
Transcriptome Dynamic Analysis Reveals New Candidate Genes Associated with Resistance to Fusarium Head Blight in Two Chinese Contrasting Wheat Genotypes

Abstract
1. Introduction
2. Results
2.1. Quality Assessment of Transcriptome Sequencing
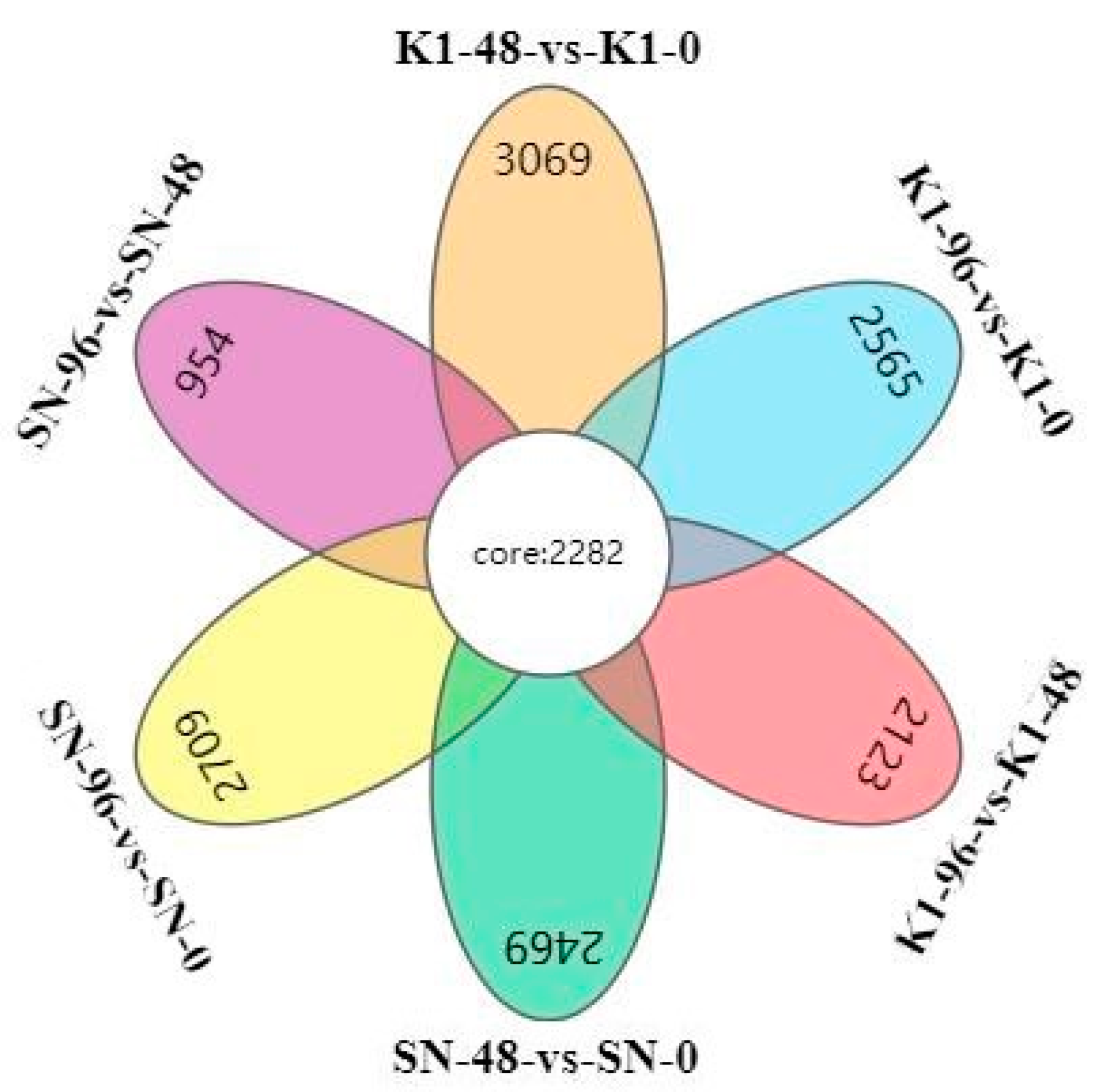
2.2. Analysis of DEGs
2.3. GO Analysis of DEGs
2.4. KEGG Analysis of DEGs
2.5. Comparing DEGs between Shannong 102 and Nankang 1 after Inoculation
2.6. DEGs Encoding Pathogenesis-Related Proteins
2.7. Distribution of DEGs on Chromosomes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Sample Preparation for RNA-Seq Analysis
4.3. RNA Extraction and Sequencing Analysis
4.4. RNA-Seq Data Analysis and Bioinformatics Analysis
4.5. Differential Expression Gene Analysis and Gene Functional Annotation
4.6. Real-Time Quantitative PCR (RT-qPCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ma, Z.Q.; Xie, Q.; Li, G.Q.; Jia, H.Y.; Zhou, J.Y.; Kong, Z.X.; Li, N.; Yuan, Y. Germplasms, genetics and genomics for better control of disastrous wheat Fusarium head blight. Theor. Appl. Genet. 2020, 133, 1541–1568. [Google Scholar] [CrossRef]
- Liu, W.C.; Liu, Z.D.; Huang, C.; Lu, M.H.; Liu, J.; Yang, Q.P. Statistics and analysis of crop yield losses caused by main diseases and insect pests in recent 10 years. Plant Prot. 2016, 42, 1–9. [Google Scholar]
- Bai, G.; Shaner, G. Scab of wheat: Prospects for control. Plant Dis. 1994, 78, 760–766. [Google Scholar] [CrossRef]
- Agriopoulou, S.; Stamatelopoulou, E.; Varzakas, T. Advances in Occurrence, Importance, and Mycotoxin Control Strategies: Prevention and Detoxification in Foods. Foods 2020, 9, 137. [Google Scholar] [CrossRef]
- Shi, J.R.; Liu, X.; Qiu, J.B.; Ji, F.; Xu, J.H.; Dong, F.; Yin, X.; Ran, J. Deoxynivalenol contamination in wheat and its management. Sci. Agric. Sinica. 2014, 47, 3641–3654. [Google Scholar] [CrossRef]
- Goswami, R.S.; Kistler, H.C. Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 2004, 5, 515–525. [Google Scholar] [CrossRef]
- McMullen, M.; Bergstrom, G.; De Wolf, E.; Dill-Macky, R.; Hershman, D.; Shaner, G.; Van Sanford, D. A unified effort to fight an enemy of wheat and barley: Fusarium head blight. Plant Dis. 2012, 96, 1712–1728. [Google Scholar] [CrossRef]
- Kriss, A.B.; Paul, P.A.; Xu, X.; Nicholson, P.; Doohan, F.M.; Hornok, L.; Rietini, A.; Edwards, S.G.; Madden, L.V. Quantification of the relationship between the environment and Fusarium head blight, Fusarium pathogen density, and mycotoxins in winter wheat in Europe. Eur. J. Plant Pathol. 2012, 133, 975–993. [Google Scholar] [CrossRef]
- Obanor, F.; Neate, S.; Simpfendorfer, S.; Sabburg, R.; Wilson, P.; Chakraborty, S. Fusarium graminearum and Fusarium pseudograminearum caused the 2010 head blight epidemics in Australia. Plant Pathol. 2013, 62, 79–91. [Google Scholar] [CrossRef]
- Van der Lee, T.; Zhang, H.; van Diepeningen, A.; Waalwijk, C. Biogeography of Fusarium graminearum species complex and chemotypes: A review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2015, 32, 453–460. [Google Scholar] [CrossRef]
- Bai, G.; Shaner, G. Management and resistance in wheat and barley to Fusarium head blight. Annu. Rev. Phytopathol. 2004, 42, 135–161. [Google Scholar] [CrossRef]
- Zhu, Z.; Hao, Y.; Mergoum, M.; Bai, G.; Humphreys, G.; Cloutier, S.; Xia, X.; He, Z. Breeding wheat for resistance to Fusarium head blight in the Global North: China, USA, and Canada. Crop J. 2019, 7, 730–738. [Google Scholar] [CrossRef]
- Mesterházy, A. Types and components of resistance to Fusarium head blight of wheat. Plant Breed. 1995, 114, 377–386. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, Z.; Rocheleau, H.; Fauteux, F.; Wang, Y.; McCartney, C.; Ouellet, T. Transcriptome dynamics associated with resistance and susceptibility against Fusarium head blight in four wheat genotypes. BMC Genom. 2018, 19, 642. [Google Scholar] [CrossRef]
- Li, X.; Zhong, S.; Chen, W.; Fatima, S.A.; Huang, Q.; Li, Q.; Tan, F.; Luo, P. Transcriptome Analysis Identifies a 140 kb Region of Chromosome 3B Containing Genes Specific to Fusarium Head Blight Resistance in Wheat. Int. J. Mol. Sci. 2018, 19, 852. [Google Scholar] [CrossRef]
- Xu, J.; Shi, S.; Wang, Z.; Ren, H.; Wang, Y.; Li, C. Identification of FHB Resistance Genes in Wheat Cultivar Xinong 979 by Transcriptome sequencing. J. Triticeae Crop 2020, 40, 665–675. [Google Scholar] [CrossRef]
- Su, P.; Zhao, L.; Li, W.; Zhao, J.; Yan, J.; Ma, X.; Li, A.; Wang, H.; Kong, L. Integrated metabolo-transcriptomics and functional characterization reveals that the wheat auxin receptor TIR1 negatively regulates defense against Fusarium graminearum. J. Integr. Plant Biol. 2021, 63, 340–352. [Google Scholar] [CrossRef]
- Cainong, J.C.; Bockus, W.W.; Feng, Y.; Chen, P.; Qi, L.; Sehgal, S.K.; Danilova, T.V.; Koo, D.-H.; Friebe, B.; Gill, B.S. Chromosome engineering, mapping, and transferring of resistance to Fusarium head blight disease from Elymus tsukushiensis into wheat. Theor. Appl. Genet. 2015, 128, 1019–1027. [Google Scholar] [CrossRef]
- Jia, H.; Zhou, J.; Xue, S.; Li, G.; Yan, H.; Ran, C.; Zhang, Y.; Shi, J.; Jia, L.; Wang, X.; et al. A journey to understand wheat Fusarium head blight resistance in the Chinese wheat landrace Wangshuibai. Crop J. 2017, 6, 48–59. [Google Scholar] [CrossRef]
- Li, G.; Zhou, J.; Jia, H.; Gao, Z.; Fan, M.; Luo, Y.; Zhao, P.; Xue, S.; Li, N.; Yuan, Y.; et al. Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat. Genet. 2019, 51, 1106–1112. [Google Scholar] [CrossRef]
- Qi, L.L.; Pumphrey, M.O.; Friebe, B.; Chen, P.D.; Gill, B.S. Molecular cytogenetic characterization of alien introgressions with gene Fhb3 for resistance to Fusarium head blight disease of wheat. Theor. Appl. Genet. 2008, 117, 1155–1166. [Google Scholar] [CrossRef]
- Su, Z.; Bernardo, A.; Tian, B.; Chen, H.; Wang, S.; Ma, H.; Cai, S.; Liu, D.; Zhang, D.; Li, T.; et al. A deletion mutation in TaHRC confers Fhb1 resistance to Fusarium head blight in wheat. Nat. Genet. 2019, 51, 1099–1105. [Google Scholar] [CrossRef]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; Lyu, Z.; Chen, L.; Xu, S.; Guo, J.; et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science. 2020, 368, eaba5435. [Google Scholar] [CrossRef]
- Xue, S.; Li, G.; Jia, H.; Xu, F.; Lin, F.; Tang, M.; Wang, Y.; An, X.; Xu, H.; Zhang, L.; et al. Fine mapping Fhb4, a major QTL conditioning resistance to Fusarium infection in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2010, 121, 147–156. [Google Scholar] [CrossRef]
- Xue, S.; Xu, F.; Tang, M.; Zhou, Y.; Li, G.; An, X.; Lin, F.; Xu, H.; Jia, H.; Zhang, L.; et al. Precise mapping Fhb5, a major QTL conditioning resistance to Fusarium infection in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 123, 1055–1063. [Google Scholar] [CrossRef]
- Xu, F.; Li, W.; Yan, S.; Zhang, C.; Zheng, J.; Du, J. Analysis of pyramiding effect of major QTLs for resistance to scab in wheat. J. Triticeae Crop 2017, 37, 585–593. [Google Scholar] [CrossRef]
- Uauy, C. Wheat genomics comes of age. Curr. Opin. Plant Biol. 2017, 36, 142–148. [Google Scholar] [CrossRef]
- Martin, L.; Fei, Z.; Giovannoni, J.; Rose, J.K.C. Catalyzing plant science research with RNA-seq. Front. Plant Sci. 2013, 4, 66. [Google Scholar] [CrossRef]
- Jia, C.L.; Zhang, Y.; Zhu, L.; Zhang, R. Application progress of transcriptome sequencing technology in biological sequencing. Mol. Plant Breed. 2015, 10, 2388–2394. [Google Scholar] [CrossRef]
- Ke, P.; Liu, Y.; Zhang, T.; Sun, Y.; He, L.; Xiao, J.; Wang, X. Function analysis of the key gene TaACS2 in ethylene synthesis pathway in resistance to Fusarium head blight of wheat. Acta Agric. Shanghai 2019, 35, 31–36. [Google Scholar] [CrossRef]
- Schweiger, W.; Steiner, B.; Ametz, C.; Siegwart, G.; Wiesenberger, G.; Berthiller, F.; Lemmens, M.; Jia, H.; Adam, G.; Muehlbauer, G.J.; et al. Transcriptomic characterization of two major F usarium resistance quantitative trait loci (QTL s), Fhb1 and Qfhs.ifa-5A, identifies novel candidate genes. Mol. Plant Pathol. 2013, 14, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Xu, H.; Yi, H.; Yang, L.; Kong, Z.; Zhang, L.; Xue, S.; Jia, H.; Ma, Z. Resistance to hemi-biotrophic F. graminearum infection is associated with coordinated and ordered expression of diverse defense signaling pathways. PLoS ONE 2011, 6, e19008. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Q.; Liu, Z.; Surendra, A.; Pan, Y.; Li, Y.; Zaharia, L.I.; Ouellet, T.; Fobert, P.R. Integrated transcriptome and hormone profiling highlight the role of multiple phytohormone pathways in wheat resistance against Fusarium head blight. PLoS ONE 2018, 13, e0207036. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Parich, A.; Bueschl, C.; Schoefbeck, D.; Neumann, N.K.N.; Kluger, B.; Schuster, K.; Krska, R.; Adam, G.; Lemmens, M.; et al. GC-MS based targeted metabolic profiling identifies changes in the wheat metabolome following deoxynivalenol treatment. Metabolomics 2015, 11, 722–738. [Google Scholar] [CrossRef]
- Biselli, C.; Bagnaresi, P.; Faccioli, P.; Hu, X.; Balcerzak, M.; Mattera, M.G.; Yan, Z.; Ouellet, T.; Cattivelli, L.; Valè, G. Comparative Transcriptome Profiles of Near-Isogenic Hexaploid Wheat Lines Differing for Effective Alleles at the 2DL FHB Resistance QTL. Front. Plant Sci. 2018, 9, 37. [Google Scholar] [CrossRef]
- Sattler, S.E.; Funnell-Harris, D.L. Modifying lignin to improve bioenergy feedstocks: Strengthening the barrier against pathogens. Front. Plant Sci. 2013, 4, 70. [Google Scholar] [CrossRef]
- Gunnaiah, R.; Kushalappa, A.C. Metabolomics deciphers the host resistance mechanisms in wheat cultivar Sumai-3, against trichothecene producing and non-producing isolates of Fusarium graminearum. Plant Physiol. Bioch. 2014, 83, 40–50. [Google Scholar] [CrossRef]
- Kosaka, A.; Manickavelu, A.; Kajihara, D.; Nakagawa, H.; Ban, T. Altered gene expression profiles of wheat genotypes against Fusarium head blight. Toxins 2015, 16, 604–620. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Zhao, M.; Zhang, W.; Li, B.; An, D.; Li, J.; Zhang, A.; Liu, R.; Liu, X. A genome-wide view of transcriptome dynamics during early spike development in bread wheat. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Pritsch, C.; Muehlbauer, G.J.; Bushnell, W.R.; Somers, D.A.; Vance, C.P. Fungal development and induction of defense response genes during early infection of wheat spikes by Fusarium graminearum. Mol. Plant Microbe Interact. 2000, 13, 159–169. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Wang, Z.; Meng, F.; Zhang, S.; Wu, X.; Zhang, Z.; Gao, Z. Overexpression of Arabidopsis Nucleotide-Binding and Leucine-Rich Repeat Genes RPS2 and RPM1 (D505V) Confers Broad-Spectrum Disease Resistance in Rice. Front. Plant Sci. 2019, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- TrapnTrapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total | Unique Mapped (%) | Multiple Mapped (%) | Total Mapped (%) | ≥Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|
| SN-0-1 | 53,437,598 | 90.57 | 5.02 | 95.58 | 94.28 | 60.01 |
| SN-0-2 | 66,258,116 | 90.27 | 5.04 | 95.31 | 93.86 | 58.98 |
| SN-0-3 | 40,958,454 | 90.49 | 4.79 | 95.28 | 93.88 | 58.60 |
| SN-48-1 | 51,240,494 | 89.45 | 3.73 | 93.17 | 94.42 | 61.37 |
| SN-48-2 | 48,617,354 | 90.54 | 4.10 | 94.64 | 93.84 | 58.50 |
| SN-48-3 | 61,489,210 | 90.13 | 4.21 | 94.34 | 94.03 | 58.89 |
| SN-96-1 | 52,242,470 | 79.70 | 3.64 | 83.34 | 94.30 | 59.72 |
| SN-96-2 | 55,219,698 | 76.89 | 3.65 | 80.54 | 94.24 | 59.16 |
| SN-96-3 | 58,356,204 | 80.23 | 3.84 | 84.07 | 94.24 | 58.85 |
| K1-0-1 | 47,633,842 | 90.62 | 5.44 | 96.05 | 94.17 | 59.00 |
| K1-0-2 | 58,276,138 | 91.05 | 5.01 | 96.06 | 94.19 | 59.03 |
| K1-0-3 | 52,976,254 | 90.64 | 5.23 | 95.87 | 93.82 | 59.12 |
| K1-48-1 | 89,254,412 | 90.04 | 4.94 | 94.98 | 93.66 | 59.46 |
| K1-48-2 | 56,188,174 | 89.32 | 4.61 | 93.93 | 94.18 | 59.19 |
| K1-48-3 | 50,330,048 | 90.52 | 5.05 | 95.57 | 94.20 | 59.52 |
| K1-96-1 | 63,193,192 | 76.02 | 3.44 | 79.47 | 94.07 | 59.21 |
| K1-96-2 | 51,992,614 | 78.60 | 3.58 | 82.18 | 94.30 | 59.25 |
| K1-96-3 | 52,012,908 | 82.05 | 3.90 | 85.95 | 94.08 | 59.18 |
| Sample | Pathway ID | Pathway | Candidate Genes with Pathway Annotation |
|---|---|---|---|
| SN-48 vs. SN-0 | ko01100 | Metabolic pathways | 2855 (49.9%) |
| ko01110 | Biosynthesis of secondary metabolites | 1844 (32.23%) | |
| ko00940 | Phenylpropanoid biosynthesis | 436 (7.62%) | |
| ko04075 | Plant hormone signal transduction | 336 (5.87%) | |
| ko01200 | Carbon metabolism | 309 (5.4%) | |
| ko01230 | Biosynthesis of amino acids | 300 (5.24%) | |
| ko04626 | Plant–pathogen interaction | 286 (5%) | |
| ko00480 | Glutathione metabolism | 243 (4.25%) | |
| ko00500 | Starch and sucrose metabolism | 240 (4.2%) | |
| ko04016 | MAPK signaling pathway—plant | 201 (3.51%) | |
| SN-96 vs. SN-0 | ko01100 | Metabolic pathways | 3005 (50.23%) |
| ko01110 | Biosynthesis of secondary metabolites | 1939 (32.41%) | |
| ko00940 | Phenylpropanoid biosynthesis | 474 (7.92%) | |
| ko04075 | Plant hormone signal transduction | 357 (5.97%) | |
| ko01200 | Carbon metabolism | 344 (5.75%) | |
| ko00480 | Glutathione metabolism | 291 (4.86%) | |
| ko04626 | Plant–pathogen interaction | 289 (4.83%) | |
| ko01230 | Biosynthesis of amino acids | 283 (4.73%) | |
| ko00500 | Starch and sucrose metabolism | 262 (4.38%) | |
| ko04016 | MAPK signaling pathway—plant | 222 (3.71%) | |
| SN-96 vs. SN-48 | ko01100 | Metabolic pathways | 1706 (52.54%) |
| ko01110 | Biosynthesis of secondary metabolites | 1138 (35.05%) | |
| ko00940 | Phenylpropanoid biosynthesis | 273 (8.41%) | |
| ko01200 | Carbon metabolism | 231 (7.11%) | |
| ko04075 | Plant hormone signal transduction | 198 (6.1%) | |
| ko01230 | Biosynthesis of amino acids | 197 (6.07%) | |
| ko00480 | Glutathione metabolism | 182 (5.61%) | |
| ko04626 | Plant–pathogen interaction | 153 (4.71%) | |
| ko04016 | MAPK signaling pathway—plant | 133 (4.1%) | |
| ko00500 | Starch and sucrose metabolism | 132 (4.07%) |
| Sample | Pathway ID | Pathway | Candidate Genes with Pathway Annotation |
|---|---|---|---|
| K1-48 vs. K1-0 | ko01100 | Metabolic pathways | 2526 (45.89%) |
| ko01110 | Biosynthesis of secondary metabolites | 1650 (29.97%) | |
| ko03010 | Ribosome | 621 (11.28%) | |
| ko00940 | Phenylpropanoid biosynthesis | 384 (6.98%) | |
| ko01200 | Carbon metabolism | 340 (6.18%) | |
| ko04626 | Plant–pathogen interaction | 321 (5.83%) | |
| ko01230 | Biosynthesis of amino acids | 314 (5.7%) | |
| ko04075 | Plant hormone signal transduction | 263 (4.78%) | |
| ko00480 | Glutathione metabolism | 227 (4.12%) | |
| ko04016 | MAPK signaling pathway—plant | 200 (3.63%) | |
| K1-96 vs. K1-0 | ko01100 | Metabolic pathways | 2877 (50.44%) |
| ko01110 | Biosynthesis of secondary metabolites | 1901 (33.33%) | |
| ko00940 | Phenylpropanoid biosynthesis | 437 (7.66%) | |
| ko01200 | Carbon metabolism | 377 (6.61%) | |
| ko04075 | Plant hormone signal transduction | 358 (6.28%) | |
| ko01230 | Biosynthesis of amino acids | 299 (5.24%) | |
| ko04626 | Plant–pathogen interaction | 286 (5.01%) | |
| ko00480 | Glutathione metabolism | 267 (4.68%) | |
| ko00500 | Starch and sucrose metabolism | 252 (4.42%) | |
| ko04016 | MAPK signaling pathway—plant | 220 (3.86%) | |
| K1-96 vs. K1-48 | ko01100 | Metabolic pathways | 2604 (46.11%) |
| ko01110 | Biosynthesis of secondary metabolites | 1594 (28.23%) | |
| ko03010 | Ribosome | 640 (11.33%) | |
| ko01200 | Carbon metabolism | 364 (6.45%) | |
| ko00940 | Phenylpropanoid biosynthesis | 337 (5.97%) | |
| ko01230 | Biosynthesis of amino acids | 303 (5.37%) | |
| ko00500 | Starch and sucrose metabolism | 249 (4.41%) | |
| ko00480 | Glutathione metabolism | 203 (3.59%) | |
| ko00010 | Glycolysis/Gluconeogenesis | 198 (3.51%) | |
| ko03013 | RNA transport | 188 (3.33%) |
| Sample | Pathway ID | Pathway | Candidate Genes with Pathway Annotation |
|---|---|---|---|
| Shannong 102 vs. Nankang 1 | ko01100 | Metabolic pathways | 281 (20.26%) |
| ko01110 | Biosynthesis of secondary metabolites | 205 (14.78%) | |
| ko00480 | Glutathione metabolism | 81 (5.84%) | |
| ko00940 | Phenylpropanoid biosynthesis | 71 (5.12%) | |
| ko01200 | Carbon metabolism | 52 (3.75%) | |
| ko04075 | Plant hormone signal transduction | 36 (2.60%) | |
| ko04626 | Plant–pathogen interaction | 29 (2.09%) | |
| ko01230 | Biosynthesis of amino acids | 29 (2.09%) | |
| ko00010 | Glycolysis/Gluconeogenesis | 27 (1.95%) | |
| ko04016 | MAPK signaling pathway—plant | 25 (1.80%) |
| Composition | Volumn (μL) |
|---|---|
| 2×ChamQ SYBR qPCR Master Mix | 10 |
| PCR Forward Primer (10μM) | 0.4 |
| PCR Reverse Primer (10μM) | 0.4 |
| cDNA | 4 |
| ddH2O | 5.2 |
| Total | 20 |
| Gene | Forward Primer (5′-3′) | Reverse Primer (3′-5′) |
|---|---|---|
| TraesCS5A02G439700 | TACTACGACCACGGCAGCAA | ATACAACTTCCATGAATCGCAACAC |
| TraesCS5B02G442900 | ACGGGGAGAACCTCTACGGA | CAGATGATGAACACACCGTC |
| TraesCS5B02G443300 | ACGGGGAGAACCTCTACGGA | CAGATGATGAACACACCGTC |
| TraesCS5B02G443400 | ACGGGGAGAACCTCTACGGA | CAGATGATGAACACACCGTC |
| TraesCS5D02G446900 | ACGGGGAGAACATCTACGGA | GCTCACCCCCTCGTAGTTG |
| ACTIN | CACTTGGTTCTCCTGCCTCT | AGGAAACTGCTGCGAGGATG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Wang, D.; Ji, M.; Tian, J.; Ding, H.; Deng, Z. Transcriptome Dynamic Analysis Reveals New Candidate Genes Associated with Resistance to Fusarium Head Blight in Two Chinese Contrasting Wheat Genotypes. Int. J. Mol. Sci. 2023, 24, 4222. https://doi.org/10.3390/ijms24044222
Zhao Y, Wang D, Ji M, Tian J, Ding H, Deng Z. Transcriptome Dynamic Analysis Reveals New Candidate Genes Associated with Resistance to Fusarium Head Blight in Two Chinese Contrasting Wheat Genotypes. International Journal of Molecular Sciences. 2023; 24(4):4222. https://doi.org/10.3390/ijms24044222
Chicago/Turabian StyleZhao, Yunzhe, Dehua Wang, Mengqi Ji, Jichun Tian, Hanfeng Ding, and Zhiying Deng. 2023. "Transcriptome Dynamic Analysis Reveals New Candidate Genes Associated with Resistance to Fusarium Head Blight in Two Chinese Contrasting Wheat Genotypes" International Journal of Molecular Sciences 24, no. 4: 4222. https://doi.org/10.3390/ijms24044222
APA StyleZhao, Y., Wang, D., Ji, M., Tian, J., Ding, H., & Deng, Z. (2023). Transcriptome Dynamic Analysis Reveals New Candidate Genes Associated with Resistance to Fusarium Head Blight in Two Chinese Contrasting Wheat Genotypes. International Journal of Molecular Sciences, 24(4), 4222. https://doi.org/10.3390/ijms24044222