1. Introduction
Since wheat (
Triticum aestivum L.) evolved into a cultivated species approximately 10,000 years ago, it has gradually become the largest crop for human rations in the world, providing approximately 35% of the population with sufficient carbohydrates and essential nutrients. However, wheat production still faces the effects of a variety of diseases and insect pests worldwide. Of these, the occurrence of scabs is becoming increasingly serious. Since 2000, there have been 17 scab outbreaks in China [
1]. In 2012, the outbreak of scab affected approximately 10 million hectares of wheat and reduced the yield by more than 2 million tons in China [
2].
Fusarium head blight (FHB) is caused by infections with
Fusarium graminearum and other types of Fusarium, which occurs during the flowering stage of wheat [
3]. After infection with Fusarium, the diseased spike becomes discoloured and withered, the rachis becomes black and necrotic, and the grains shrink and become shrivelled, resulting in a significant reduction in wheat yields [
1]. At the same time, the affected part accumulates a variety of toxic secondary metabolites, such as trichothecene mycotoxins and zearalenone mycotoxins [
4]. Among them, deoxynivalenol is recognised as a strong carcinogenic toxin worldwide. After being consumed by humans and animals, it causes a decline in the body’s immunity and a variety of toxic reactions, which will seriously threaten human and animal health [
5]. FHB brought on by
F. graminearum was originally discovered in England in 1884 [
6]. With climate warming and changes in farming systems, FHB has become a common occurrence in Asia, Europe, Oceania, and South America [
7,
8,
9,
10,
11,
12]. In China’s main wheat-producing areas, FHB seriously hinders the development of the wheat industry and has become an issue of great concern to wheat scientists.
Generally, FHB resistance is classified into five types, as follows: resistance to initial spike infection (type I), resistance to spike spread infection (type II), resistance to mycotoxin accumulation (type III), resistance to kernel infection (type IV), and resistance to yield reduction (type V) [
13]. Of these, type II has been widely studied because of its stable resistance phenotype [
3]. Wheat FHB resistance is a quantitative trait controlled by multiple genes, which are easily affected by the environment, genotype–environment interactions, and other factors. QTL/gene mapping of FHB resistance has been extensively studied, and more than 400 QTLs are distributed on all chromosomes of wheat [
1]. However, there are only seven clear resistance genes known, namely
Fhb1 to
Fhb7; therefore, new genes/QTLs and new varieties with FHB resistance still need to be studied.
Transcriptome sequencing (RNA-Seq) technology has rapidly developed in recent years. This technology can quickly and comprehensively obtain the complete expression information of a sample in a certain state through high-throughput sequencing of the transcriptome product mRNA. It has been widely used in all aspects of plant research, especially abiotic and biotic stresses. A few studies have reported on wheat FHB resistance using this method. Transcriptome analysis showed that protein serine/threonine kinase and LRR-RK are associated with susceptibility to FHB, whereas several ethylene-responsive,
WRKY,
Myb,
bZIP, and
NAC-domains containing transcription factors were also found to be associated with susceptibility [
14]. Li et al. [
15] conducted a comparative transcriptome analysis to identify genes that are differentially expressed in FHB-resistant and FHB-susceptible wheat lines grown under field conditions for various periods after
F. graminearum infection and determined the chromosomal distribution of the differentially expressed genes (DEGs). XU et al. [
16] identified the FHB resistance genes of wheat variety Xinong 979 by transcriptome sequencing and screened detoxification-related protein genes, such as UDP-glucosyltransferase genes. Su et al. [
17] used transcriptomics and metabolomics to track and analyse the infection process of
F. graminearum and found that a large number of secondary metabolites, such as flavonoids, plant hormones, tryptamines, phenolic amines, and alkaloids, were involved in wheat scab resistance.
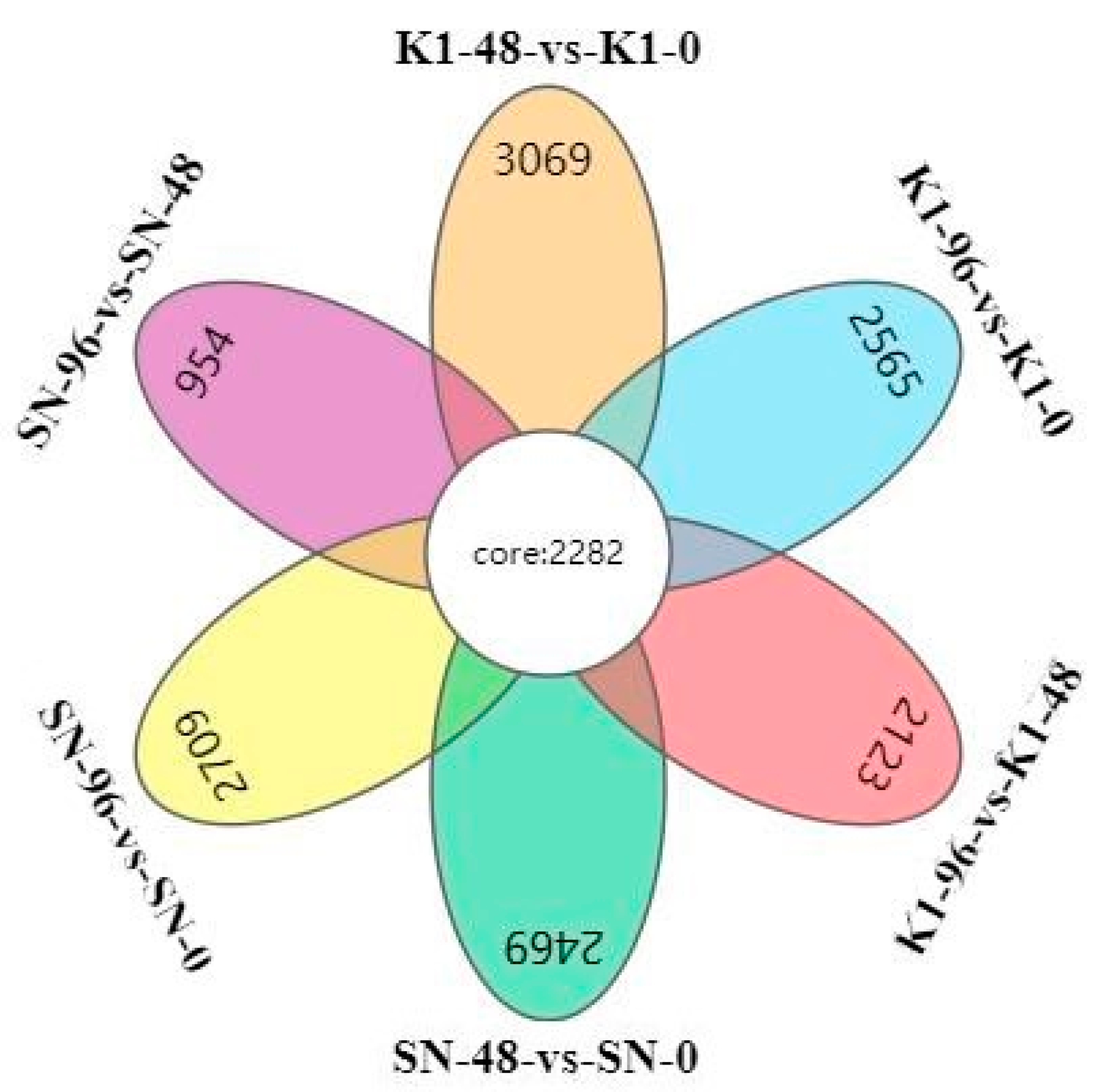
Although a few studies have been conducted on FHB resistance using transcriptome sequencing, the disease resistance mechanism is still not very clear because of the huge wheat genome and the complicated traits of FHB resistance. Therefore, further studies are required. Moreover, most previous studies used materials with high resistance and high sensitivity because of their ease of study, few studies used medium-sensitivity (MS) and medium-resistance (MR) materials. In fact, there are few varieties with high and medium resistance in production, most of which are highly susceptible and moderately susceptible. Therefore, the mechanism of resistance should be further studied. Meanwhile, the release of the complete wheat genome sequence and detailed annotations allows for the exploratory analysis of DEGs, specifically in known FHB resistance QTL/gene regions. Therefore, to explore the molecular differences in FHB resistance between MS and MR wheat strains, two special genotypes, Shannong 102 (MS) and Nankang 1 (MR), were inoculated with F. graminearum. The inoculated spikelets were analysed at 0, 48, and 96 h using RNA sequencing technology. The results will provide insight into the genetic basis of moderate FHB resistance and suggest a molecular strategy for breeding wheat cultivars with this trait. Furthermore, this study provides new potential genes for breeding applications.
3. Discussion
Facing the increasingly serious epidemic of wheat scab, the scab-resistant varieties have become the focus of wheat breeding research. Wheat scab resistance is a quantitative trait controlled by multiple genes, and there are some major disease resistance genes [
13]. Using different types of genetic mapping techniques, the main QTL/genes, including
Fhb1 to
Fhb7, associated with resistance to scab, have been finely mapped, and a series of molecular markers has been developed for assisted selection breeding [
18,
19,
20,
21,
22,
23,
24,
25]. Using marker-assisted backcross, Ma et al. [
1] transferred
Fhb1,
Fhb2,
Fhb4, and
Fhb5 from Wangshuibai to 40 excellent varieties introduced from different wheat-producing areas and cultivated more than 70 FHB-resistant lines with different QTL combinations. Compared with their recurrent parents, the lines carrying
Fhb4 and/or
Fhb5 showed significantly better type I resistance, and the lines carrying
Fhb1 and/or
Fhb2 showed significantly better type II resistance. The breeding lines carrying these four genes showed significant improvements in resistance, which was equivalent to that of Wangshuibai and Sumai 3 [
1]. Four FHB resistance genes,
Fhb1,
Fhb2,
Fhb4, and
Fhb5, were polymerised into the same susceptible variety, Aikang 58, and the disease resistance of some lines was close to that of Sumai 3 and Wangshuibai [
26]. Molecular marker detection showed that Shannong 102 contained
Fhb2 and
Fhb4, and Nankang 1 contained
Fhb1,
Fhb4, and
Fhb5 in our study. Some lines from the recombinant inbred line (RIL) population derived from Shannong 102 and Nankang 1 showed high resistance, and the type II resistance was close to that of Sumai 3 and Wangshuibai. Elite lines with high resistance to scabs from the RIL population were identified (unpublished data). These results indicate that the gene aggregation of FHB resistance can improve FHB resistance, but the mechanism by which the genes interact to increase resistance remains unclear.
At present, most popularised cultivated varieties are moderately or highly susceptible to scabs. Highly resistant varieties, such as Sumai 3 and Wangshuibai, have failed to be popularised in a large area because of their poor agronomic characteristics. Although the agronomic characteristics of Yangmai 158 and other moderately resistant varieties are good, disease is still inevitable in years of severe FHB epidemics. Among them, Shannong 102 was selected for this experiment, with good agronomic characteristics and medium sensitivity to scab. The average yield per hectare in a 3-year regional experiment was 8316 kg. The genetic population constructed by crossing Shannong 102 and Nankang 1, with medium resistance to scab, combined with molecular marker-assisted selection and agronomic characters, can obtain lines with excellent resistance and good agronomic characteristics. Therefore, the combination of molecular marker-assisted breeding and traditional breeding will play an increasingly important role in wheat breeding in the future [
2]. At present, gratifying progress has been made in the improvement of wheat scab resistance, but the final solution to this problem is limited because its resistance mechanism is not clear [
1]. In the future, we should not only increase the breeding of scab resistant varieties, but also strengthen research on the resistance mechanism to provide a theoretical basis for the improvement of wheat scab resistance [
2].
Wheat is allohexaploid and contains three genomes, A, B, and D. The genome is huge (three ancestral genomes, approximately 17,000 Mbp [
27]. Therefore, it is not easy to carry out genetic research on complex traits, which also greatly hinders the research on FHB resistance mechanisms. In recent years, RNA-seq technology based on the Illumina sequencing platform has become a powerful tool for the rapid and comprehensive establishment of plant basic molecular platforms [
28,
29] and also provides convenience for exploring the mechanism of wheat scab resistance [
30,
31]. In this study, the transcriptomes of Shannong 102 and Nankang 1 at 0, 48, and 96 h after inoculation were sequenced, and the gene expression in response to scab was obtained.
Plant disease resistance is a fine molecular process regulated by a gene network that involves cellular and molecular events and signalling pathways. Functional annotation of the DEGs was performed. The most common classifications are the defence-related plant hormone synthesis and pathways [
32,
33], phenylpropionic acid synthesis pathway [
34], production and clearance of reactive oxygen species, synthesis of antibacterial compounds [
35], detoxification, and cell wall reinforcement [
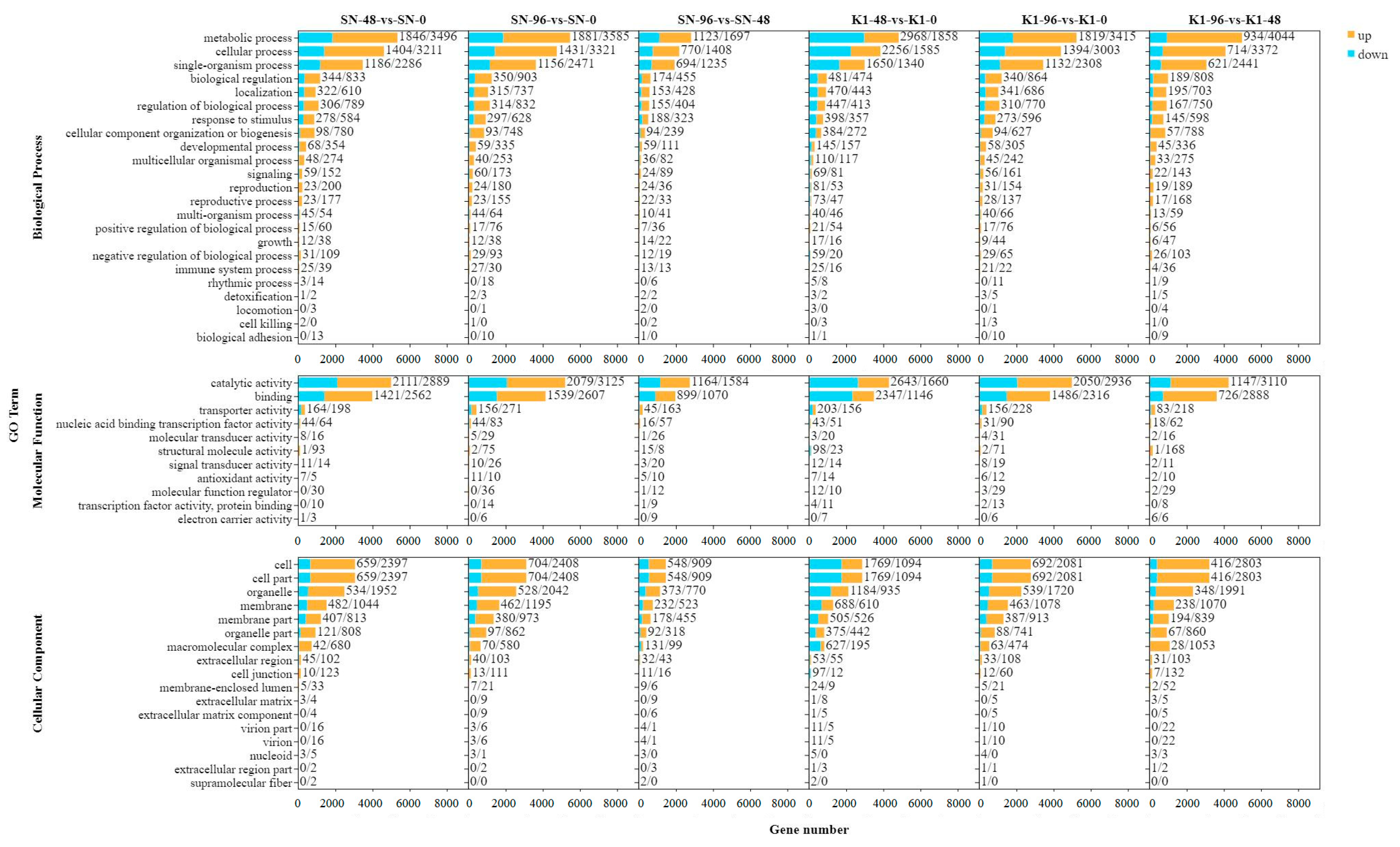
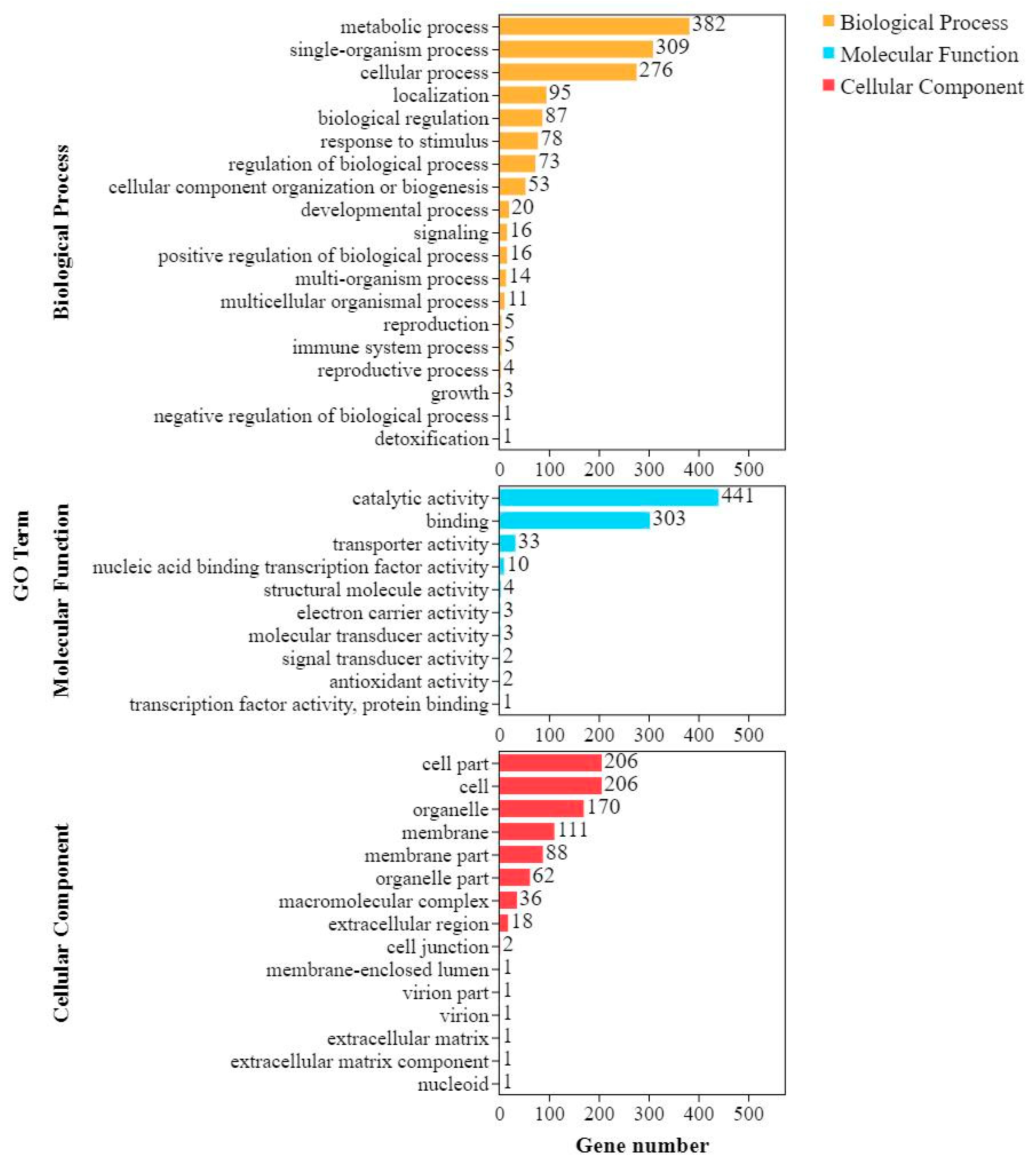
36], among others. After infection, many of these pathway products were upregulated or inhibited, indicating that they are involved in the defense response. The pathways of the upregulated genes of Shannong 102 and Nankang 1 were consistent. BPs included biological regulation, response to stimulus, immune system processes, and the positive regulation of biological processes. The classification of CCs included cells, organelles, and membranes, and the MFs included catalytic activity, binding, and transporter activity. Some of these were similar to those in previous research.
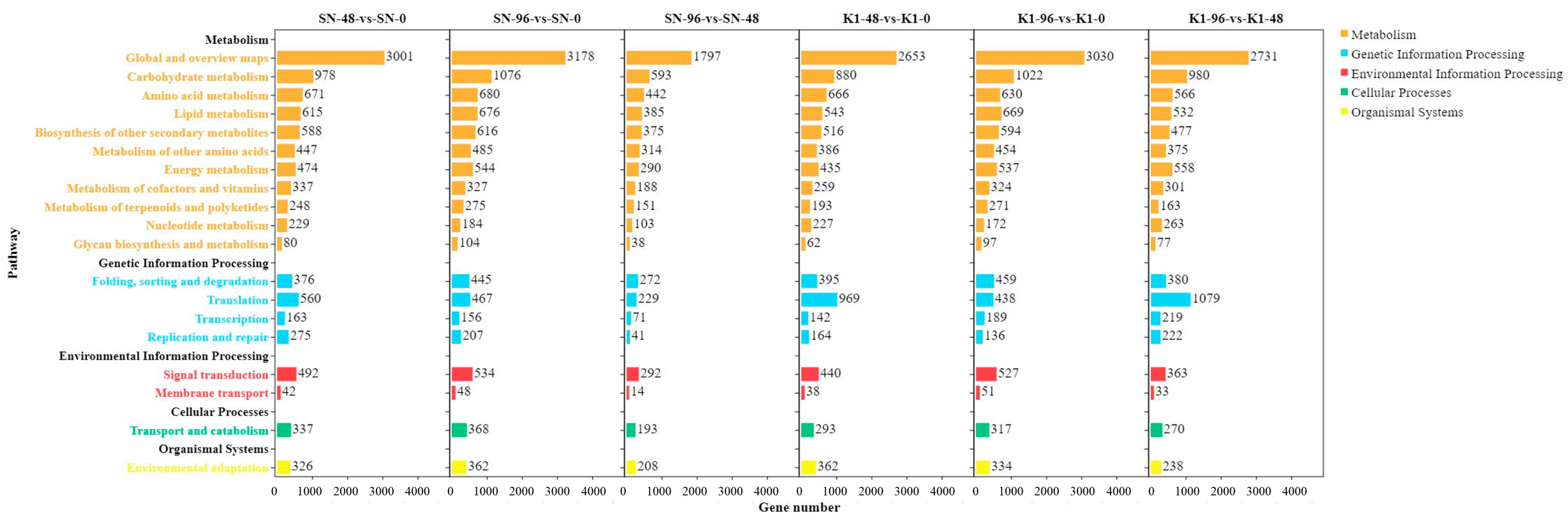
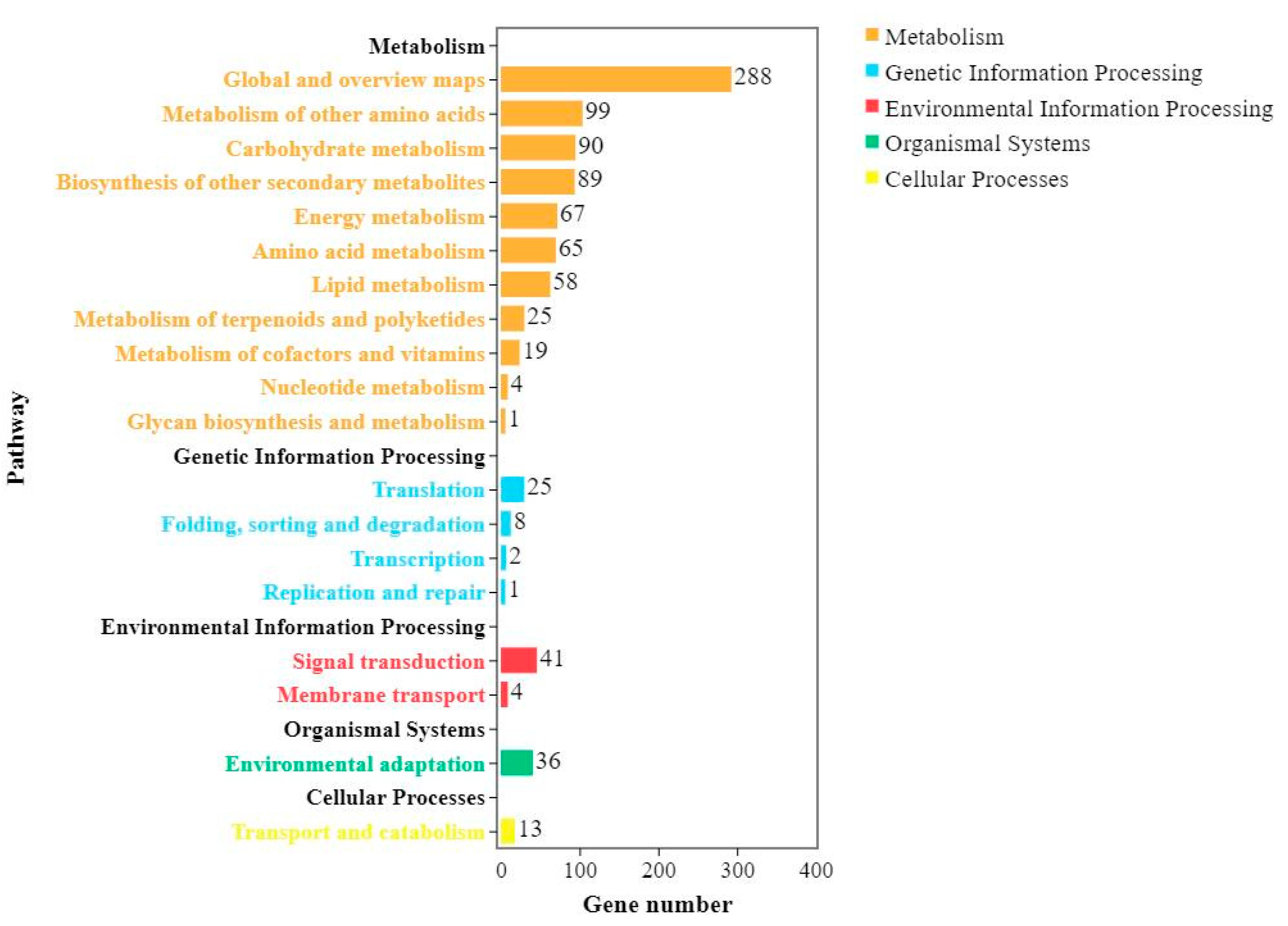
The pathways involved in the KEGG analysis of differential expression might be related to the resistance of Shannong 102 and Nankang 1 to scab, and both contained two highly enriched pathways, that is, biosynthesis of secondary metabolites (ko01110) and metabolic pathways (ko01100). This provides a reference for research on the FHB resistance mechanism of Shannong 102 and Nankang 1. A previous study showed that 70 pathways are involved in metabolism after
F. graminearum infection using Xinong 979, based on transcriptome analysis [
8]. The first four main pathways are the protein processing pathways in the endoplasmic reticulum, photosynthesis-antenna protein pathway, starch and sucrose metabolism pathway, and plant hormone signal transduction pathway [
16]. This is similar to our results. However, the differences in the first few pathways with the highest enrichment led to differences in the FHB resistance mechanisms between Nankang 1 and Xinong 979.
Some different pathways that accumulated in one of the two genotypes were upregulated or downregulated, which is more likely to be related to disease resistance or susceptibility [
1]. Therefore, in the transcriptome analysis of Shannong 102 and Nankang 1, we focused on the differences in DEGs between the two strains and found that Nankang 1 showed alterations in glutathione metabolism (ko00480), phenylpropanoid biosynthesis (ko00940), plant–pathogen interaction (ko04626), plant hormone signal transduction (ko04075), and other pathways, with respect to the number of upregulated genes. Transcription products, proteins, and metabolites related to the phenylpropanoid pathway, such as phenylalanine ammonia lyase (PAL) and hydroxycinnamic acid amides, are only induced in disease-resistant strains or induced with higher abundance. Gunnaiah and Kushalappa [
37] showed that the phenylpropanols related to disease resistance that accumulate in Sumai 3 mainly consist of syringyl-rich monomeric alcohols and their glycosides, which are lignin and antibacterial plant antitoxins. Biosynthetic precursors, therefore, might play an important role in inhibiting the penetration and growth of fungi. In addition, because SA can be synthesised through the PAL pathway to respond to pathogen attacks, the phenylpropanoid pathway is related to the SA signalling pathway.
A pathogenesis-related protein refers to a type of water-soluble protein produced by plants after infection by pathogens or stimulation with non-biological factors. In recent years, researchers have discovered that PR protein genes also play an important role in the disease resistance of crops. In a study of FHB, glucanase and chitinase were found to play an important role in the resistance and defense response of wheat to FHB, and they can play a direct role in the “damage of fungal structural barrier” or indirectly through the “activity of fungal cell wall damage products” [
38]. Li et al. [
39] used the resistant variety Sumai 3 and susceptible variety Y1193-6 as experimental materials to study their difference of FHB resistance and found that some PR genes of the two materials were upregulated after inoculation.
In this study, many PR protein genes were differentially upregulated, and these were enriched in the plant–pathogen interaction (ko04626) pathway. The gene expression showed an increasing trend with the inoculation time, indicating that PR protein genes play an important role in regulating plant disease resistance after
F. graminearum infection. The expression levels of PR-1 and PR-2 (β-1,3-glucanase), PR-3 (chitinase), PR-4 (he-vein-like protein), and PR-5 (thaumatin-like protein) peaked at 36 to 48 h using Sumai 3 and Wheaton as experimental materials. However, there was no significant difference in the expression of PR protein genes between Sumai 3 and Wheaton [
40]. However, in this study, it was found that most of the disease course-related proteins or disease resistance genes screened from the two wheat materials, Nankang 1 and Shannong 102, were upregulated and differentially expressed between the two strains, and there was a large difference in the gene expression between these two genotypes, which was not consistent with the results of Pritsch et al. [
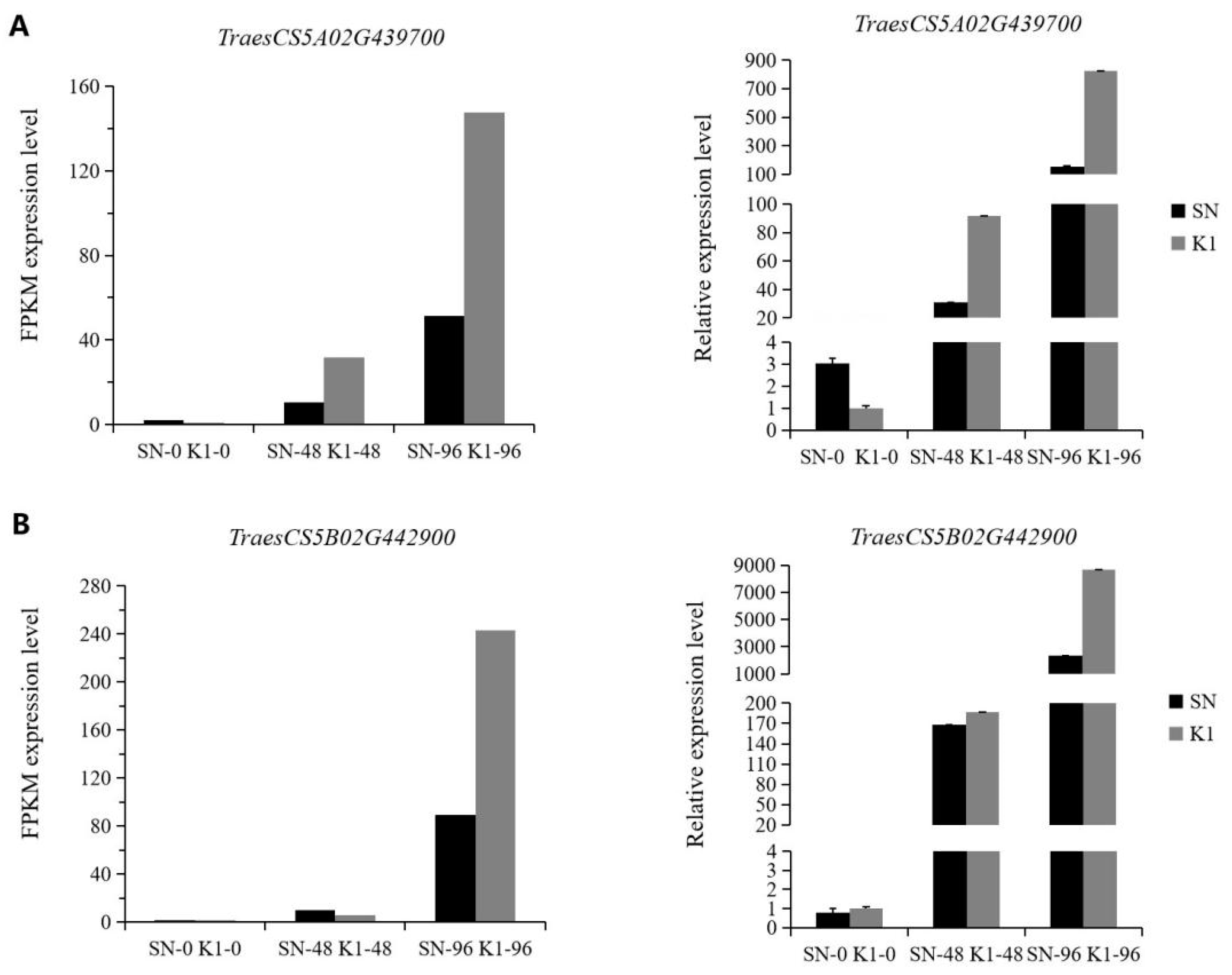
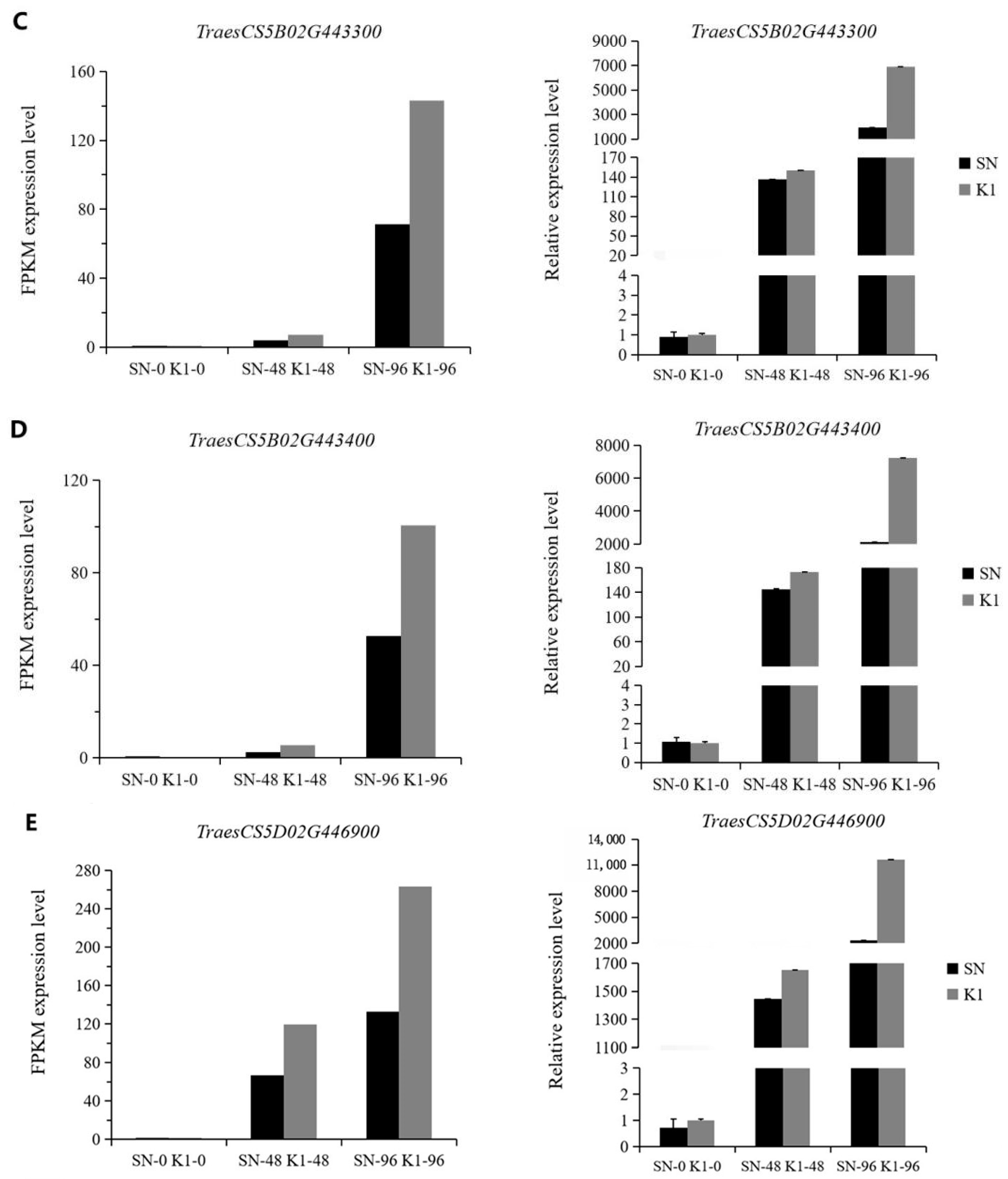
40]. To verify the accuracy of the transcriptome data, we selected five PRMS (pathogenesis-related protein 1)-related genes in the plant–pathogen interaction (ko04626) pathway for RT-qPCR analysis. The results of this were consistent with the transcriptome results, that is, the upregulated or downregulated gene expression trend was the same between them. This indicated that the transcriptome test results were more reliable. Pan et al. [
14] found that all PR1, PR1-1, and PR-4 genes were upregulated by
F. graminearum, but none of them were expressed at higher levels in any resistant genotype, relative to levels in the susceptible Shaw strain. However, in our study, PR-related genes were also upregulated, but the expression levels were different between the two genotypes, which was different from the previous results [
14]. This could be caused by the difference in strains with different known FHB QTLs/genes or gene interactions. PR genes can be expressed, or not, which might be closely related to the genetic background of the plant lines. In addition to finding that PRM-related genes are closely related to wheat disease resistance, the overexpression of RPS2 (disease resistance protein RPS2) and RPM1 (disease resistance protein RPM1) were found to provide broad-spectrum disease resistance in rice [
41]. In this study, it was found that two RPS2 protein gene upregulation events were identified in the plant–pathogen interaction pathway, indicating that they might be related to resisting invasion by
F. graminearum and play a positive regulatory role in this process. However, the specific mechanism needs to be further studied in the future.
4. Materials and Methods
4.1. Plant Materials
Two wheat (
T. aestivum L.) genotypes were used in this study, Shannong 102 (a cultivar with MS to FHB) and Nankang 1 (a line with MR to FHB). Seeds were kindly provided by Dr. Jichun Tian (Shandong Agricultural University). After screening two genotypes using the special molecular markers for Fhb1, Fhb2, Fhb4, and Fhb5 (
Figure S1), it was found that Nankang 1 had the QTL/gene regions of Fhb1, Fhb4, and Fhb5, whereas Shannong 102 had the QTL/gene regions of Fhb2 and Fhb4 (
Table S1).
The seeds of the two genotypes were germinated and vernalised for an additional 4 weeks (4 °C, 12 h light/dark regime) before being transferred to the greenhouse. Plants were potted in a mixture of compost, sand chalk, and common soil. Each sample was planted in an individual pot (30 cm in diameter and 25 cm in depth, with three seedlings) and with 10 replicates (pots). The temperature of the greenhouse was gradually increased from 15 °C/13 °C during the day/night to 20 °C/18 °C, and a 16 h/day photoperiod was used at the time of anthesis.
In this study, mixed conidiospore suspensions of 7136, F301, F609, and F15 virulent strains of F. graminearum were obtained from the courtesy of Nanjing Agricultural University. The pathogen was inoculated in mung bean medium and vortexed at 150 rpm at 25 °C for 4–5 days. After culturing and filtering, the mass of conidia was examined under a microscope. Then, the four pathogen strains were mixed equally and stored at 4 °C for later use. Wheat was inoculated with 20 μL of the F. graminearum conidia suspension (4 × 105 spores/mL), applied to a pair of florets in the middle of the spike (or 1/2 position of the spike) during flowering. The entire wheat spike was then covered with a self-sealing bag to retain moisture and sprayed with water 1–2 times per day, and the self-sealing bag was removed after 3 days.
4.2. Sample Preparation for RNA-Seq Analysis
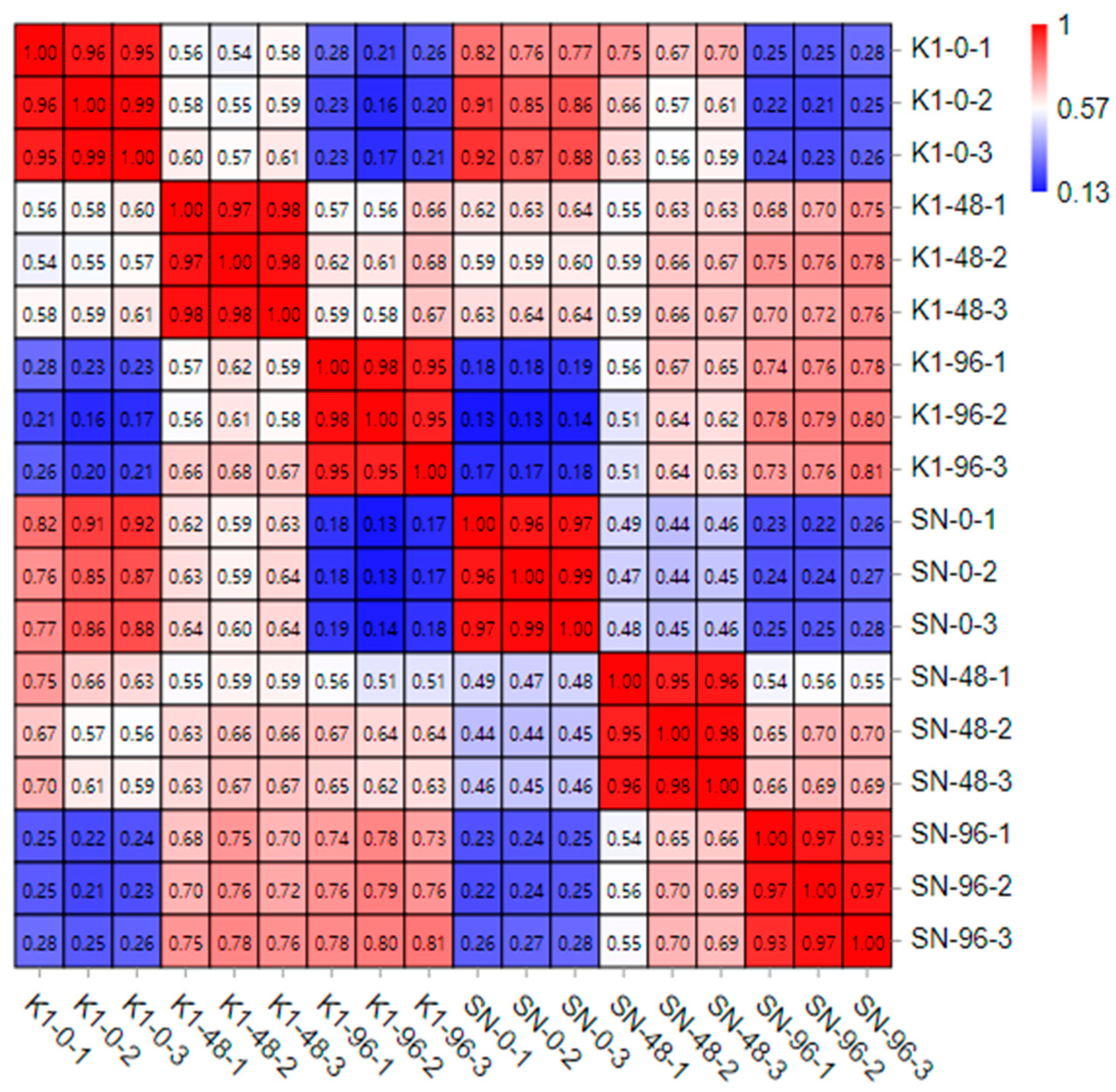
The entire spikes of the two genotypes were collected at 0 h (inoculated with fresh sterile water), 48 h, and 96 h after inoculation with three biological replicates, and these were named SN-0, SN-48, and SN-96 for Shannong 102 and K1-0, K1-48, and K1-96 for Nankang 1. In total, 18 samples of the two genotypes were collected.
4.3. RNA Extraction and Sequencing Analysis
Total RNA was extracted using TRIzol reagent (Life Technologies, Carlsbad, CA, USA), followed by treatment with DNase I (Ambion, Austin, TX, USA), according to the manufacturer’s protocol.
The Illumina Novaseq 6000 (Illumina, Inc. 9885 Towne Centre Drive, San Diego, CA, USA) was used to sequence the cDNA library. Single-end sequencing ensured that the sequencing depth of each sample exceeded 10 Gb. RNA-Seq was performed by Gene Denovo Co. Ltd. (Guangzhou, China).
4.4. RNA-Seq Data Analysis and Bioinformatics Analysis
FASTP (version 0.18.0) [
42] was used to filter the raw data to obtain clean data. The BWA software (version 0.7.12) [
43] was used with the mem algorithm to compare the filtered reads to the Chinese spring wheat reference genome (IWGSC_RefSeq_v1.1). After the comparison, the results were marked with the software Picard (version 1.129), as well as statistical marking of the depth and coverage of the reads.
4.5. Differential Expression Gene Analysis and Gene Functional Annotation
The fragments per kilobase of transcript per million fragments mapped value [
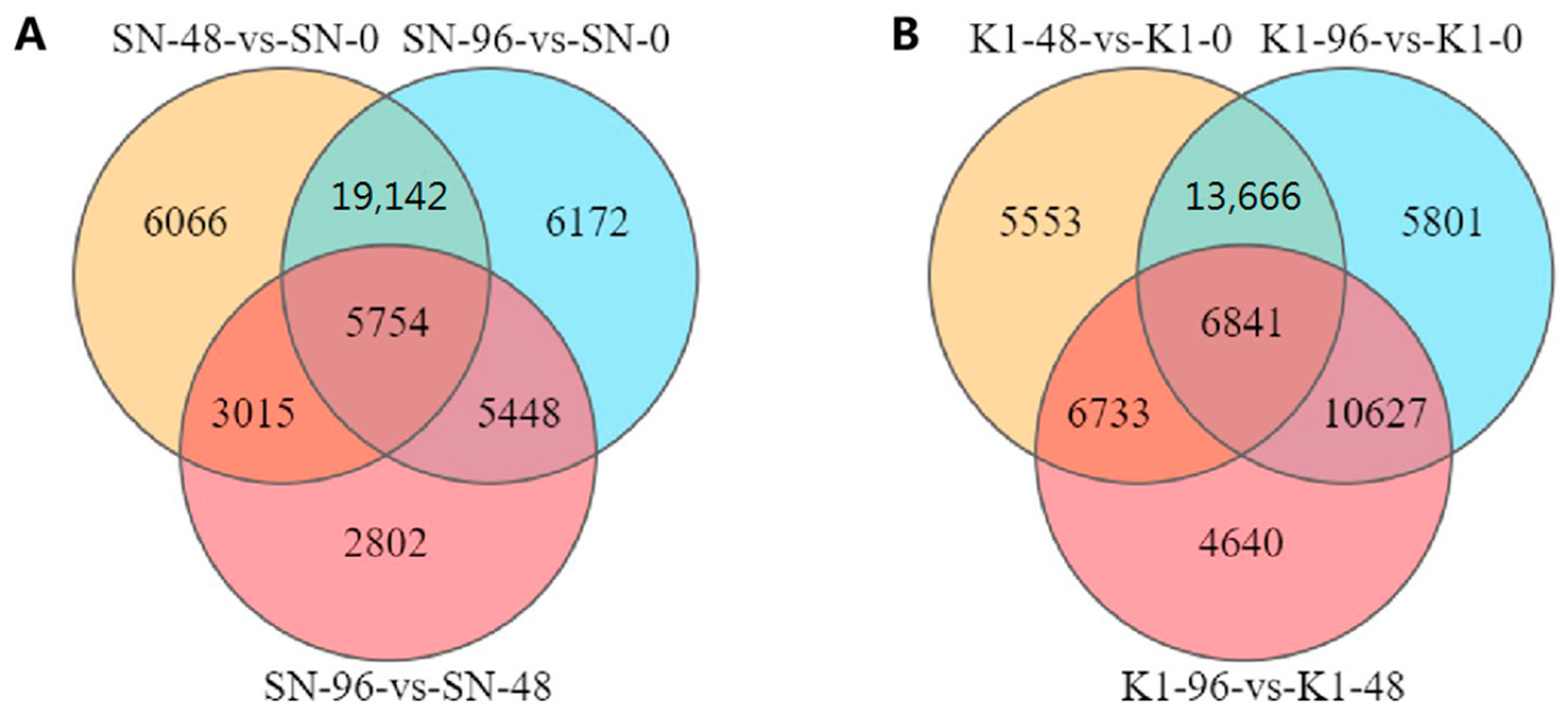
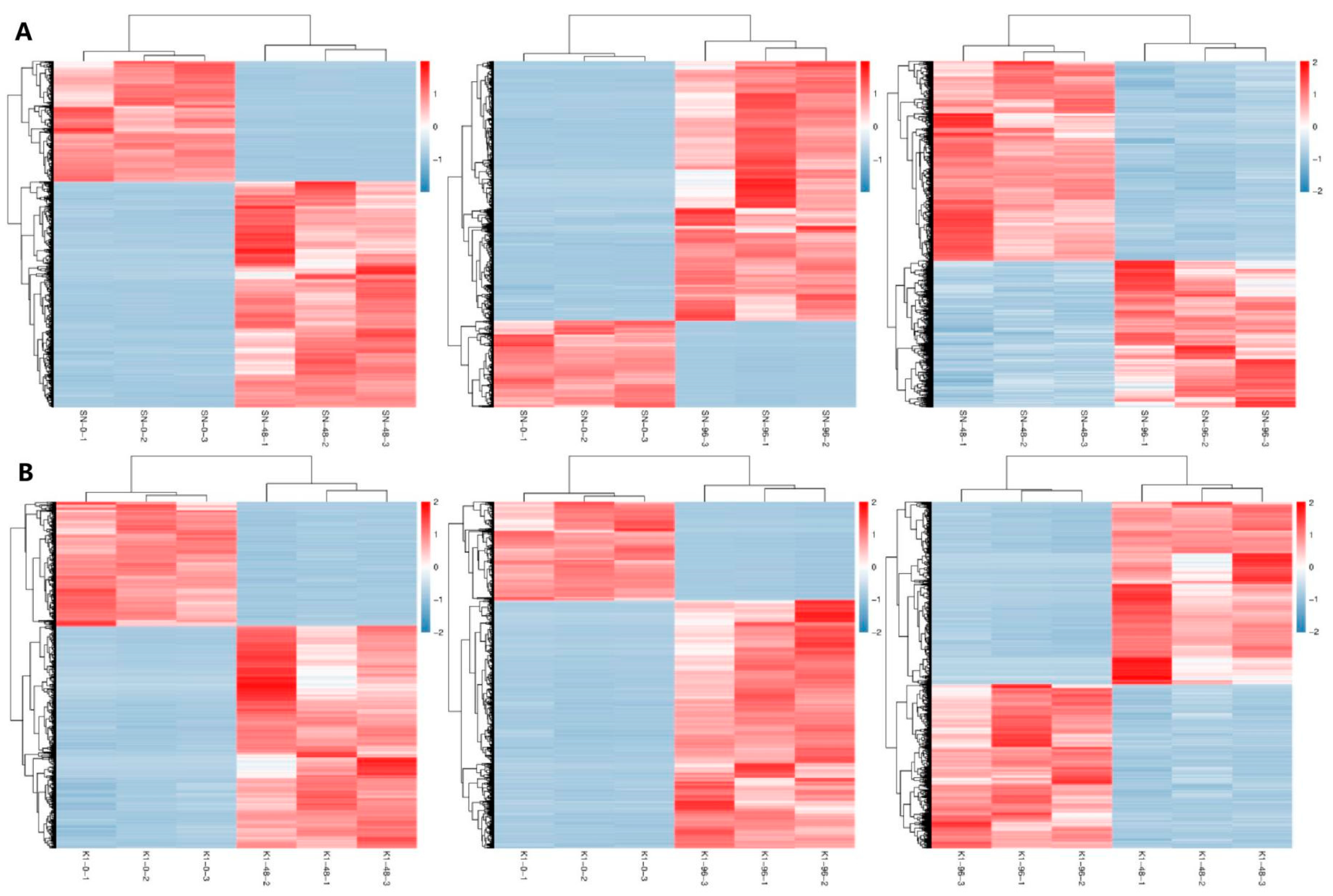
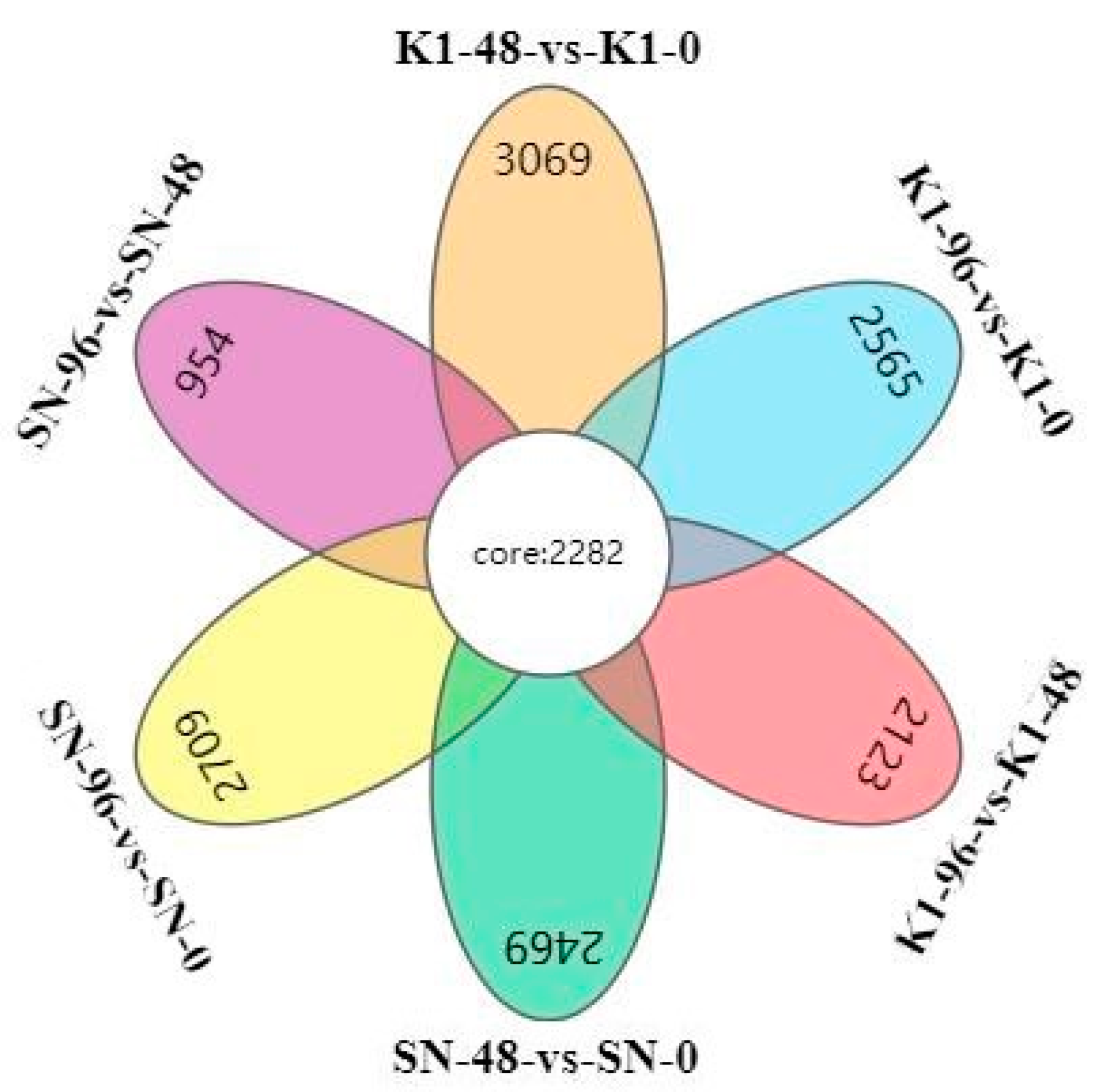
44] was used to reflect the gene expression and analyse the differential genes. The differential expression of genes between the two groups was analysed using the DESeq2 (version 1.30.0). A false detection rate (FDR) < 0.05, and |log2fold-change (FC)| > 1 were used as the screening criteria to obtain the DEGs between the two samples.
The DEG sequences were compared with the Gene Ontology (GO) database (
http://www.geneontology.org/; accessed on 28 March 2022), and the function was annotated. The DEGs were also compared with the Kyoto Encyclopedia of Genes and Genes (KEGG) database using blastx, and the corresponding pathway annotation information was obtained.
4.6. Real-Time Quantitative PCR (RT-qPCR) Analysis
The DEGs in pathways related to plant disease resistance and immunity were selected for real-time fluorescent quantitative PCR verification (
Table 5). Actin (
https://www.ncbi.nlm.nih.gov/nuccore/AB181991, accessed on 16 December 2022) was used as the internal reference gene, and the RT-PCR kit from Vazyme Company (Q341) was used. The PCR reaction system is presented in
Table 5. PCR reaction conditions were 95 °C for 90 s, and 95 °C for 5 s, 60 °C for 15 s, and 72 °C for 20 s, for 40 cycles. Melting curve analysis was performed with a temperature gradient of 65–95 °C. The primers used for RT-qPCR are listed in
Table 6. The data were processed according to the 2
−ΔΔCt method, and each sample was subjected to four technical replicates.


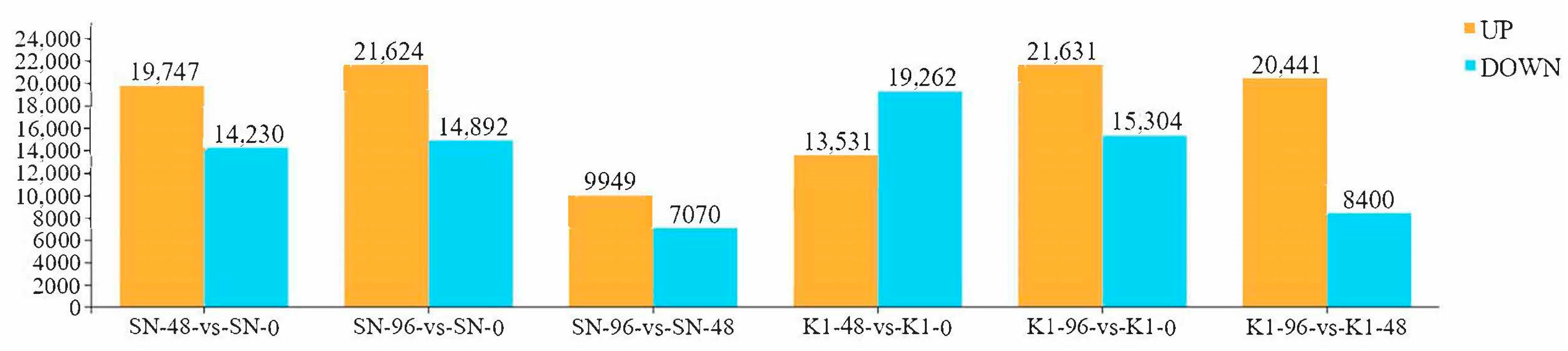

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}