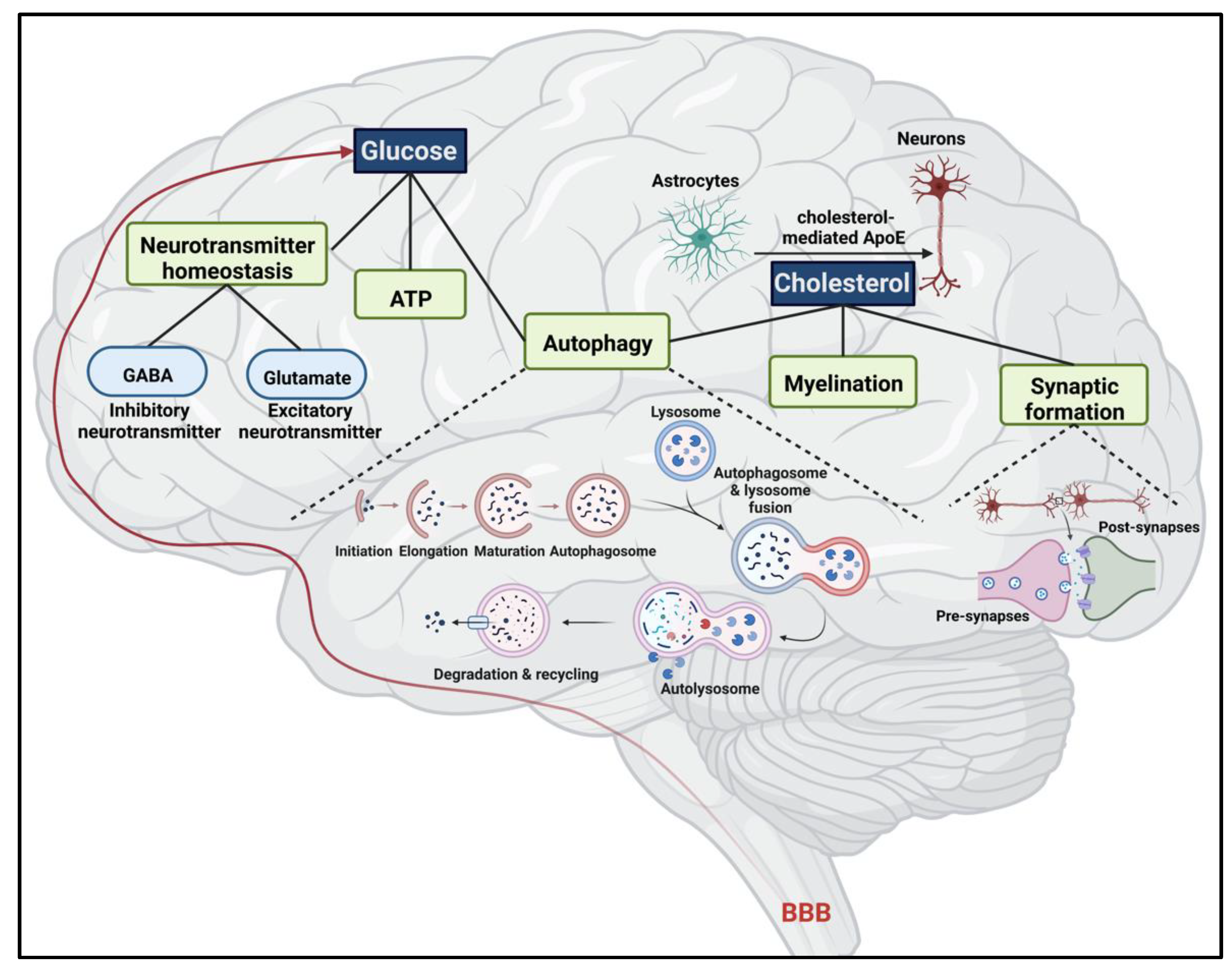
3. Brain Metabolism
3.1. Glucose
3.1.1. ATP Production
3.1.2. Neurotransmitter Homeostasis
3.1.3. Glucose Metabolism in the Brain and the Link with Autophagy
3.2. Cholesterol
3.2.1. Cholesterol Homeostatic Mechanism in the Brain
3.2.2. Major Role of Cholesterol in Myelin and Synaptic Formation
3.2.3. Cholesterol and Autophagy
4. The Interplay between MeCP2 and Brain Metabolism
4.1. MeCP2 and Glucose Metabolism
4.1.1. Glucose Transporters
4.1.2. Brain-Derived Neurotrophic Factor (BDNF) Involvement in Glucose Homeostasis
4.1.3. mTOR, a Principal Regulator of Glucose Metabolism
4.1.4. AMPK: A Key Regulator in Energy Homeostasis
4.2. MeCP2 and Cholesterol Metabolism
4.2.1. Involvement of BDNF in Cholesterol Homeostasis in the Brain
4.2.2. mTOR: A Principal Regulator of Cholesterol-Related Cellular Pathways
4.2.3. PI3K-PKB/Akt Signaling Pathway: A Companion to Cholesterol Metabolism
4.2.4. Cholesterol-24 Hydroxylase: A Critical Enzyme in Brain Cholesterol Homeostasis
4.2.5. AMPK: A Critical Regulator in Cholesterol Metabolism
4.3. Advancements in Research Targeting MeCP2 in Neuronal Pathologies Involving the Link between Brain Metabolism and DNA Methylation
5. Conclusions and Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Liyanage, V.R.B.; Zachariah, R.M.; Delcuve, G.P.; Davie, J.R.; Rastegar, M. Chromatin Structure and Epigenetics. In Advances in Genetics Research; Urbano, K.V., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2015; Volume 13, pp. 57–88. [Google Scholar]
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell. Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Spadafora, C. The epigenetic basis of evolution. Prog. Biophys. Mol. Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Cheema, M.S.; Ausio, J. The Structural Determinants behind the Epigenetic Role of Histone Variants. Genes 2015, 6, 685–713. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, V.R.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA modifications: Function and applications in normal and disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef]
- Rastegar, M. Epigenetics and Cerebellar Neurodevelopmental Disorders. In Development of the Cerebellum from Molecular Aspects to Diseases; Marzban, H., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 197–218. [Google Scholar]
- Rastegar, M. Editorial (Thematic Issue: NeuroEpigenetics and Neurodevelopmental Disorders: From Molecular Mechanisms to Cell Fate Commitments of the Brain Cells and Human Disease). Curr. Top. Med. Chem. 2017, 17, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M.; Yasui, D.H. Editorial: Epigenetic Mechanisms and Their Involvement in Rare Diseases. Front. Genet. 2021, 12, 755076. [Google Scholar] [CrossRef]
- Begum, N.; Mandhare, A.; Tryphena, K.P.; Srivastava, S.; Shaikh, M.F.; Singh, S.B.; Khatri, D.K. Epigenetics in depression and gut-brain axis: A molecular crosstalk. Front. Aging Neurosci. 2022, 14, 1048333. [Google Scholar] [CrossRef] [PubMed]
- Bin Akhtar, G.; Buist, M.; Rastegar, M. MeCP2 and transcriptional control of eukaryotic gene expression. Eur. J. Cell. Biol. 2022, 101, 151237. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Curtis, K.; Zachariah, R.M.; Chudley, A.E.; Rastegar, M. Overview of the Genetic Basis and Epigenetic Mechanisms that Contribute to FASD Pathobiology. Curr. Top. Med. Chem. 2017, 17, 808–828. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Rastegar, M. Rett syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef] [Green Version]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Bower, C.; English, D. The prevalence and incidence of Rett syndrome in Australia. Eur. Child Adolesc. Psychiatry 1997, 6 (Suppl. S1), 8–10. [Google Scholar] [PubMed]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural. Plast 2012, 2012, 415825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Buchovecky, C.M.; Turley, S.D.; Brown, H.M.; Kyle, S.M.; McDonald, J.G.; Liu, B.; Pieper, A.A.; Huang, W.; Katz, D.M.; Russell, D.W.; et al. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013, 45, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Cooke, D.W.; Naidu, S.; Plotnick, L.; Berkovitz, G.D. Abnormalities of Thyroid Function and Glucose Control in Subjects with Rett Syndrome. Horm. Res. 1995, 43, 273–278. [Google Scholar] [CrossRef]
- Justice, M.J.; Buchovecky, C.M.; Kyle, S.M.; Djukic, A. A role for metabolism in Rett syndrome pathogenesis: New clinical findings and potential treatment targets. Rare Dis. 2013, 1, e27265. [Google Scholar] [CrossRef] [Green Version]
- Matsuishi, T.; Urabe, F.; Percy, A.K.; Komori, H.; Yamashita, Y.; Schultz, R.S.; Ohtani, Y.; Kuriya, N.; Kato, H. Abnormal Carbohydrate Metabolism in Cerebrospinal Fluid in Rett Syndrome. J. Child Neurol. 1994, 9, 26–30. [Google Scholar] [CrossRef]
- Villemagne, P.M.; Naidu, S.; Villemagne, V.L.; Yaster, M.; Wagner, H.N.; Harris, J.C.; Moser, H.W.; Johnston, M.V.; Dannals, R.F.; Wong, D.F. Brain glucose metabolism in Rett Syndrome. Pediatr. Neurol. 2002, 27, 117–122. [Google Scholar] [CrossRef]
- Wakai, S.; Kameda, K.; Ishikawa, Y.; Miyamoto, S.; Nagaoka, M.; Okabe, M.; Minami, R.; Tachi, N. Rett syndrome: Findings suggesting axonopathy and mitochondrial abnormalities. Pediatr. Neurol. 1990, 6, 339–343. [Google Scholar] [CrossRef]
- Can, K.; Menzfeld, C.; Rinne, L.; Rehling, P.; Kügler, S.; Golubiani, G.; Dudek, J.; Müller, M. Neuronal Redox-Imbalance in Rett Syndrome Affects Mitochondria as Well as Cytosol, and Is Accompanied by Intensified Mitochondrial O2 Consumption and ROS Release. Front. Physiol. 2019, 10, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampson, K.; Woods, C.G.; Latif, F.; Webb, T. Mutations in the MECP2 gene in a cohort of girls with Rett syndrome. J. Med. Genet. 2000, 37, 610–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.N.A.; Malhotra, S.; Bhattacharya, A.; Grover, S.; Batra, Y.K. Regional Cerebral Glucose Metabolism and its Association with Phenotype and Cognitive Functioning in Patients with Autism. Indian J. Psychol. Med. 2017, 39, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manco, M.; Guerrera, S.; Ravà, L.; degli Atti, M.C.; Di Vara, S.; Valeri, G.; Vicari, S. Cross-sectional investigation of insulin resistance in youths with autism spectrum disorder. Any role for reduced brain glucose metabolism? Transl. Psychiatry 2021, 11, 229. [Google Scholar] [CrossRef]
- Usui, N.; Iwata, K.; Miyachi, T.; Takagai, S.; Wakusawa, K.; Nara, T.; Tsuchiya, K.J.; Matsumoto, K.; Kurita, D.; Kameno, Y.; et al. VLDL-specific increases of fatty acids in autism spectrum disorder correlate with social interaction. Ebiomedicine 2020, 58, 102917. [Google Scholar] [CrossRef]
- Weeks, O.; Bossé, G.D.; Oderberg, I.M.; Akle, S.; Houvras, Y.; Wrighton, P.J.; Labella, K.; Iversen, I.; Tavakoli, S.; Adatto, I.; et al. Fetal alcohol spectrum disorder predisposes to metabolic abnormalities in adulthood. J. Clin. Investig. 2020, 130, 2252–2269. [Google Scholar] [CrossRef] [Green Version]
- Geerdink, N.; Rotteveel, J.J.; Lammens, M.; Sistermans, E.A.; Heikens, G.T.; Gabreëls, F.J.M.; Mullaart, R.A.; Hamel, B.C.J. MECP2 Mutation in a Boy with Severe Neonatal Encephalopathy: Clinical, Neuropathological and Molecular Findings. Neuropediatrics 2002, 33, 33–36. [Google Scholar] [CrossRef]
- Kankirawatana, P.; Leonard, H.; Ellaway, C.; Scurlock, J.; Mansour, A.; Makris, C.M.; Dure, L.S.; Friez, M.; Lane, J.; Kiraly-Borri, C.; et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006, 67, 164–166. [Google Scholar] [CrossRef]
- Johnson, T.B.; Coghill, R.D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus1. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Wyatt, G.R. Occurrence of 5-Methyl-Cytosine in Nucleic Acids. Nature 1950, 166, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Shied, B.; Srinivasan, P.R.; Borek, E. Deoxyribonucleic acid methylase of mammalian tissues. Biochemistry 1968, 7, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The Epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Lederberg, S. Suppression of the multiplication of heterologous bacteriophages in lysogenic bacteria. Virology 1957, 3, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, G.R.; Cohen, S.S. A New Pyrimidine Base from Bacteriophage Nucleic Acids. Nature 1952, 170, 1072–1073. [Google Scholar] [CrossRef]
- Barber, B.A.; Rastegar, M. Epigenetic control of Hox genes during neurogenesis, development, and disease. Ann. Anat. Anat. Anz. 2010, 192, 261–274. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Pevsner, J.; Sabunciyan, S.; Yolken, R.H.; Webster, M.J.; Dinkins, T.; Callinan, P.A.; Fan, J.-B.; Potash, J.B.; Feinberg, A. DNA Methylation Signatures within the Human Brain. Am. J. Hum. Genet. 2007, 81, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Olynik, B.M.; Rastegar, M. The Genetic and Epigenetic Journey of Embryonic Stem Cells into Mature Neural Cells. Front. Genet. 2012, 3, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zocher, S.; Toda, T. Epigenetic aging in adult neurogenesis. Hippocampus 2023, 1–13. [Google Scholar] [CrossRef]
- Amiri, S.; Davie, J.R.; Rastegar, M. Chronic Ethanol Exposure Alters DNA Methylation in Neural Stem Cells: Role of Mouse Strain and Sex. Mol. Neurobiol. 2020, 57, 650–667. [Google Scholar] [CrossRef]
- Xu, W.; Liyanage, V.R.B.; MacAulay, A.; Levy, R.D.; Curtis, K.; Olson, C.O.; Zachariah, R.M.; Amiri, S.; Buist, M.; Hicks, G.G.; et al. Genome-Wide Transcriptome Landscape of Embryonic Brain-Derived Neural Stem Cells Exposed to Alcohol with Strain-Specific Cross-Examination in BL6 and CD1 Mice. Sci. Rep. 2019, 9, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.B.; Rastegar, M. Brain Region-Specific Expression of MeCP2 Isoforms Correlates with DNA Methylation within Mecp2 Regulatory Elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol. 2015, 265, 102–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, V.R.B.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism 2013, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, V.R.; Olson, C.O.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. DNA Methylation Contributes to the Differential Expression Levels of Mecp2 in Male Mice Neurons and Astrocytes. Int. J. Mol. Sci. 2019, 20, 1845. [Google Scholar] [CrossRef] [Green Version]
- Teter, B.; Rozovsky, I.; Krohn, K.; Anderson, C.; Osterburg, H.; Finch, C. Methylation of the glial fibrillary acidic protein gene shows novel biphasic changes during brain development. Glia 1996, 17, 195–205. [Google Scholar] [CrossRef]
- Muñoz, P.; Estay, C.; Díaz, P.; Elgueta, C.; Ardiles, O.; Lizana, P.A. Inhibition of DNA Methylation Impairs Synaptic Plasticity during an Early Time Window in Rats. Neural Plast. 2016, 2016, 4783836. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Sweatt, J.D. Covalent Modification of DNA Regulates Memory Formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhou, J.; Tian, W.; Luo, C.; Bartlett, A.; Aldridge, A.; Lucero, J.; Osteen, J.K.; Nery, J.R.; Chen, H.; et al. DNA methylation atlas of the mouse brain at single-cell resolution. Nature 2021, 598, 120–128. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Schermelleh, L.; Walter, J.; Cardoso, M.C.; Leonhardt, H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc. Natl. Acad. Sci. USA 2005, 102, 8905–8909. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.; Bacolla, A.; Wells, R.D.; Roberts, R.J. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999, 274, 33002–33010. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Suetake, I.; Tada, T.; Tajima, S. Stage- and cell-specific expression of Dnmt3a and Dnmt3b during embryogenesis. Mech. Dev. 2002, 118, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Brenner, C.; Burgers, W.A.; Putmans, P.; Kouzarides, T.; de Launoit, Y.; Fuks, F. Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res. 2002, 30, 3831–3838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Pollex, T.; Hanna, K.; Tuorto, F.; Meusburger, M.; Helm, M.; Lyko, F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010, 24, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Mayer, W.; Niveleau, A.; Walter, J.; Fundele, R.; Haaf, T. Demethylation of the zygotic paternal genome. Nature 2000, 403, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Zachariah, R.M.; Delcuve, G.P.; Davie, J.R.; Rastegar, M. New Developments in Chromatin Research: An Epigenetic Perspective. In New Developments in Chromatin Research; Simpson, N.M., Stewart, V.J., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 29–58. [Google Scholar]
- Arioka, Y.; Watanabe, A.; Saito, K.; Yamada, Y. Activation-Induced Cytidine Deaminase Alters the Subcellular Localization of Tet Family Proteins. PLoS ONE 2012, 7, e45031. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.-L.; Song, H. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-C.; Wang, K.-Y.; Shen, C.-K.J. The Mammalian de Novo DNA Methyltransferases DNMT3A and DNMT3B Are Also DNA 5-Hydroxymethylcytosine Dehydroxymethylases. J. Biol. Chem. 2012, 287, 33116–33121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusmintratip, V.; Sowers, L.C. An unexpectedly high excision capacity for mispaired 5-hydroxymethyluracil in human cell extracts. Proc. Natl. Acad. Sci. USA 2000, 97, 14183–14187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, D.; Estève, P.-O.; Pradhan, S. Targeting the SET and RING-associated (SRA) domain of ubiquitin-like, PHD and ring finger-containing 1 (UHRF1) for anti-cancer drug development. Oncotarget 2018, 9, 26243–26258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taka, N.; Asami, S.; Sakamoto, M.; Matsui, T.; Yoshida, W. Quantification of Global DNA Hydroxymethylation Level Using UHRF2 SRA-Luciferase Based on Bioluminescence Resonance Energy Transfer. Anal. Chem. 2022, 94, 8618–8624. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.P.; Zhenilo, S.; Salozhin, S.; Yamada, D.; Prokhortchouk, E.; Defossez, P.-A. A Family of Human Zinc Finger Proteins That Bind Methylated DNA and Repress Transcription. Mol. Cell. Biol. 2006, 26, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Coelho, F.S.; Sangi, S.; Moraes, J.L.; Santos, W.D.S.; Gamosa, E.A.; Fernandes, K.V.S.; Grativol, C. Methyl-CpG binding proteins (MBD) family evolution and conservation in plants. Gene 2022, 824, 146404. [Google Scholar] [CrossRef]
- Balmer, D.; Goldstine, J.; Rao, Y.M.; LaSalle, J.M. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J. Mol. Med. 2003, 81, 61–68. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MeCP2 Isoform-Specific Antibody Reveals the Endogenous MeCP2E1 Expression in Murine Brain, Primary Neurons and Astrocytes. PLoS ONE 2012, 7, e49763. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to Methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M.; Hotta, A.; Pasceri, P.; Makarem, M.; Cheung, A.Y.L.; Elliott, S.; Park, K.J.; Adachi, M.; Jones, F.S.; Clarke, I.D.; et al. MECP2 Isoform-Specific Vectors with Regulated Expression for Rett Syndrome Gene Therapy. PLoS ONE 2009, 4, e6810. [Google Scholar] [CrossRef] [PubMed]
- Shevkoplyas, D.; Vuu, Y.M.; Davie, J.R.; Rastegar, M. The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. Int. J. Mol. Sci. 2022, 23, 15643. [Google Scholar] [CrossRef]
- de Paz, A.M.; Khajavi, L.; Martin, H.; Claveria-Gimeno, R.; Dieck, S.T.; Cheema, M.S.; Sanchez-Mut, J.V.; Moksa, M.M.; Carles, A.; Brodie, N.I.; et al. MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claveria-Gimeno, R.; Lanuza, P.M.; Morales-Chueca, I.; Jorge-Torres, O.C.; Vega, S.; Abian, O.; Esteller, M.; Velazquez-Campoy, A. The intervening domain from MeCP2 enhances the DNA affinity of the methyl binding domain and provides an independent DNA interaction site. Sci. Rep. 2017, 7, srep41635. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.L.; McNae, I.W.; Schmiedeberg, L.; Klose, R.J.; Bird, A.P.; Walkinshaw, M.D. MeCP2 Binding to DNA Depends upon Hydration at Methyl-CpG. Mol. Cell 2008, 29, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, T.; Shi, X.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Woodcock, C.L. Multiple Modes of Interaction between the Methylated DNA Binding Protein MeCP2 and Chromatin. Mol. Cell. Biol. 2007, 27, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Klose, R.J.; Bird, A.P. MeCP2 Behaves as an Elongated Monomer That Does Not Stably Associate with the Sin3a Chromatin Remodeling Complex. J. Biol. Chem. 2004, 279, 46490–46496. [Google Scholar] [CrossRef] [Green Version]
- Li, C.H.; Coffey, E.L.; Dall’Agnese, A.; Hannett, N.M.; Tang, X.; Henninger, J.E.; Platt, J.M.; Oksuz, O.; Zamudio, A.V.; Afeyan, L.K.; et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 2020, 586, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Lagger, S.; Connelly, J.C.; Schweikert, G.; Webb, S.; Selfridge, J.; Ramsahoye, B.H.; Yu, M.; He, C.; Sanguinetti, G.; Sowers, L.C.; et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017, 13, e1006793. [Google Scholar] [CrossRef]
- Tillotson, R.; Cholewa-Waclaw, J.; Chhatbar, K.; Connelly, J.C.; Kirschner, S.A.; Webb, S.; Koerner, M.V.; Selfridge, J.; Kelly, D.A.; De Sousa, D.; et al. Neuronal non-CG methylation is an essential target for MeCP2 function. Mol. Cell 2021, 81, 1260–1275.e12. [Google Scholar] [CrossRef] [PubMed]
- Sperlazza, M.J.; Bilinovich, S.M.; Sinanan, L.M.; Javier, F.R.; Williams, D.C. Structural Basis of MeCP2 Distribution on Non-CpG Methylated and Hydroxymethylated DNA. J. Mol. Biol. 2017, 429, 1581–1594. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Ng, H.-H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Mahgoub, M.; Adachi, M.; Suzuki, K.; Liu, X.; Kavalali, E.T.; Chahrour, M.H.; Monteggia, L.M. MeCP2 and histone deacetylases 1 and 2 in dorsal striatum collectively suppress repetitive behaviors. Nat. Neurosci. 2016, 19, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- Ellis, J.; Hotta, A.; Rastegar, M. Retrovirus Silencing by an Epigenetic TRIM. Cell 2007, 131, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Kavalali, E.T.; Nelson, E.D.; Monteggia, L.M. Role of MeCP2, DNA methylation, and HDACs in regulating synapse function. J. Neurodev. Disord. 2011, 3, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, P.; Schultz, R.M. HDAC1 and HDAC2 in mouse oocytes and preimplantation embryos: Specificity versus compensation. Cell Death Differ. 2016, 23, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayne, S.; Zwartjes, C.G.M.; Van Schaik, F.M.A.; Timmers, H.T.M. Involvement of the SMRT/NCoR–HDAC3 complex in transcriptional repression by the CNOT2 subunit of the human Ccr4–Not complex. Biochem. J. 2006, 398, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.-C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, J.A.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 2011, 18, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, A.; Buchholz, D.R.; Shi, Y.-B. Recruitment of N-CoR/SMRT-TBLR1 Corepressor Complex by Unliganded Thyroid Hormone Receptor for Gene Repression during Frog Development. Mol. Cell. Biol. 2004, 24, 3337–3346. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 Nuclear Receptor Corepressor Complex Inhibits the JNK Pathway through the Integral Subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.; Renthal, W.; Greben, A.W.; Whitwam, T.; Silberfeld, A.; Stroud, H.; Li, E.; Yang, M.G.; Kinde, B.; Griffith, E.; et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol. Cell 2020, 77, 294–309.e299. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Siu, V.M.; Ang, L.C.; Del Bigio, M.R.; Rastegar, M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: A distinct grey-white matter variation. Neuropathol. Appl. Neurobiol. 2020, 46, 735–750. [Google Scholar] [CrossRef]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev. Biol. 2020, 8, 763. [Google Scholar] [CrossRef]
- Buist, M.; El Tobgy, N.; Shevkoplyas, D.; Genung, M.; Sher, A.A.; Pejhan, S.; Rastegar, M. Differential Sensitivity of the Protein Translation Initiation Machinery and mTOR Signaling to MECP2 Gain- and Loss-of-Function Involves MeCP2 Isoform-Specific Homeostasis in the Brain. Cells 2022, 11, 1442. [Google Scholar] [CrossRef]
- Buist, M.; Fuss, D.; Rastegar, M. Transcriptional Regulation of MECP2E1-E2 Isoforms and BDNF by Metformin and Simvastatin through Analyzing Nascent RNA Synthesis in a Human Brain Cell Line. Biomolecules 2021, 11, 1253. [Google Scholar] [CrossRef]
- Su, M.; Hong, J.; Zhao, Y.; Liu, S.; Xue, X. MeCP2 controls hippocampal brain-derived neurotrophic factor expression via homeostatic interactions with microRNA-132 in rats with depression. Mol. Med. Rep. 2015, 12, 5399–5406. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W.; Autry, A.E.; Na, E.S.; Adachi, M.; Björkholm, C.; Kavalali, E.T.; Monteggia, L.M. Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat. Neurosci. 2021, 24, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; He, L.; Ma, R.; Ding, W.; Zhou, C.; Lin, S.; Zhang, J.-C.; Chen, J.; Yao, W. The role of MeCP2 and the BDNF/TrkB signaling pathway in the stress resilience of mice subjected to CSDS. Psychopharmacology 2022, 239, 2921–2929. [Google Scholar] [CrossRef]
- Buchthal, B.; Lau, D.; Weiss, U.; Weislogel, J.-M.; Bading, H. Nuclear Calcium Signaling Controls Methyl-CpG-binding Protein 2 (MeCP2) Phosphorylation on Serine 421 following Synaptic Activity. J. Biol. Chem. 2012, 287, 30967–30974. [Google Scholar] [CrossRef] [Green Version]
- Ezeonwuka, C.D.; Rastegar, M. MeCP2-Related Diseases and Animal Models. Diseases 2014, 2, 45–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, E.; Monteggia, L.M. The role of MeCP2 in CNS development and function. Horm. Behav. 2011, 59, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef]
- Marzban, H.; Del Bigio, M.R.; Alizadeh, J.; Ghavami, S.; Zachariah, R.M.; Rastegar, M. Cellular commitment in the developing cerebellum. Front. Cell. Neurosci. 2015, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Schanen, N.C.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Klauck, S.M.; Lindsay, S.; Beyer, K.S.; Splitt, M.; Burn, J.; Poustka, A. A Mutation Hot Spot for Nonspecific X-Linked Mental Retardation in the MECP2 Gene Causes the PPM-X Syndrome. Am. J. Hum. Genet. 2002, 70, 1034–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, L.K.; Schousboe, A.; Sonnewald, U.; Waagepetersen, H.S. Glucose is Necessary to Maintain Neurotransmitter Homeostasis during Synaptic Activity in Cultured Glutamatergic Neurons. J. Cereb. Blood Flow Metab. 2006, 26, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incardona, J.P.; Eaton, S. Cholesterol in signal transduction. Curr. Opin. Cell Biol. 2000, 12, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Koudinov, A.R.; Koudinova, N.V. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 2001, 15, 1858–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saher, G.; Brügger, B.; Lappe-Siefke, C.; Möbius, W.; Tozawa, R.-I.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.-A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.G.; Gill, M.; Lemkey-Johnston, N.; Larramendi, L.M.H. Cholesterol and phospholipid composition of cerebellar fractions enriched in unmyelinated axons and synaptosomes. Exp. Brain Res. 1971, 13, 504–513. [Google Scholar] [CrossRef]
- Du, F.; Zhu, X.-H.; Zhang, Y.; Friedman, M.; Zhang, N.; Uğurbil, K.; Chen, W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. USA 2008, 105, 6409–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, H.F.; Thomas, A.J. Metabolism of glucose and glutamate by synaptosomes from mammalian cerebral cortex. J. Neurochem. 1969, 16, 1495–1504. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Gerónimo-Olvera, C.; Montiel, T.; Rincon-Heredia, R.; Castro-Obregón, S.; Massieu, L. Autophagy fails to prevent glucose deprivation/glucose reintroduction-induced neuronal death due to calpain-mediated lysosomal dysfunction in cortical neurons. Cell Death Dis. 2017, 8, e2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.-H.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kulas, J.A.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef] [PubMed]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.; Ramaekers, F.C.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24(S)-Hydroxycholesterol Participates in a Liver X Receptor-controlled Pathway in Astrocytes That Regulates Apolipoprotein E-mediated Cholesterol Efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.S.; Sampson, E.L.; Stern, M.B. Lipid composition of myelin from the peripheral nervous system. Intradural spinal roots. J. Neurochem. 1967, 14, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fester, L.; Zhou, L.; Bütow, A.; Huber, C.; von Lossow, R.; Prange-Kiel, J.; Jarry, H.; Rune, G.M. Cholesterol-promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus 2009, 19, 692–705. [Google Scholar] [CrossRef]
- Goritz, C.; Mauch, D.H.; Pfrieger, F.W. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol. Cell. Neurosci. 2005, 29, 190–201. [Google Scholar] [CrossRef]
- Barbero-Camps, E.; Roca-Agujetas, V.; Bartolessis, I.; de Dios, C.; Fernández-Checa, J.C.; Marí, M.; Morales, A.; Hartmann, T.; Colell, A. Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 2018, 14, 1129–1154. [Google Scholar] [CrossRef] [Green Version]
- Nóbrega, C.; Mendonça, L.; Marcelo, A.; Lamazière, A.; Tomé, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; Dunnen, W.D.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 138, 837–858. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.M.; Sickmann, H.M.; Fosgerau, K.; Lund, T.M.; Schousboe, A.; Waagepetersen, H.S.; Ransom, B.R. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 2005, 79, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Vergara, R.C.; Jaramillo-Riveri, S.; Luarte, A.; Moënne-Loccoz, C.; Fuentes, R.; Couve, A.; Maldonado, P.E. The Energy Homeostasis Principle: Neuronal Energy Regulation Drives Local Network Dynamics Generating Behavior. Front. Comput. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piot, V.M.; Verrijcken, A.; Vanhoof, M.; Mertens, I.; Soetens, F. Full Neurological Recovery after Extreme Hypoglycemia during Intensive Insulin Therapy: A Case Report. J. Diabetes Sci. Technol. 2012, 6, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Root, D.H.; Mejias-Aponte, C.A.; Zhang, S.; Wang, H.-L.; Hoffman, A.F.; Lupica, C.R.; Morales, M. Single rodent mesohabenular axons release glutamate and GABA. Nat. Neurosci. 2014, 17, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Rothman, D.L. Glucose, Lactate, β-Hydroxybutyrate, Acetate, GABA, and Succinate as Substrates for Synthesis of Glutamate and GABA in the Glutamine–Glutamate/GABA Cycle. Glutamate/GABA-Glutamine Cycle Amino Acid Neurotransm. Homeost. 2016, 13, 9–42. [Google Scholar] [CrossRef]
- Okuda, T.; Haga, T.; Kanai, Y.; Endou, H.; Ishihara, T.; Katsura, I. Identification and characterization of the high-affinity choline transporter. Nat. Neurosci. 2000, 3, 120–125. [Google Scholar] [CrossRef]
- Akaaboune, M.; Culican, S.M.; Turney, S.G.; Lichtman, J.W. Rapid and Reversible Effects of Activity on Acetylcholine Receptor Density at the Neuromuscular Junction in Vivo. Science 1999, 286, 503–507. [Google Scholar] [CrossRef]
- Olshansky, B.; Sabbah, H.N.; Hauptman, P.J.; Colucci, W.S. Parasympathetic nervous system and heart failure: Pathophysiology and potential implications for therapy. Circulation 2008, 118, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Kessler, G.; Friedman, J. Metabolism of Fatty Acids and Glucose. Circulation 1998, 98, 1351. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Reiser, G. Why does Brain Metabolism not Favor Burning of Fatty Acids to Provide Energy?—Reflections on Disadvantages of the Use of Free Fatty Acids as Fuel for Brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef]
- Mathews, E.S.; Appel, B. Cholesterol Biosynthesis Supports Myelin Gene Expression and Axon Ensheathment through Modulation of P13K/Akt/mTor Signaling. J. Neurosci. 2016, 36, 7628–7639. [Google Scholar] [CrossRef] [Green Version]
- Björkhem, I.; Lütjohann, D.; Diczfalusy, U.; Ståhle, L.; Ahlborg, G.; Wahren, J. Cholesterol homeostasis in human brain: Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J. Lipid Res. 1998, 39, 1594–1600. [Google Scholar] [CrossRef]
- Lütjohann, D.; Breuer, O.; Ahlborg, G.; Nennesmo, I.; Sidén, A.; Diczfalusy, U.; Björkhem, I. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 1996, 93, 9799–9804. [Google Scholar] [CrossRef] [Green Version]
- Muse, E.D.; Jurevics, H.; Toews, A.D.; Matsushima, G.K.; Morell, P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J. Neurochem. 2001, 76, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Saher, G.; Quintes, S.; Möbius, W.; Wehr, M.; Krämer-Albers, E.-M.; Brügger, B.; Nave, K.-A. Cholesterol Regulates the Endoplasmic Reticulum Exit of the Major Membrane Protein P0 Required for Peripheral Myelin Compaction. J. Neurosci. 2009, 29, 6094–6104. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Borbat, P.P.; Freed, J.H.; Shin, Y.-K. A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. USA 2009, 106, 5141–5146. [Google Scholar] [CrossRef] [Green Version]
- Lang, T.; Bruns, D.; Wenzel, D.; Riedel, D.; Holroyd, P.; Thiele, C.; Jahn, R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001, 20, 2202–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, G.; Cheung, S.; Galeano, J.; Ji, A.X.; Qin, Q.; Bi, X. Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1−/− mouse brain. Brain Res. 2009, 1270, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kacher, R.; Mounier, C.; Caboche, J.; Betuing, S. Altered Cholesterol Homeostasis in Huntington’s Disease. Front. Aging Neurosci. 2022, 14, 797220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shin, B.-C.; Thamotharan, S.; Devaskar, S.U. Creb1-Mecp2-mCpG Complex Transactivates Postnatal Murine Neuronal Glucose Transporter Isoform 3 Expression. Endocrinology 2013, 154, 1598–1611. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF Transcription Involves Calcium-Dependent Phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tsuchida, A.; Itakura, Y.; Nonomura, T.; Ono, M.; Hirota, F.; Inoue, T.; Nakayama, C.; Taiji, M.; Noguchi, H. Brain-derived neurotrophic factor regulates glucose metabolism by modulating energy balance in diabetic mice. Diabetes 2000, 49, 436–444. [Google Scholar] [CrossRef] [Green Version]
- Sakagami, H.; Makino, Y.; Mizumoto, K.; Isoe, T.; Takeda, Y.; Watanabe, J.; Fujita, Y.; Takiyama, Y.; Abiko, A.; Haneda, M. Loss of HIF-1α impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am. J. Physiol. Metab. 2014, 306, E1065–E1076. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Gomes, A.P.; Wang, X.; Yoon, S.O.; Lee, G.; Nagiec, M.J.; Cho, S.; Chavez, A.; Islam, T.; Yu, Y.; et al. mTORC1 Promotes Metabolic Reprogramming by the Suppression of GSK3-Dependent Foxk1 Phosphorylation. Mol. Cell 2018, 70, 949–960.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Del Rey, M.J.; Valín, Á.; Usategui, A.; García-Herrero, C.M.; Sánchez-Aragó, M.; Cuezva, J.M.; Galindo, M.; Bravo, B.; Cañete, J.D.; Blanco, F.J.; et al. Hif-1α Knockdown Reduces Glycolytic Metabolism and Induces Cell Death of Human Synovial Fibroblasts Under Normoxic Conditions. Sci. Rep. 2017, 7, 3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Krishnan, K.; Connors, C.R.; Choy, M.S.; Page, R.; Peti, W.; Van Aelst, L.; Shea, S.; Tonks, N.K. PTP1B inhibition suggests a therapeutic strategy for Rett syndrome. J. Clin. Investig. 2015, 125, 3163–3177. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, M.S.; Donizetti, A.; Iannotta, L.; Aliperti, V.; Cupidi, C.; Bruni, A.C.; Cigliano, L. Brain-derived neurotrophic factor modulates cholesterol homeostasis and Apolipoprotein E synthesis in human cell models of astrocytes and neurons. J. Cell. Physiol. 2018, 233, 6925–6943. [Google Scholar] [CrossRef]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef] [Green Version]
- Henin, N.; Vincent, M.; Gruber, H.E.; Berghe, G.V.D. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995, 9, 541–546. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, P.-G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brûlé, S.; Turcotte, V.; Côté, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.-N.; Ho, H.-Y.H.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-Specific Phosphorylation of MeCP2 Regulates Activity-Dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Sher, A.A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin–mTOR–P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangasamy, S.; Olfers, S.; Gerald, B.; Hilbert, A.; Svejda, S.; Narayanan, V. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Research 2016, 5, 2269. [Google Scholar] [CrossRef] [Green Version]
- Tsujimura, K.; Irie, K.; Nakashima, H.; Egashira, Y.; Fukao, Y.; Fujiwara, M.; Itoh, M.; Uesaka, M.; Imamura, T.; Nakahata, Y.; et al. miR-199a Links MeCP2 with mTOR Signaling and Its Dysregulation Leads to Rett Syndrome Phenotypes. Cell Rep. 2015, 12, 1887–1901. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, S.; Boggio, E.M.; Grosso, S.; Lonetti, G.; Forlani, G.; Stefanelli, G.; Calcagno, E.; Morello, N.; Landsberger, N.; Biffo, S.; et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 2011, 20, 1182–1196. [Google Scholar] [CrossRef] [Green Version]
- Toloe, J.; Mollajew, R.; Kügler, S.; Mironov, S. Metabolic differences in hippocampal ‘Rett’ neurons revealed by ATP imaging. Mol. Cell. Neurosci. 2014, 59, 47–56. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Mao, J.; DeMayo, F.J.; Li, H.; Abu-Elheiga, L.; Gu, Z.; Shaikenov, T.E.; Kordari, P.; Chirala, S.S.; Heird, W.C.; Wakil, S.J. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8552–8557. [Google Scholar] [CrossRef] [Green Version]
- Heh, C.W.; Smith, R.; Wu, J.; Hazlett, E.; Russell, A.; Asarnow, R.; Tanguay, P.; Buchsbaum, M.S. Positron emission tomography of the cerebellum in autism. Am. J. Psychiatry 1989, 146, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Chakravarthy, H.; Suresh, G.; Devanathan, V. Adult Goat Retinal Neuronal Culture: Applications in Modeling Hyperglycemia. Front. Neurosci. 2019, 13, 983. [Google Scholar] [CrossRef] [PubMed]
- Sevastre-Berghian, A.C.; Ielciu, I.; Mitre, A.O.; Filip, G.A.; Oniga, I.; Vlase, L.; Benedec, D.; Gheldiu, A.-M.; Toma, V.A.; Mihart, B.; et al. Targeting Oxidative Stress Reduction and Inhibition of HDAC1, MECP2, and NF-kB Pathways in Rats with Experimentally Induced Hyperglycemia by Administration of Thymus marshallianus Willd. Extracts. Front. Pharmacol. 2020, 11, 581470. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Bondy, C.A.; Lee, W.-H.; Zhou, J. Ontogeny and cellular distribution of brain glucose transporter gene expression. Mol. Cell. Neurosci. 1992, 3, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Burant, C.F.; Bell, G.I. Mammalian facilitative glucose transporters: Evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 1992, 31, 10414–10420. [Google Scholar] [CrossRef]
- Morgello, S.; Uson, R.R.; Schwartz, E.J.; Haber, R.S. The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia 1995, 14, 43–54. [Google Scholar] [CrossRef]
- Stolarczyk, E.; Guissard, C.; Michau, A.; Even, P.C.; Grosfeld, A.; Serradas, P.; Lorsignol, A.; Pénicaud, L.; Brot-Laroche, E.; Leturque, A.; et al. Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Am. J. Physiol. Metab. 2010, 298, E1078–E1087. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.M.; Lew, P.S.; Jhanji, G. Regulation of the Fructose Transporter Gene Slc2a5 Expression by Glucose in Cultured Microglial Cells. Int. J. Mol. Sci. 2021, 22, 12668. [Google Scholar] [CrossRef]
- Payne, J.; Maher, F.; Simpson, I.; Mattice, L.; Davies, P. Glucose transporter glut 5 expression in microglial cells. Glia 1997, 21, 327–331. [Google Scholar] [CrossRef]
- Carayannopoulos, M.O.; Chi, M.M.-Y.; Cui, Y.; Pingsterhaus, J.M.; McKnight, R.A.; Mueckler, M.; Devaskar, S.U.; Moley, K.H. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc. Natl. Acad. Sci. USA 2000, 97, 7313–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holman, G.D.; Kozka, I.J.; Clark, A.E.; Flower, C.J.; Saltis, J.; Habberfield, A.D.; Simpson, I.A.; Cushman, S.W. Cell surface labeling of glucose transporter isoform GLUT4 by bis-mannose photolabel. Correlation with stimulation of glucose transport in rat adipose cells by insulin and phorbol ester. J. Biol. Chem. 1990, 265, 18172–18179. [Google Scholar] [CrossRef] [PubMed]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef]
- Raychaudhuri, N.; Raychaudhuri, S.; Thamotharan, M.; Devaskar, S.U. Histone Code Modifications Repress Glucose Transporter 4 Expression in the Intrauterine Growth-restricted Offspring. J. Biol. Chem. 2008, 283, 13611–13626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Cory, S.; Fraser, S.E. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature 1995, 378, 192–196. [Google Scholar] [CrossRef]
- Rauti, R.; Cellot, G.; D’Andrea, P.; Colliva, A.; Scaini, D.; Tongiorgi, E.; Ballerini, L. BDNF impact on synaptic dynamics: Extra or intracellular long-term release differently regulates cultured hippocampal synapses. Mol. Brain 2020, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Hauser, C.; Kahn, B.B.; Haj, F.G.; Neel, B.G.; Tonks, N.K.; Tiganis, T. Coordinated Regulation of Insulin Signaling by the Protein Tyrosine Phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 2005, 25, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.; Yeo, G.S.; Cox, J.J.; Morton, J.; Adlam, A.-L.R.; Keogh, J.M.; Yanovski, J.A.; El Gharbawy, A.; Han, J.C.; Tung, Y.L.; et al. Hyperphagia, Severe Obesity, Impaired Cognitive Function, and Hyperactivity Associated with Functional Loss of One Copy of the Brain-Derived Neurotrophic Factor (BDNF) Gene. Diabetes 2006, 55, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef] [Green Version]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for Cell Cycle Arrest by the Immunosuppressant Rapamycin in Yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Akt Maintains Cell Size and Survival by Increasing mTOR-dependent Nutrient Uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, V.; Svensson, K.; Shimobayashi, M.; Colombi, M.; Muñoz, S.; Jimenez, V.; Handschin, C.; Bosch, F.; Hall, M.N. mTORC 2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol. Med. 2016, 8, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Cornu, M.; Cybulski, N.; Polak, P.; Betz, C.; Trapani, F.; Terracciano, L.; Heim, M.H.; Rüegg, M.A.; Hall, M.N. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012, 15, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Pino, E.; Wu, L.; Kacergis, M.; Soukas, A.A. Identification of Akt-independent Regulation of Hepatic Lipogenesis by Mammalian Target of Rapamycin (mTOR) Complex 2. J. Biol. Chem. 2012, 287, 29579–29588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-Activated Protein Kinase Regulates GLUT4 Transcription by Phosphorylating Histone Deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- Lira, V.A.; Soltow, Q.A.; Long, J.H.D.; Betters, J.L.; Sellman, J.E.; Criswell, D.S. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am. J. Physiol. Metab. 2007, 293, E1062–E1068. [Google Scholar] [CrossRef] [Green Version]
- Golubiani, G.; Lagani, V.; Solomonia, R.; Müller, M. Metabolomic Fingerprint of Mecp2-Deficient Mouse Cortex: Evidence for a Pronounced Multi-Facetted Metabolic Component in Rett Syndrome. Cells 2021, 10, 2494. [Google Scholar] [CrossRef]
- Segatto, M.; Trapani, L.; Di Tunno, I.; Sticozzi, C.; Valacchi, G.; Hayek, J.; Pallottini, V. Cholesterol Metabolism Is Altered in Rett Syndrome: A Study on Plasma and Primary Cultured Fibroblasts Derived from Patients. PLoS ONE 2014, 9, e104834. [Google Scholar] [CrossRef] [Green Version]
- Sticozzi, C.; Belmonte, G.; Pecorelli, A.; Cervellati, F.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C.; Hayek, J.; Valacchi, G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 2013, 587, 2199–2204. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.M.; Chuang, J.-C.; Posey, K.S.; Turley, S.D. Suppression of brain cholesterol synthesis in male Mecp2-deficient mice is age dependent and not accompanied by a concurrent change in the rate of fatty acid synthesis. Brain Res. 2017, 1654, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Kiyosue, K.; Hazama, S.; Ogura, A.; Kashihara, M.; Hara, T.; Koshimizu, H.; Kojima, M. Brain-Derived Neurotrophic Factor Regulates Cholesterol Metabolism for Synapse Development. J. Neurosci. 2007, 27, 6417–6427. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.-A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001, 15, 1206–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, G.; Xie, C.; Dietschy, J.M.; Turley, S.D. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Dev. Brain Res. 2003, 146, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Valdez, C.M.; Smith, M.A.; Perry, G.; Phelix, C.; Santamaria, F. Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus 2010, 20, 902–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, A.B.; Thiele, C.; Tenedini, F.; Richard, M.; Leyendecker, P.; Hoermann, A.; Soba, P.; Tavosanis, G. Cell-Autonomous Control of Neuronal Dendrite Expansion via the Fatty Acid Synthesis Regulator SREBP. Cell Rep. 2017, 21, 3346–3353. [Google Scholar] [CrossRef] [Green Version]
- Ferris, H.A.; Perry, R.J.; Moreira, G.V.; Shulman, G.I.; Horton, J.D.; Kahn, C.R. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Schüle, M.; Butto, T.; Dewi, S.; Schlichtholz, L.; Strand, S.; Gerber, S.; Endres, K.; Schweiger, S.; Winter, J. mTOR Driven Gene Transcription Is Required for Cholesterol Production in Neurons of the Developing Cerebral Cortex. Int. J. Mol. Sci. 2021, 22, 6034. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, J.N.; Paik, Y.-K. Cholesterol biosynthesis from lanosterol. A concerted role for Sp1 and NF-Y-binding sites for sterol-mediated regulation of rat 7-dehydrocholesterol reductase gene expression. J. Biol. Chem. 2001, 276, 18153–18160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, S.P.; Hartley, J.W.; Rowe, W.P. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc. Natl. Acad. Sci. USA 1977, 74, 3065–3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porstmann, T.; Griffiths, B.; Chung, Y.-L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, C.; Assmus, B.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arter. Thromb. Vasc. Biol. 2000, 20, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of Proline-rich Akt Substrate of 40 kDa (PRAS40) Function by Mammalian Target of Rapamycin Complex 1 (mTORC1)-mediated Phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.-L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the Nuclear Receptor LXR by Oxysterols Defines a New Hormone Response Pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Lee, T.S.; Zhu, M.; Gu, C.; Wang, Y.; Zhu, Y.; Shyy, J.Y. Statins Activate AMP-Activated Protein Kinase In Vitro and In Vivo. Circulation 2006, 114, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-T.; Ho, H.J.; Lin, J.-T.; Shieh, J.-J.; Wu, C.-Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [Green Version]
- Sheikholeslami, K.; Sher, A.A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [Green Version]
- Zuliani, I.; Urbinati, C.; Valenti, D.; Quattrini, M.C.; Medici, V.; Cosentino, L.; Pietraforte, D.; Di Domenico, F.; Perluigi, M.; Vacca, R.A.; et al. The Anti-Diabetic Drug Metformin Rescues Aberrant Mitochondrial Activity and Restrains Oxidative Stress in a Female Mouse Model of Rett Syndrome. J. Clin. Med. 2020, 9, 1669. [Google Scholar] [CrossRef]
- Amadasu, E.; Kang, R.; Usmani, A.; Borlongan, C.V. Effects of Lovastatin on Brain Cancer Cells. Cell Transplant. 2022, 31, 9636897221102903. [Google Scholar] [CrossRef] [PubMed]
- Takhwifa, F.; Aninditha, T.; Setiawan, H.; Sauriasari, R. The potential of metformin as an antineoplastic in brain tumors: A systematic review. Heliyon 2021, 7, e06558. [Google Scholar] [CrossRef]
- Frieden, C. ApoE: The role of conserved residues in defining function. Protein Sci. 2014, 24, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Rall, S.C., Jr.; Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E. The complete amino acid sequence. J. Biol. Chem. 1982, 257, 4171–4178. [Google Scholar] [CrossRef]
- Zahorakova, D.; Jachymova, M.; Kemlink, D.; Baxova, A.; Martasek, P. APOE ε4: A Potential Modulation Factor in Rett Syndrome. J. Child Neurol. 2010, 25, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-G.; Tulloch, J.; Chen, S.; Leong, L.; Saxton, A.D.; Kraemer, B.; Darvas, M.; Keene, C.D.; Shutes-David, A.; Todd, K.; et al. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer’s disease. PLoS ONE 2020, 15, e0227667. [Google Scholar] [CrossRef]
- Kyle, S.M.; Saha, P.K.; Brown, H.M.; Chan, L.C.; Justice, M.J. MeCP2 co-ordinates liver lipid metabolism with the NCoR1/HDAC3 corepressor complex. Hum. Mol. Genet. 2016, 25, 3029–3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, N.L.; Heaven, M.R.; Holt, L.M.; Crossman, D.K.; Boggio, K.J.; Shaffer, S.A.; Flint, D.L.; Olsen, M.L. RNA sequencing and proteomics approaches reveal novel deficits in the cortex of Mecp2-deficient mice, a model for Rett syndrome. Mol. Autism 2017, 8, 56. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diseases | Metabolic Impairments | References |
|---|---|---|
| Rett Syndrome | Abnormal systemic and brain glucose utilization as well as insulin resistance Impaired systemic and brain cholesterol metabolism Abnormal mitochondrial function | [18,19,20,21,22,23,24,25] |
| Autism Spectrum Disorders | Altered brain glucose utilization and insulin resistance dyslipidemia | [26,27,28] |
| Fetal Alcohol Spectrum Disorders | Impaired glucose tolerance Increase plasma triglyceride level | [29] |
| Severe Neonatal Encephalopathy | Impaired mitochondrial oxidative phosphorylation | [30,31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuu, Y.M.; Roberts, C.-T.; Rastegar, M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int. J. Mol. Sci. 2023, 24, 4218. https://doi.org/10.3390/ijms24044218
Vuu YM, Roberts C-T, Rastegar M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. International Journal of Molecular Sciences. 2023; 24(4):4218. https://doi.org/10.3390/ijms24044218
Chicago/Turabian StyleVuu, Yen My, Chris-Tiann Roberts, and Mojgan Rastegar. 2023. "MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism" International Journal of Molecular Sciences 24, no. 4: 4218. https://doi.org/10.3390/ijms24044218