Abstract
NFIX, a member of the nuclear factor I (NFI) family of transcription factors, is known to be involved in muscle and central nervous system embryonic development. However, its expression in adults is limited. Similar to other developmental transcription factors, NFIX has been found to be altered in tumors, often promoting pro-tumorigenic functions, such as leading to proliferation, differentiation, and migration. However, some studies suggest that NFIX can also have a tumor suppressor role, indicating a complex and cancer-type dependent role of NFIX. This complexity may be linked to the multiple processes at play in regulating NFIX, which include transcriptional, post-transcriptional, and post-translational processes. Moreover, other features of NFIX, including its ability to interact with different NFI members to form homodimers or heterodimers, therefore allowing the transcription of different target genes, and its ability to sense oxidative stress, can also modulate its function. In this review, we examine different aspects of NFIX regulation, first in development and then in cancer, highlighting the important role of NFIX in oxidative stress and cell fate regulation in tumors. Moreover, we propose different mechanisms through which oxidative stress regulates NFIX transcription and function, underlining NFIX as a key factor for tumorigenesis.
1. Introduction
The nuclear factor I (NFI) family of transcription factors controls the expression of several genes that play a role in various cellular processes (e.g., proliferation, migration, and differentiation) during normal development, as well as in the context of disease, including cancer [1,2]. This family includes four closely related genes, NFIA, NFIB, NFIC, and NFIX, present in human chromosomes 1p31.2-p31.3 (NFIA), 9p24.1 (NFIB) and 19p13.3 (NFIX, NFIC), whose proteins share a highly conserved N-terminal DNA-binding and a dimerization domain [3]. The binding of NFI proteins to DNA occurs in the form of homodimers or heterodimers, which increases the diversity of targets for these transcriptional regulators [4]. Additionally, a recent study on the interaction between a large universe of transcription factors showed that 118 out of the 202 interactions analyzed involved members of the NFI family [5]. This suggests that NFI family members may play a variety of roles in the regulation of transcription, either acting directly as activators or repressors or by interacting with other proteins, namely transcription factors, to modulate their function [1].
While NFI family members share several common features, allowing for compensatory roles [6,7], they also have specific regulatory functions [8,9]. Important information about these specific roles comes from the analysis of knockout mice for different NFI family members [10]. For example, NFIA, NFIB, and NFIX play an important role in glial and neuronal differentiation in the central nervous system [10], while NFIC plays a specific role in tooth development [8], and only NFIX plays a role in muscle development [9]. Not surprisingly, alterations in the expression of these genes can lead to several pathologies, including developmental defects and cancer [2,10]. This multi-faceted family of transcription factors has also been implicated in the regulation of epigenetic modifications in various ways, possibly due to their transactivation domain that interacts with histones H1 and H3 or through the binding and modulation of the activity of different chromatin modifiers [11,12,13]. The global effect of NFI-chromatin interactions seems to be the increase in chromatin accessibility and gene expression [14,15]. Likewise, NFI proteins were shown to positively regulate transcription by recruiting histone acetylases and nucleosome remodeling enzymes (e.g., NURF) and to drive an increase in active chromatin modifications, such as H3K4me3 and H3K36me3 [16].
Apart from the role that NFI transcription factors play in regulating gene expression, they are themselves regulated at different levels. This regulation may occur at (i) the transcriptional level, (ii) the post-transcriptional level via alternative splicing, (iii) the mRNA stability and translational level, regulated by different non-coding RNAs, and (iv) the post-translational level [1,10,17,18,19]. Transcriptional regulation may occur, for example, through the action of paired box gene 6 (PAX6) or empty spiracles homolog 2 (EMX2), two transcription factors that allow the transcription of NFI family members [1,17,18]. NFI family regulation by non-coding RNAs has been addressed particularly in the context of cancer and includes: (i) microRNAs (miRNAs), which are 18–25 nucleotide long abundant non-coding RNAs that inhibit translation or promote degradation of messenger RNAs (mRNAs) and can stimulate proliferation and migration [20,21,22,23,24,25]; (ii) circular RNA (circRNA), which are non-coding RNAs, most of them originated from protein-coding exons that sponge and regulate the activity of miRNAs or serve as protein decoys to recruit and modulate the transcription and translation of downstream target genes [26,27,28]; and (iii) long non-coding RNAs (lncRNAs), which are functional 200 nucleotide transcripts that mainly modulate transcription through a variety of epigenetic mechanisms, post-transcriptional processing via cross-talk with other RNA species, or modulate gene expression through lncRNA-protein interactions [29,30]. Finally, an example of a post-translational modification of the NFI family transcription factors is the conserved cysteine residue that has been shown to undergo oxidative inactivation [19]. This oxidative inactivation of NFI transcription factors, proposed as important for cellular responses to oxidative stress, can be reverted by the glutathione antioxidant pathway [31]. Although more and more examples of this multilevel regulation of NFI transcription factors are being discovered, the choice of downstream target genes and pathways, which are decisive for developmental and disease processes, still needs to be clarified.
Cancer is a complex disease characterized by multiple events known as hallmarks [32,33]. These hallmarks are associated with a profound change in the cell’s expression profile, allowing cancer cells to acquire the ability to proliferate, migrate and regain certain characteristics of stem cells. The increased plasticity of cancer cells often correlates with a blockage in differentiation, which, in its turn, depends on alterations in the expression of transcription factors that play key roles during development, such as the HOX, SOX, and PAX families [32]. The co-option of developmental pathways during cancer onset and progression has been described [32,34,35,36], raising the possibility that NFI proteins may play a relevant role in cancer [2,7,10,11,17].
In this review, we build on the existing knowledge about the role of NFIX during development to examine its role in cancer. Thereafter, in the context of cancer, we will focus on how NFIX relates to oxidative stress and alters cell fate and how that impacts tumor progression.
2. NFIX Roles in Development
To understand the role being played by NFIX in cancer, it is important to characterize its function during development. During mouse fetal myogenesis, NFIX activates specific fetal muscle genes, such as those encoding enolase-β (Eno3) and muscle creatine kinase (Ckm), both known downstream targets of the NFIX pathway [9] (Figure 1A). The transcription factor PAX7 (a key muscle stem cell marker) binds to the Nfix promotor to activate its expression. Nfix transcription also occurs through the action of JUNB (a member of the AP-1 family of transcription factors), which binds to the Nfix promotor via an unknown mechanism [37]. JUNB is activated downstream of ERK kinase signaling, which is low during embryonic myogenesis but increases at the beginning of fetal myogenesis due to a decrease in the RhoA/ROCK axis [37]. The activation of NFIX is, thus, downstream of a switch between RhoA/ROCK and ERK signaling, which occurs precisely at the onset of fetal myogenesis (Figure 1A) [37]. NFIX, in its turn, activates the expression of the fetal muscle-specific genes and inhibits the transcription of embryonic muscle-specific genes, such as slow myosin heavy chain (slow MHC, encoded by Myh7), marking the transition between embryonic and fetal muscle development [9].
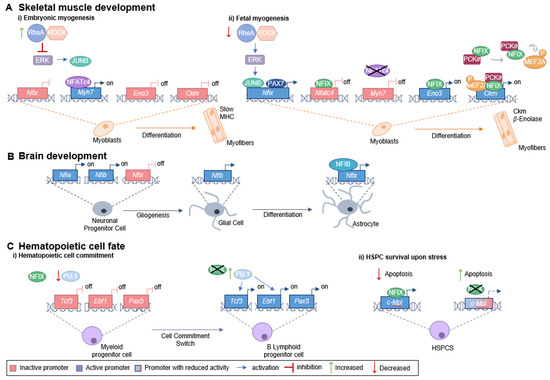
Figure 1.
Role of NFIX in development. (A) NFIX promotes the switch between embryonic and fetal myogenesis: (i) During embryonic development, embryonic muscle-specific genes are expressed, including Myh7 (encoding slow MHC), which is activated by NFATc4 that binds to the Myh7 promoter. The RhoA/ROCK axis promotes the embryonic identity of myoblasts through the repression of ERK kinases, JUNB and NFIX. (ii) In the transition from embryonic to fetal muscle development, RhoA/ROCK activity decreases, which leads to increased ERK activity and subsequent activation of JUNB. The transcription factor PAX7 also binds to the Nfix promoter activating its transcription. NFIX binds to the Nfatc4 promotor inhibiting Nfatc4 expression and, consequently, slow MHC is not produced. On the other hand, NFIX activates fetal-specific genes, such as Ckm and Eno3 (which encodes β-enolase), its downstream targets. NFIX binds directly to the Eno3 promoter, activating its transcription, while activation of the Ckm promoter involves a MEF2A/NFIX/PKC𝜃 complex. (B) Glial cell differentiation is promoted by NFIX: the expression of Nfia and Nfib by neuronal progenitor cells leads to a gliogenic switch. Then, NFIB binds the Nfix promoter region activating its transcription, and, in its turn, NFIX activates the astrocytic genes leading to astrocyte differentiation. (C) NFIX regulates hematopoietic cell fate: (i) During a stressful event, NFIX activates the c-Mpl promoter directly, leading to a reduction in the apoptosis of hematopoietic stem and progenitor cells. (ii) NFIX also prevents early B cell development and favors myeloid differentiation. PU.1 regulates the transcription factors E2A (encoded by Tcf3), EBF (encoded by Ebf1), and PAX5. In the presence of NFIX, PU.1 levels are lower, and therefore, the expression of Tcf3, Ebf1, and Pax5 decreases, favoring myeloid fate. When NFIX is absent, the expression of PU.1 increases, enabling the activation of key genes required for B cell lymphoid lineage commitment (Tcf3, Ebf1, and Pax5).
NFIX is normally not expressed in adult muscle stem cells (satellite cells), but its abnormal activation is associated with disease progression, namely in the context of muscular dystrophies [38]. Muscular dystrophies are a group of diseases characterized by loss of muscle mass, fibrosis, and chronic inflammation, which are frequently associated with an increase in oxidative stress [39,40,41,42]. The deleterious role played by NFIX in the context of muscular dystrophies has been associated with consecutive cycles of regeneration and degeneration [38]. Additionally, NFIX is thought to contribute to increased oxidative stress both by driving regeneration in dystrophic muscles and by countering the switch of myofibers towards oxidative slow-twitch fibers, which are thought to reduce oxidative stress [9,38,43]. Consequently, this supports the idea that NFIX has a role in regulating oxidative stress levels in the muscle. During muscle regeneration, there is a close interaction between myogenic cells and macrophages, where NFIX regulates macrophage differentiation [44]. Under this scenario, injury-activated satellite cells attract blood monocytes that infiltrate into the damaged muscle and differentiate into pro-inflammatory macrophages, which, in their turn, stimulate myoblast proliferation [44]. Macrophages then switch to an anti-inflammatory phenotype, which sustains myogenic differentiation [45]. This phenotypic change is controlled by NFIX, which becomes activated in response to the inhibition of the RhoA/ROCK axis and by the induction of phagocytosis, a necessary feature for the acquisition of anti-inflammatory phenotype [44]. The anti-inflammatory phenotype enhances tissue repair and promotes fibroblast proliferation, which may lead to fibrosis and is highly detrimental to dystrophic muscles [44,46,47,48,49,50].
In addition to the crucial role in skeletal muscle development, Nfix is also expressed within the nervous system throughout embryogenesis [51]. In mouse cortical development, NFIX promotes the timely generation of intermediate progenitor cells that will originate cortical neurons through the transcriptional activation of the Insc (encoding inscuteable protein) [52]. Moreover, during mouse spinal cord development, the transition from producing neurons to producing glial cells (gliogenic switch) occurs via sequential action of NFI transcription factors [53]. In particular, during this gliogenic switch, NFIX has been shown to act downstream of NFIA and NFIB [53] (Figure 1B). NFIX has also been described to promote the differentiation of neural progenitor cells within the developing neocortex and hippocampus, triggering cell cycle exit via the transcriptional repression of SOX9 [54], a transcription factor required for the self-renewal of cortical neural progenitors [55]. Additionally, NFIX is an important regulator of proliferation and migration in the subventricular zone of the neurogenic niche during mouse embryonic development, a region that continuously generates neurons throughout adult life [54]. NFIX also plays a key role postnatally, by maintaining proliferative progenitor cells in this region, such as those expressing Pax6, Sox2, Hes1 and Hes5, and Mash1, markers for progenitor or transit-amplifying cells [54]. The regulation of proliferation and migration, mediated by NFIX, may be associated with the neuroblast chemoattractant GDNF (glial cell-derived neurotrophic factor), whose gene has been shown to be a potential target for transcriptional activation by NFIX [56]. In addition, NFIX has been implicated in the regulation of post-mitotic cell migration within the hippocampus [54] and rostral migratory stream [56].
NFIX is also involved in the proliferation and repopulation activity of hematopoietic stem and progenitor cells [2]. This transcription factor has been implicated as a modulator of hematopoietic cell fate since its expression prevents early B cell development and favors myeloid differentiation [57]. The transcription factor PU.1, known to control myeloid and early B and T-cell development, and the transcription factors E2A (encoded by Tcf3), EBF (encoded by Ebf1), and PAX5 [58], necessary for B cell lineage commitment and development into mature B cells, are altered in the presence of NFIX [57] (Figure 1C). Moreover, during a stressful event, such as a hematopoietic stem and progenitor cells transplant, NFIX regulates c-MPL (thrombopoietin receptor or myeloproliferative leukemia protein) signaling pathway, promoting the survival of the hematopoietic stem cells [59]. It does so by directly activating the c-MPL promoter, which regulates the maintenance of hematopoietic stem cells in the bone marrow niche, promotes their survival via JAK/STAT and MAPK/ERK signaling cascades, and prevents apoptosis (Figure 1C) [59].
NFIX has also been shown to play a key role in meiosis during spermatogenesis, with NFIX deficiency leading to a blockage in prophase 1 (diplotene), possibly associated with a defect in the synaptonemal complex and accumulation of DNA damage in mouse spermatocytes [60]. The possible role of NFIX as a cell cycle checkpoint regulator during human spermatogenesis was further suggested by another study where the regulation of NFIX expression during spermatogenesis was shown to be controlled by the microRNA miR-663 [23]. Silencing NFIX stimulated proliferation, possibly by increasing the expression of Cyclin A2, Cyclin B1, and Cyclin E1, and DNA synthesis, and inhibited apoptosis of human spermatogonia stem cells [23].
NFIX may also play a role in heart development, even though the data available is still scarce [61]. Interestingly, circRNAs have been indicated in several studies as playing a key role in physiological processes in various diseases, including the initiation and progression of cardiovascular diseases [62,63]. One such example is circNFIX (a circRNA derived from NFIX), which has been suggested to play a role in cardiac development and disease [64,65,66]. Moreover, the downregulation of circNFIX has been shown to lead to increased cardiomyocyte proliferation and angiogenesis [65], supporting the idea that circNFIX downregulation could be important for cardiac regeneration after injury. In addition, circNFIX counters heart hypertrophy by indirectly targeting activating transcription factor 3 (ATF3) in cardiomyocytes through binding to the microRNA miR-145-5p [64]. ATF3, a member of the cAMP response element-binding protein/ATF family, has been linked to heart hypertrophy [67,68]. Research has shown that, by regulating the miR-145-5p/ATF3 axis, circNFIX can attenuate pressure overload-induced cardiac hypertrophy [64]. Additionally, circNFIX expression is altered in response to increased levels of oxidative stress [28]. For example, in the fetal cardiomyocyte-derived H9c2 cell line, circNFIX was found to be downregulated after treatment with the pro-oxidant agent hydrogen peroxide, which correlated with reduced apoptosis [28]. Furthermore, overexpression of circNFIX promoted apoptosis in this model, possibly by reducing the cellular response to oxidative stress [28]. These results suggest that regulation of circNFIX or NFIX may impact heart development and disease through a mechanism that is linked to the oxidative stress response. The crosstalk between NFIX and oxidative stress extends beyond the heart. For example, its expression contributes to increased oxidative stress in response to optic nerve crush in the retina [69].
Taken altogether, these studies allow us to conclude that NFIX plays multiple roles during the development of a variety of tissues, influencing cell proliferation, cell fate, and differentiation. It also affects cell migration, apoptosis, and oxidative stress. These cellular processes are either well-documented hallmarks or emerging hallmarks of cancer [32,33], opening the possibility of an important role of NFIX in cancer.
3. Roles of NFIX in Cancer
Tumorigenesis is characterized by the gain of malignant properties, including sustained proliferative signaling, phenotypic plasticity, and epigenetic reprogramming, all features also observed during embryonic development [32]. Not surprisingly, several pathways that play central roles during development are also altered during tumorigenesis. This is the case of RhoA/ROCK and JUNB signaling pathways that regulate NFIX expression during myogenesis and are involved in cancer cell proliferation and invasion [70,71]. In prostate cancer cells, SOX4, a transcription factor involved in the development of various tissues and which is commonly overexpressed in tumors [72], is overexpressed and activates NFIX [73]. Moreover, the overexpression of acyl-CoA synthetase 4 in the MCF-7 breast cancer cell line leads to changes in various developmental pathways, including the overactivation of NFIX and its target gene ENO3 [74].
Apart from its positive and negative transcriptional regulation, genomic analysis of the NFIX gene in various tumors has revealed several mutations, including gene fusions [75]. Gene fusions are chromosomal rearrangements, usually involving insertions, deletions, inversions, or translocations, where two independent genes fuse together to form a hybrid gene [76]. These fusions have been studied primarily in the context of hematological and mesenchymal malignancies, but they also contribute to epithelial tumors [76]. Even though the role of NFIX in gene fusions is still not fully understood, it is likely that most of the gene fusions involving this gene have oncogenic properties (Table 1). This is the case of NFIX-MAST1 [77] fusions in breast cancer and may also include the NFIX–PKN1 translocation, described in carcinoma of the skin [78], the BSG-NFIX fusion identified in breast cancer [79] and the NFIX–STAT6 gene fusion, which was identified in a tumor lesion with histological features of a solitary fibrous tumor [75].

Table 1.
Oncogenic and tumor suppressor roles of NFIX.
To understand the role of NFIX in cancer, it is essential to know how the gene fusions, epigenetic changes, non-coding RNAs targets, and mutations in NFIX and in its regulatory elements contribute to specific pathways that drive tumor progression. This can reveal when NFIX acts as an oncogene and when it acts as a tumor suppressor (Table 1).
3.1. NFIX and Oxidative Stress
Tumors are characterized by increased levels of oxidative stress, which impact tumorigenesis in different ways, including by (i) triggering DNA damage; (ii) altering signaling pathways involved in cell proliferation and tumor growth; (iii) leading to chronic inflammation in the tumor environment; and (iv) changing the composition of the extracellular matrix, which impacts cell survival, proliferation, migration, and adhesion [41,83,84,85,86].
Members of the NFI family are thought to be pro-oxidants, and their inactivation is crucial for proper oxidative stress response [19]. NFIX may act as an oxidative stress producer, for example, by activating the transcription of CYP1A1 (encoding cytochrome P450 1A1), which has an NFI binding site in the promoter region [87]. CYP1A1 is known to be pro-carcinogenic [88] and, similarly to other monooxygenases, leads to the generation of reactive oxygen species (ROS) as part of its catalytic activity [89,90]. Under normal conditions, the expression of CYP1A1 is suppressed, possibly due to an autoregulatory loop that controls the expression of CYP1A1 via CYP1A1-based hydrogen peroxide production and the NFI family [89,91]. However, when deregulated, the increased production of ROS and the production of pro-oncogenic metabolites may contribute to tumor progression [88]. Studies have shown that CYP1A1 is upregulated in breast [91], bladder [92], and colon cancers [92]. Accordingly, the knockdown of CYP1A1 has been found to downregulate ERK and PI3K/AKT pathways and to induce the AMPK pathway, leading to a reduction in tumor progression and cancer cell survival [91]. Supporting the idea that oxidative stress impacts the function of NFI family members, hepatoma cell lines treated with the pro-oxidant hydrogen peroxide or L-buthionine- (S,R)-sulfoximine showed impaired NFI binding to its DNA binding site due to increased oxidative stress, resulting in the inhibition of its function as a transcription factor [87].
Analysis of oxidative stress-related differentially expressed genes using data from 594 lung adenocarcinoma patients revealed that NFIX is downregulated in this type of cancer and has a direct correlation with poor prognosis [93]. This study proposed that NFIX downregulation serves as a mechanism for cancer cells to reduce ROS production, thus, increasing their fitness [93]. Similarly, another study found that NFIX upregulation is associated with poor prognosis in breast cancer because of its role in ROS status [94]. This indicates that NFIX may be used as a key gene in a ROS scoring system to predict prognosis and therapeutic efficiency. NFIX has also been identified as part of the common mitochondrial defect signature genes in hepatocellular carcinoma, which are genes activated in response to mitochondrial dysfunction, a major source of ROS in organisms [95], and associated with poor prognosis and reduced overall survival [96].
Besides NFIX protein being associated with oxidative stress in different contexts, circNFIX has also been shown to have an impact on both tumor progression and oxidative stress [28,97,98]. For example, circNFIX was found to promote cancer progression by upregulating glycolysis, as well as glucose uptake in glioma [99] and in non-small cell lung cancer [100], which can lead to overproduction of ROS in the context of diabetes [101]. In glioma, tumor progression was associated with the suppression of miR-378e and consequent expression of ribophorin-II (RPN2) [99], a target of miR-378e that promotes increased ROS and glycolysis [99,102]. Similarly, in non-small cell lung cancer, tumor progression was associated with the suppression of miR-212-3p and upregulation of ADAM10 [100], a protein that has been shown to be involved in oxidative stress-related conditions, such as cancer, Alzheimer, neurodegeneration, and inflammation [103]. Further research is needed in order to understand whether NFIX’s role as a pro-oxidant contributes to ROS accumulation in tumors and therefore promotes genomic instability, increased proliferation, and differentiation. In support of this notion, studies are recognizing NFIX and its target genes/proteins that are involved in oxidative stress as potential therapeutic targets for cancer therapy [91,93,94,99].
3.2. NFIX and Cell Fate
Given the pleiotropic role of NFIX during development, it is not surprising that changes in NFIX expression can significantly influence proliferation and differentiation. Apart from NFIX’s indirect role in proliferation through its involvement in oxidative stress, NFIX has also been shown to be involved in cell cycle regulation and cell fate decisions, which are closely linked to proliferation. For example, NFIX downregulation has been shown to reduce proliferation and cell viability in lung cancer [80] but to lead to increased proliferation in the context of endometrial carcinoma [21] and colorectal cancer [20]. On the other hand, overexpression of NFIX in esophageal squamous cell carcinoma has been shown to reduce cell proliferation and induce cell cycle arrest in G1/G0 phase [25].
The role of NFIX in cancer proliferation, migration, and invasion has been linked to the expression of non-coding RNAs, namely miRNA and lncRNA (Table 1). One example is the regulation of NFIX mediated by miR-1290, which has a target site on the NFIX 3′-UTR [25] (Figure 2A). An inverse correlation between the levels of miR-1290 and NFIX protein and mRNA was observed in esophageal squamous cell carcinoma tissue samples, suggesting that miR-1290 is an oncogene that downregulates NFIX and promotes proliferation, migration, and invasion in this type of tumor [25]. Moreover, analysis of the genetic profile of colorectal cancer tissue through screening of genes that were upregulated or downregulated identified increased expression of two miRNAs, miR-1914 and miR-647, in colorectal cancer specimens and cell lines [20]. These miRNAs were shown to promote the proliferation and migration of colorectal cancer cells, functioning as oncogenes, possibly by directly targeting and downregulating NFIX (Figure 2B).
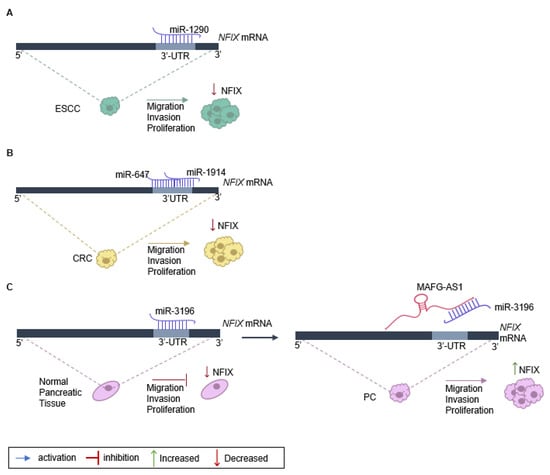
Figure 2.
Regulation of NFIX expression in cancer. (A) NFIX regulation by miR-1290 promotes esophageal squamous cell carcinoma (ESCC) progression: miR-1290 directly targets the 3′UTR sites of NFIX mRNA, negatively regulating its expression. The decrease in NFIX expression leads to ESCC cell proliferation, migration, and invasion. (B) NFIX co-regulation by miR-647 and miR-1914 in colorectal cancer (CRC): NFIX mRNA is co-targeted by miR-647 and miR-1914 in the 3′UTR. The negative regulation of NFIX expression leads to CRC cell migration and invasion. (C) ceRNA network of MAFG-AS1/miR-3196/NFIX in pancreatic cancer (PC): in normal pancreatic tissue, miR-3196 directly binds to 3′UTR sites of NFIX mRNA and silences its expression. The lncRNA MAFG-AS1, highly expressed in PC cells, binds directly to the miR-3196, promoting NFIX upregulation and, as a consequence leading to proliferation, migration, and invasion of PC cells.
The impact of NFIX on proliferation has also been associated with lncRNAs that play diverse roles in regulating gene expression [104]. Numerous lncRNAs can act as competing endogenous RNAs (ceRNAs) to regulate the expression of coding genes that have common miRNA response elements [105], with pancreatic cancer being one example. In normal pancreatic tissue, miRNA-3196 is expressed, leading to a downregulation of NFIX [30]. However, in pancreatic cancer tissue, the lncRNA MAFG-AS1 acts as a ceRNA and binds to the miR-3196, resulting in the neutralization of miR-3196 and the upregulation of NFIX [30]. Functional assays have shown that MAFG-AS1 knockdown suppresses cell proliferation and migration while promoting cell apoptosis in pancreatic cancer [30]. Additionally, when miR-3196 is up-regulated, the proliferative and migratory capacities of pancreatic cancer cells are inhibited (Figure 2C).
In addition to cell cycle regulation and cell proliferation, NFIX may also play a role in other cell fates. Apoptosis is a central pathway that is rendered inactive in cancer cells [22,59,106]. It was recently shown that NFIX overactivation has an anti-apoptotic effect via the STAT5 signaling pathway leading to a reduction in apoptosis levels in hematopoietic stem and progenitor cells [59]. This is supported by the observation that the overactivation of NFIX leads to increased expression of the anti-apoptotic factor Bcl2l1 (encoding BCL-XL) in these cells [59]. Additionally, NFIX downregulation through overexpression of miR-744-5p in ovarian cancer has been shown to decrease the expression of BCL2, an anti-apoptotic factor, leading to an increase in apoptosis levels [22]. Moreover, hematopoietic stem and progenitor cells that lack NFIX cannot survive in the bone marrow after transplantation due to an increase in apoptosis [107]. Nevertheless, NFIX silencing in the context of human spermatogonia stem cells seems to suppress early apoptosis [23], suggesting that its role in apoptosis may be tissue and/or cell-type-dependent.
Considering the important role of the NFI family in neuronal development, several studies have analyzed NFIX’s role in glioblastomas as a potential tumor-promoter [81,107,108]. One such study found that NFIX promotes glioblastoma cell migration by directly upregulating the expression of EZR (encoding ezrin), which is involved in linking the actin cytoskeleton and the plasma membrane and plays a role in cell migration [81] (Table 1). In accordance with the role of NFIX promoting cell migration, NFIX has been identified as a potential oncogene that plays a role in the development of metastasis. NFIX was recently described as a master regulator activating the expression of 17 genes that are involved in migration and invasion in lung cancer [80]. Using two different cell lines for lung cancer, it was shown that NFIX regulates interleukin-6 receptor subunit β (IL6ST), metalloproteinase inhibitor 1 (TIMP1), and integrin β-1 (ITGB1) genes, all of which are involved in cell proliferation, migration, and invasion [80] (Table 1). Altogether these studies suggest that NFIX may be a key player during cancer onset and progression, modulating several pathways implicated in tumorigenesis.
4. Discussion
NFIX has been established as a central transcription factor during development, for example, by promoting the switch between embryonic and fetal myogenesis [9] and in adulthood, being required for muscle regeneration [38,43]. The role of NFIX in mediating the switch between different cellular differentiation stages is not unique to muscle. For example, it occurs during the production of glial cells [53] or during hematopoietic cell fate [57]. This raises the possibility that NFIX is a critical factor for cell differentiation, which may be critical during tumor progression and metastasis. While some studies have suggested that NFIX may have a putative role as a tumor suppressor, most studies have identified NFIX as an oncogene (Table 1). However, the exact mechanisms that contribute to the alterations of NFIX mRNA or protein expression in cancer have not yet been fully described. One possible mechanism explaining the importance of NFIX in cancer might be dedifferentiation, which has recently been proposed as an emerging cancer hallmark [32]. In this scenario, it is possible that the control of NFIX expression leads to changes in the differentiation status and promotes a stem cell-like phenotype in cancer cells. This is in line with the role of NFIX in development, where it has been shown to control the differentiation stage of various cell types, including muscle [9,38], nervous system [54], and hematopoietic lineages [57,106,109]. The mechanisms controlling NFIX are diverse, and it is possible that several of these mechanisms may be regulated, or be regulated by, the production of ROS. Apart from the direct regulation of NFIX by ROS through the oxidative inactivation of its cysteine residues [19,31] (Figure 3i), other pathways that control NFIX activation may also be modulated by oxidative stress. For example, RhoA/ROCK and JUNB signaling pathways [70,71] or SOX4 overexpression [73], which control NFIX expression both during embryonic development and in the context of cancer, have been linked to oxidative stress. RhoA/ROCK pathway has been shown to have a bi-directional inhibitory effect of the RAC GTPase RAC1, which leads to ROS generation via NADPH oxidase [110,111,112] (Figure 3ii). This can occur during the phagocytosis of pathogens and apoptotic cells by macrophages, where ROS production may control NFIX expression [110,111,112]. Another RAC GTPase and catalytic subunit of NADPH oxidase, RAC2, has also been described to promote transcriptional activation of JUNB in lung cancer [71] (Figure 3iii), which could be yet another mechanism that leads to NFIX activation. In the scenario of high levels of ROS produced via NADPH oxidase, RhoA becomes activated and subsequently leads to the activation of the downstream targets, such as ROCK. This then allows ROCK to phosphorylate LIM kinase, leading to F-actin stabilization. With LIM kinase upregulation, MAL (megakaryocytic acute leukemia) can no longer be sequestered by actin monomers and translocates to the nucleus, where it activates SRF (serum response factor), a factor that responds to morphological changes in the actin cytoskeleton. RAC GTPases are well known for their role in cancer progression [110,113]. It is possible that NFIX is one of the downstream targets promoting tumorigenesis through increased proliferation, migration, and metastasis. SOX4 was shown to be activated via TGF-β and ROS, promoting cell senescence [114] (Figure 3iv). Another piece of evidence supporting the important role of NFIX in oxidative stress regulation is the fact that NFI I/CCAAT box transcription factor (NFI/CTF1) domain, present in the NFIX family members, was shown to interact with pirin [115] (Figure 3v). Pirin is an iron-binding protein, which is involved in iron metabolism, one of the sources of oxidative stress in organisms, and is regulated by NRF2 (nuclear factor erythroid 2-related factor 2), a major regulator of antioxidant response [116,117]. The role of pirin in cancer has been widely studied in the last decades [118]. Pirin is overexpressed in various types of cancer, such as colorectal cancer [116] and melanoma [119]. Therefore, it is possible that oxidative stress-dependent pirin activation allows its interaction with NFIX, modulating the activation of its downstream target genes. In addition, the important role that miRNA and lncRNA play in regulating NFIX expression may also be linked to oxidative stress (Figure 3vi). There is a growing body of evidence showing that miRNA and lncRNA lead to either a pro-oxidant or antioxidant response [120,121,122,123,124]. This is the case of miR-212-3p, which contributes to oxidative stress [125,126]. Finally, another link between NFIX and oxidative stress comes from circNFIX [64,65,66] (Figure 3vii). The expression of circNFIX leads to glioma progression through the increase in ROS. RPN2, which is part of an N-oligosaccharyl transferase complex, is considered oncogenic and a ROS inducer. In gliomas, miR-378e targets RPN2, suppressing its oncogenic functions, for example, by inhibiting ROS production. circNFIX can sponge the miR-378e action, allowing RPN2 activity (as part of the ceRNA network). By doing so, circNFIX alters glucose metabolism, reduces proliferation, and consequently contributes to glioma progression. [99,102]. These data, therefore, suggest a putative oncogenic role for NFIX.
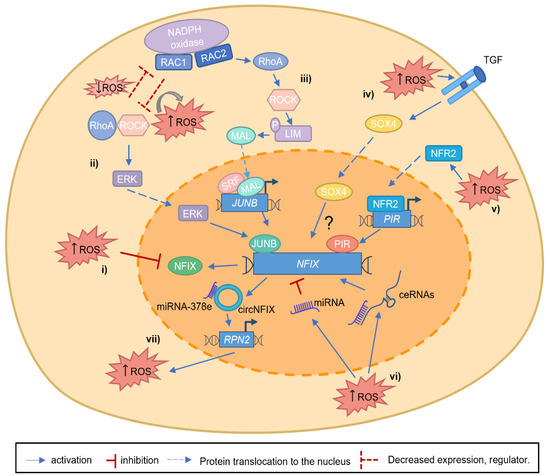
Figure 3.
Putative mechanisms linking NFIX and increased ROS levels. (i) During tissue development, ROS can regulate NFIX expression, oxidizing specific NFIX cysteine residues. (ii). RAC1, a catalytic subunit of NADPH oxidase, can generate ROS and interact with RhoA/ROCK axis leading to its activation or inhibition. On the other hand, RhoA, through its target gene ROCK, can inhibit RAC1 activity, decreasing ROS levels. When RAC1 is able to activate the RhoA/ROCK axis and increase ROS levels, ERK kinases are activated, leading to JUNB activation, and consequently, JUNB activates NFIX expression. (iii) The increased expression of RAC2, and subsequent ROS production, lead to the activation and increase in RhoA and its targets, such as ROCK, that will phosphorylate LIM kinase. With LIM kinase phosphorylation, MAL translocates to the nucleus activating SRF. Consequently, SRF activates JUNB expression, and JUNB protein binds to the NFIX promoter, activating its transcription. (iv) NFIX is a putative target gene of SOX4, but the exact mechanism remains unknown (question mark). SOX4 activation is mediated by ROS/TGFβ, and it is possible that this protein can translocate to the nucleus activating NFIX expression. (v). The oxidative stress sensor NRF2 is activated by increased ROS levels, allowing its translocation to the nucleus, where it activates the Pirin (PIR) promoter. Consequently, NFIX expression is induced through the binding of Pirin to the NFI I/CCAAT box transcription factor (NFI/CTF1) domain. (vi) ROS can lead to the activation of miRNAs or ceRNAs (network composed by miRNA and the inhibitory binding through lncRNA), which lead to a decrease or an increase in NFIX expression, respectively. (vii) In gliomas, miR-378e functionally targets RPN2, an oncogene, and ROS inducer, inhibiting its oncogenic functions. circNFIX can sponge the action of miR-378e, allowing RPN2 activity (as part of the ceRNA network), contributing to glioma progression.
Together, these mechanisms contribute to the regulation of NFIX and ROS levels, which may influence the outcome of cell fate decisions. This is supported by the important role that oxidative stress plays, for example, in proliferation, cell migration and metastasis, and apoptosis [84,86,127,128]. In addition, there is an important link between oxidative stress and glucose metabolism, which has been described not only in cancer [84,86] but also in the context of several other pathologies [129,130,131,132]. In keeping with this notion, studies have suggested that NFIX and circNFIX play an important role in regulating glucose metabolism [99,133]. One example is the above-mentioned activation of RPN2 by circNFIX [99], which has been suggested to promote glycolysis [102]. A recent study has also shown that Nfix was downregulated in a mouse model of obesity in response to glucokinase deficiency, a glycolytic enzyme possibly associated with a reduction in oxidative stress levels [133].
The work being reviewed highlights the role of NFIX in cancer, particularly by showing its strict association with increased oxidative stress. Previous studies suggest that NFIX may be a promising prognostic marker [80,94,134], and even though more research is needed to fully understand the relationship between NFIX and ROS in the context of cancer, it is possible that targeting NFIX offers a means of modulating ROS levels in cancer. When designing new therapeutic strategies, it is crucial to consider that targeting NFIX can impact vital cell mechanisms in various cell types, such as hematopoietic, neuronal, or germ cells. Strategies, such as using adeno-associated virus (AAV)-based gene therapy for efficient and tissue/cell-specific delivery of NFIX silencing molecules (including miRNA or lncRNA), could provide a successful approach. Additionally, a growing number of therapeutic approaches are currently being established to increase ROS levels in cancer cells to a point that surpasses the cells’ redox tolerance, triggering an overt oxidative stress response that ultimately may lead to cancer cell death [135]. A better understanding of how NFIX crosstalks with the oxidative stress response may provide yet another application for the modulation of NFIX levels in cancer treatment.
5. Conclusions
Over the past few decades, NFIX has primarily been studied in the context of skeletal muscle development and muscle dystrophies, as well as in relation to neuronal and hematopoietic cell differentiation and fate. In this review, we explored the role of NFIX in cancer and its crosstalk with oxidative stress pathways. Given the crucial function of NFIX in cell differentiation during embryonic development, it is possible that a potential link between NFIX, oxidative stress, and cancer cell dedifferentiation might be a pivotal factor in tumor progression. Collectively, this review increases our understanding of the involvement of NFIX in both development and cancer, which is essential for the establishment of targeted cancer therapies.
Author Contributions
V.R. and A.R.C. conceptualized the manuscript and figures. V.R., S.G.M., A.S.L., R.Z., and A.R.C. performed the literature review and wrote the manuscript. S.T. contributed with active discussion and revision. All authors contributed to the article and approved the final submission. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by Fundação para a Ciência e a Tecnologia (2022.10813.BD) to S.G.M., Association Française contre les Myopathies (AFM) Téléthon (contract no. 23049) to S.T., Fundação para a Ciência e Tecnologia (FCT, Portugal; CEECIND/01589/2017), and L’Oréal Portugal Medals of Honor for Women in Science 2019 to A.R.C. The donor Henrique Meirelles who chose to support the MATRIHEALTH Project (CC1036), Fundação para a Ciência e a Tecnologia Project (Ref. PTDC/BTM-ORG/1383/2020) and Unit Funding (Ref. UIDB/00329/2020) supports all authors.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
| Abbreviation | Definition |
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 10 |
| ATF3 | Activating transcription factor 3 |
| BCL2 | B-cell leukemia/lymphoma 2 |
| Bcl2l1/BCL-XL | Bcl-2-like protein 1 or B-cell lymphoma-extra large |
| c-MLP | Thrombopoietin receptor or myeloproliferative leukemia protein |
| ceRNAs | Competing endogenous RNAs |
| circRNA | Circular RNA |
| CKM | Muscle creatine kinase |
| CYP1A1 | Cytochrome P450 1A1 |
| EMX2 | Empty spiracles homolog 2 |
| E2A | Immunoglobulin enhancer-binding factors |
| EBF | Early B cell factor |
| ENO3 | β-enolase |
| ERK | Extracellular signal-regulated kinases |
| EZR | Ezrin |
| GDNF | Glial cell derived neurotrophic factor |
| IL6ST | Interleukin-6 receptor subunit β |
| HES | Hairy and enhancer of split-1 |
| HOX | Homeobox genes |
| Insc | Inscuteable |
| ITGB1 | Integrin β-1 |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription |
| JUNB | AP-1 transcription factor subunit |
| lncRNA | Long non-coding RNA |
| MHC | Myosin heavy chain |
| MAL | Megakaryocytic acute leukemia |
| MAPK | Mitogen-activated protein kinase |
| MASH1 | Homologue of ASCL1, Achaete-scute homolog 1 |
| MAST1 | Microtubule Associated Serine/Threonine Kinase 1 |
| miRNA | microRNA |
| NFI | Nuclear factor I |
| NFIX | Nuclear factor I X |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NURF | Nucleosome remodeling factor |
| PAX6/7 | Paired box gene 6/7 |
| PI3K/AKT | Phosphoinositide 3-kinase/protein kinase B |
| Pir | Pirin |
| PKN1 | Protein Kinase N1 |
| RAC1/2 | Rac Family Small GTPase 1/2 |
| RhoA | Ras Homolog Family Member A |
| RPN2 | Ribophorin-II |
| ROCK | Rho associated coiled-coil containing protein kinase 1 |
| ROS | Reactive oxygen species |
| SRF | Serum response factor |
| STAT3/6 | Signal transducer and activator of transcription 3/6 |
| SOX | SRY-Box Transcription Factor |
| TGF-β | Transforming growth factor-beta |
| TIMP1 | Metalloproteinase inhibitor 1 |
References
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.; Gronostajski, R.; Messina, G. Nuclear factor one X in development and disease. Trends Cell Biol. 2018, 29, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Kruse, U.; Lichter, P.; Sippel, A.E. Chromosomal localization of the four genes (NFIA, B, C, and X) for the human transcription factor nuclear factor I by fish. Genomics 1995, 28, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kruse, U.; Sippel, A.E. Transcription factor nuclear factor I proteins form stable homo- and heterodimers. FEBS Lett. 1994, 348, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Göös, H.; Kinnunen, M.; Salokas, K.; Tan, Z.; Liu, X.; Yadav, L.; Zhang, Q.; Wei, G.-H.; Varjosalo, M. Human transcription factor protein interaction networks. Nat. Commun. 2022, 13, 1–16. [Google Scholar] [CrossRef]
- Fraser, J.; Essebier, A.; Brown, A.S.; Davila, R.A.; Harkins, D.; Zalucki, O.; Shapiro, L.P.; Penzes, P.; Wainwright, B.J.; Scott, M.P.; et al. Common regulatory targets of NFIA, NFIX and NFIB during postnatal cerebellar development. Cerebellum 2019, 19, 89–101. [Google Scholar] [CrossRef]
- Chen, K.-S.; Lim, J.W.; Richards, L.J.; Bunt, J. The convergent roles of the nuclear factor I transcription factors in development and cancer. Cancer Lett. 2017, 410, 124–138. [Google Scholar] [CrossRef]
- Steele-Perkins, G.; Butz, K.G.; Lyons, G.E.; Zeichner-David, M.; Kim, H.-J.; Cho, M.-I.; Gronostajski, R.M. Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol. Cell. Biol. 2003, 23, 1075–1084. [Google Scholar] [CrossRef]
- Messina, G.; Biressi, S.; Monteverde, S.; Magli, A.; Cassano, M.; Perani, L.; Roncaglia, E.; Tagliafico, E.; Starnes, L.; Campbell, C.E.; et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell 2010, 140, 554–566. [Google Scholar] [CrossRef]
- Harris, L.; Genovesi, L.A.; Gronostajski, R.M.; Wainwright, B.J.; Piper, M. Nuclear factor one transcription factors: Divergent functions in developmental versus adult stem cell populations. Dev. Dyn. 2014, 244, 227–238. [Google Scholar] [CrossRef]
- Fane, M.; Harris, L.; Smith, A.G.; Piper, M. Nuclear factor one transcription factors as epigenetic regulators in cancer. Int. J. Cancer 2017, 140, 2634–2641. [Google Scholar] [CrossRef]
- Alevizopoulos, A.; Dusserre, Y.; Tsai-Pflugfelder, M.; von der Weid, T.; Wahli, W.; Mermod, N. A proline-rich TGF-beta-responsive transcriptional activator interacts with histone H3. Genes Dev. 1995, 9, 3051–3066. [Google Scholar] [CrossRef] [PubMed]
- Dusserre, Y.; Mermod, N. Purified cofactors and histone H1 mediate transcriptional regulation by CTF/NF-I. Mol. Cell. Biol. 1992, 12, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Pjanic, M.; Schmid, C.D.; Gaussin, A.; Ambrosini, G.; Adamcik, J.; Pjanic, P.; Plasari, G.; Kerschgens, J.; Dietler, G.; Bucher, P.; et al. Nuclear Factor I genomic binding associates with chromatin boundaries. BMC Genom. 2013, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Denny, S.K.; Yang, D.; Chuang, C.-H.; Brady, J.J.; Lim, J.S.; Grüner, B.M.; Chiou, S.-H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef]
- Pjanic, M.; Pjanic, P.; Schmid, C.; Ambrosini, G.; Gaussin, A.; Plasari, G.; Mazza, C.; Bucher, P.; Mermod, N. Nuclear factor I revealed as family of promoter binding transcription activators. BMC Genom. 2011, 12, 181. [Google Scholar] [CrossRef] [PubMed]
- Becker-Santos, D.D.; Lonergan, K.M.; Gronostajski, R.M.; Lam, W.L. Nuclear factor I/B: A Master regulator of cell differentiation with paradoxical roles in cancer. EBioMedicine 2017, 22, 2–9. [Google Scholar] [CrossRef]
- Mason, S.; Piper, M.; Gronostajski, R.M.; Richards, L.J. Nuclear factor one transcription factors in cns development. Mol. Neurobiol. 2008, 39, 10–23. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Gronostajski, R.M. Identification of a conserved oxidation-sensitive cysteine residue in the NFI family of DNA-binding proteins. J. Biol. Chem. 1994, 269, 29949–29955. [Google Scholar] [CrossRef]
- Liu, S.; Qu, D.; Li, W.; He, C.; Li, S.; Wu, G.; Zhao, Q.; Shen, L.; Zhang, J.; Zheng, J. miR-647 and miR-1914 promote cancer progression equivalently by downregulating nuclear factor IX in colorectal cancer. Mol. Med. Rep. 2017, 16, 8189–8199. [Google Scholar] [CrossRef]
- Yang, L.; Yang, Z.; Yao, R.; Li, Y.; Liu, Z.; Chen, X.; Zhang, G. miR-210 promotes progression of endometrial carcinoma by regulating the expression of NFIX. Int. J. Clin. Exp. Pathol. 2018, 11, 5213–5222. [Google Scholar]
- Kleemann, M.; Schneider, H.; Unger, K.; Sander, P.; Schneider, E.M.; Fischer-Posovszky, P.; Handrick, R.; Otte, K. MiR-744-5p inducing cell death by directly targeting HNRNPC and NFIX in ovarian cancer cells. Sci. Rep. 2018, 8, 9020. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yuan, Q.; Zhang, W.; Niu, M.; Fu, H.; Qiu, Q.; Mao, G.; Wang, H.; Wen, L.; Wang, H.; et al. MiR-663a Stimulates proliferation and suppresses early apoptosis of human spermatogonial stem cells by targeting NFIX and regulating cell cycle. Mol. Ther. Nucleic Acids 2018, 12, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of MicroRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Liu, J.; Zhang, D.; Li, B. MiR-1290 promotes cancer progression by targeting nuclear factor I/X(NFIX) in esophageal squamous cell carcinoma (ESCC). Biomed. Pharmacother. 2015, 76, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, Y.; Qi, L.; Ding, L.; Jiang, H.; Yu, H. NFIX circular RNA promotes glioma progression by regulating miR-34a-5p via notch signaling pathway. Front. Mol. Neurosci. 2018, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Veno, M.T.; Hansen, T.B.; Venø, S.T.; Clausen, B.H.; Grebing, M.; Finsen, B.; Holm, I.E.; Kjems, J. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015, 16, 245. [Google Scholar] [CrossRef]
- Cui, X.; Dong, Y.; Li, M.; Wang, X.; Jiang, M.; Yang, W.; Liu, G.; Sun, S.; Xu, W. A circular RNA from NFIX facilitates oxidative stress-induced H9c2 cells apoptosis. Vitr. Cell. Dev. Biol. Anim. 2020, 56, 715–722. [Google Scholar] [CrossRef]
- Zhao, L.; Song, X.; Guo, Y.; Ding, N.; Wang, T.; Huang, L. Long non-coding RNA SNHG3 promotes the development of non-small cell lung cancer via the miR-1343-3p/NFIX pathway. Int. J. Mol. Med. 2021, 48, 1–12. [Google Scholar] [CrossRef]
- Ye, L.; Feng, W.; Weng, H.; Yuan, C.; Liu, J.; Wang, Z. MAFG-AS1 aggravates the progression of pancreatic cancer by sponging miR-3196 to boost NFIX. Cancer Cell Int. 2020, 20, 591. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Starke, D.W.; Mieyal, J.J.; Gronostajski, R.M. Thioltransferase (glutaredoxin) reactivates the DNA-binding activity of oxidation-inactivated nuclear factor I. J. Biol. Chem. 1998, 273, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G. Similarities between embryo development and cancer process suggest new strategies for research and therapy of tumors: A new point of view. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, P.; Wang, F.; Yang, J.; Yang, Z.; Qin, H. The relationship between early embryo development and tumourigenesis. J. Cell. Mol. Med. 2010, 14, 2697–2701. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Brivanlou, A.H. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007, 3, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Angelini, G.; Mura, G.; Bonfanti, C.; Caruso, E.; Monteverde, S.; Le Carrou, G.; Tajbakhsh, S.; Relaix, F.; Messina, G. Rhoa and erk signalling regulate the expression of the transcription factor nfix in myogenic cells. Development 2018, 145, dev163956. [Google Scholar] [CrossRef]
- Rossi, G.; Taglietti, V.; Messina, G. Targeting Nfix to fix muscular dystrophies. Cell Stress 2018, 2, 17–19. [Google Scholar] [CrossRef]
- Rando, T.A. Oxidative stress and the pathogenesis of muscular dystrophies. Am. J. Phys. Med. Rehabil. 2002, 81, S175–S186. [Google Scholar] [CrossRef]
- Moore, T.; Lin, A.J.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; Ho, T.; Lee, J.L.; Rucker, D.H.; Nguyen, C.Q.; et al. Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol. 2020, 11, 690. [Google Scholar] [CrossRef]
- Martins, S.G.; Zilhão, R.; Thorsteinsdóttir, S.; Carlos, A.R. Linking Oxidative stress and DNA Damage to changes in the expression of extracellular matrix components. Front. Genet. 2021, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef]
- Rossi, G.; Antonini, S.; Bonfanti, C.; Monteverde, S.; Vezzali, C.; Tajbakhsh, S.; Cossu, G.; Messina, G. Nfix Regulates temporal progression of muscle regeneration through modulation of myostatin expression. Cell Rep. 2016, 14, 2238–2249. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Lapi, M.; Bonfanti, C.; Rossi, G.; Antonini, S.; Messina, G. The transcription factor Nfix requires RhoA-ROCK1 dependent phagocytosis to mediate macrophage skewing during skeletal muscle regeneration. Cells 2020, 9, 708. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; Van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; de Lagrán, M.M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68, 81–93. [Google Scholar] [CrossRef]
- Smith, L.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68, 602–615. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. Shifts in macrophage cytokine production drive muscle fibrosis. Nat. Med. 2015, 21, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.Z.; Lyons, G.E.; Gronostajski, R.M. Expression patterns of the four nuclear factor I genes during mouse embryogenesis indicate a potential role in development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1997, 208, 313–325. [Google Scholar] [CrossRef]
- Harris, L.; Zalucki, O.; Gobius, I.; McDonald, H.; Osinki, J.; Harvey, T.J.; Essebier, A.; Vidovic, D.; Gladwyn-Ng, I.; Burne, T.H.; et al. Transcriptional regulation of intermediate progenitor cell generation during hippocampal development. Development 2016, 143, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- Matuzelski, E.; Bunt, J.; Harkins, D.; Lim, J.W.; Gronostajski, R.M.; Richards, L.J.; Harris, L.; Piper, M. Transcriptional regulation of Nfix by NFIB drives astrocytic maturation within the developing spinal cord. Dev. Biol. 2017, 432, 286–297. [Google Scholar] [CrossRef]
- Heng, Y.H.E.; McLeay, R.C.; Harvey, T.J.; Smith, A.G.; Barry, G.; Cato, K.; Plachez, C.; Little, E.; Mason, S.; Dixon, C.; et al. NFIX Regulates neural progenitor cell differentiation during hippocampal morphogenesis. Cereb. Cortex 2012, 24, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.E.; Wynn, S.L.; Sesay, A.; Cruz, C.; Cheung, M.; Gaviro, M.V.G.; Booth, S.; Gao, B.; Cheah, K.S.; Lovell-Badge, R.; et al. SOX9 induces and maintains neural stem cells. Nat. Neurosci. 2010, 13, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Heng, Y.H.E.; Zhou, B.; Harris, L.; Harvey, T.; Smith, A.; Horne, E.; Martynoga, B.; Andersen, J.; Achimastou, A.; Cato, K.; et al. NFIX Regulates proliferation and migration within the murine SVZ neurogenic niche. Cereb. Cortex 2014, 25, 3758–3778. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Campos, J.; Osinski, J.M.; Gronostajski, R.M.; Michie, A.M.; Keeshan, K. Nfix Expression critically modulates early B lymphopoiesis and myelopoiesis. PLoS ONE 2015, 10, e0120102. [Google Scholar] [CrossRef]
- Singh, H.; Medina, K.L.; Pongubala, J.M.R. Contingent gene regulatory networks and B cell fate specification. Proc. Natl. Acad. Sci. USA 2005, 102, 4949–4953. [Google Scholar] [CrossRef]
- Hall, T.; Walker, M.; Ganuza, M.; Holmfeldt, P.; Bordas, M.; Kang, G.; Bi, W.; Palmer, L.E.; Finkelstein, D.; McKinney-Freeman, S. Nfix promotes survival of immature hematopoietic cells via regulation of C-MPL. Stem Cells 2018, 36, 943–950. [Google Scholar] [CrossRef]
- Davila, R.A.; Spiller, C.; Harkins, D.; Harvey, T.; Jordan, P.W.; Gronostajski, R.M.; Piper, M.; Bowles, J. Deletion of NFIX results in defective progression through meiosis within the mouse testis. Biol. Reprod. 2022, 106, 1191–1205. [Google Scholar] [CrossRef]
- Landi, S.; Barbuti, A.F. The Transcription Factor Nfix as a Novel Modulator of Heart Rate. Ph.D. Thesis, The University of Milan, Milano, Italy, 2018. [Google Scholar]
- Jaé, N.; Heumüller, A.W.; Fouani, Y.; Dimmeler, S. Long non-coding RNAs in vascular biology and disease. Vasc. Pharmacol. 2019, 114, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mester-Tonczar, J.; Hašimbegović, E.; Spannbauer, A.; Traxler, D.; Kastner, N.; Zlabinger, K.; Einzinger, P.; Pavo, N.; Goliasch, G.; Gyöngyösi, M. Circular RNAs in cardiac regeneration: Cardiac cell proliferation, differentiation, survival, and reprogramming. Front. Physiol. 2020, 11, 580465. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xu, Z.; Guo, G.; Xu, C.; Song, Z.; Li, K.; Zhong, K.; Wang, D. Circ_nuclear factor I X (circNfix) attenuates pressure overload-induced cardiac hypertrophy via regulating miR-145-5p/ATF3 axis. Bioengineered 2021, 12, 5373–5385. [Google Scholar] [CrossRef]
- Huang, S.; Li, X.; Zheng, H.; Si, X.; Li, B.; Wei, G.; Li, C.; Chen, Y.; Chen, Y.; Liao, W.; et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 2019, 139, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.W.; Lim, B.T.; Anene-Nzelu, C.G.; Ackers-Johnson, M.; Dashi, A.; See, K.; Tiang, Z.; Lee, D.P.; Chua, W.W.; Luu, T.D.; et al. A landscape of circular RNA expression in the human heart. Cardiovasc. Res. 2016, 113, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Koren, L.; Elhanani, O.; Kehat, I.; Hai, T.; Aronheim, A. Adult cardiac expression of the activating transcription factor 3, ATF3, promotes ventricular hypertrophy. PLoS ONE 2013, 8, e68396. [Google Scholar] [CrossRef] [PubMed]
- Soraya, A.-S.; Tali, H.; Rona, S.; Tom, F.; Roy, K.; Ami, A. ATF3 expression in cardiomyocytes and myofibroblasts following transverse aortic constriction displays distinct phenotypes. IJC Hear. Vasc. 2021, 32, 100706. [Google Scholar] [CrossRef]
- Victorino, P.; Marra, C.; Iacobas, D.; Iacobas, S.; Spray, D.; Linden, R.; Adesse, D.; Petrs-Silva, H. Retinal Genomic fabric remodeling after optic nerve injury. Genes 2021, 12, 403. [Google Scholar] [CrossRef]
- Yuan, B.; Cui, J.; Wang, W.; Deng, K. Gα12/13 signaling promotes cervical cancer invasion through the RhoA/ROCK-JNK signaling axis. Biochem. Biophys. Res. Commun. 2016, 473, 1240–1246. [Google Scholar] [CrossRef]
- Pei, H.; Guo, Z.; Wang, Z.; Dai, Y.; Zheng, L.; Zhu, L.; Zhang, J.; Hu, W.; Nie, J.; Mao, W.; et al. RAC2 promotes abnormal proliferation of quiescent cells by enhanced JUNB expression via the MAL-SRF pathway. Cell Cycle 2018, 17, 1115–1123. [Google Scholar] [CrossRef]
- Moreno, C.S. SOX4: The unappreciated oncogene. Semin. Cancer Biol. 2020, 67, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; McCabe, C.D.; Ali-Seyed, M.; Berger, M.F.; Bulyk, M.L.; Moreno, C.S. Genome-wide promoter analysis of the SOX4 transcriptional network in prostate cancer cells. Cancer Res. 2009, 69, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.F.; Orlando, U.D.; López, P.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Gene expression profile and signaling pathways in MCF-7 breast cancer cells mediated by acyl-coa synthetase 4 overexpression. Transcr. Open Access 2015, 3, 1–9. [Google Scholar] [CrossRef]
- Moura, D.; Díaz-Martín, J.; Bagué, S.; Orellana-Fernandez, R.; Sebio, A.; Mondaza-Hernandez, J.; Salguero-Aranda, C.; Rojo, F.; Hindi, N.; Fletcher, C.; et al. A novel NFIX-STAT6 gene fusion in solitary fibrous tumor: A case report. Int. J. Mol. Sci. 2021, 22, 7514. [Google Scholar] [CrossRef]
- Prensner, J.R.; Chinnaiyan, A.M. Oncogenic gene fusions in epithelial carcinomas. Curr. Opin. Genet. Dev. 2009, 19, 82–91. [Google Scholar] [CrossRef]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef]
- Kastnerova, L.; Luzar, B.; Goto, K.; Grishakov, V.; Gatalica, Z.; Kamarachev, J.; Martinek, P.; Hájková, V.; Grossmann, P.; Imai, H.; et al. Secretory carcinoma of the skin. Am. J. Surg. Pathol. 2019, 43, 1092–1098. [Google Scholar] [CrossRef]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.-L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12, R6. [Google Scholar] [CrossRef]
- Rahman, N.I.; Abdul Murad, N.A.; Mollah, M.M.; Jamal, R.; Harun, R. NFIX as a master regulator for lung cancer progression. Front. Pharmacol. 2017, 8, 540. [Google Scholar] [CrossRef]
- Liu, Z.; Ge, R.; Zhou, J.; Yang, X.; Cheng, K.K.-Y.; Tao, J.; Wu, D.; Mao, J. Nuclear factor IX promotes glioblastoma development through transcriptional activation of Ezrin. Oncogenesis 2020, 9, 39. [Google Scholar] [CrossRef]
- Grabowska, M.M.; Elliott, A.D.; DeGraff, D.J.; Anderson, P.D.; Anumanthan, G.; Yamashita, H.; Sun, Q.; Friedman, D.B.; Hachey, D.L.; Yu, X.; et al. NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol. Endocrinol. 2014, 28, 949–964. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Freudenthal, B.D.; Whitaker, A.M.; Schaich, M.A.; Smith, M.S.; Flynn, T.S. Base excision repair of oxidative DNA damage from mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Morel, Y.; Barouki, R. Down-regulation of cytochrome P450 1A1 gene promoter by oxidative stress. Critical contribution of nuclear factor 1. J. Biol. Chem. 1998, 273, 26969–26976. [Google Scholar] [CrossRef] [PubMed]
- Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Cytochrome P450 CYP1A1: Wider roles in cancer progression and prevention. BMC Cancer 2009, 9, 187. [Google Scholar] [CrossRef]
- Morel, Y.; Mermod, N.; Barouki, R. An autoregulatory loop controlling CYP1A1 gene expression: Role of H2O2 and NFI. Mol. Cell. Biol. 1999, 19, 6825–6832. [Google Scholar] [CrossRef]
- Taverne, Y.J.; Merkus, D.; Bogers, A.J.; Halliwell, B.; Duncker, D.J.; Lyons, T.W. Reactive oxygen species: Radical factors in the evolution of animal life. Bioessays 2018, 40, 1700158. [Google Scholar] [CrossRef]
- Rodriguez, M.; Potter, D.A. CYP1A1 regulates breast cancer proliferation and survival. Mol. Cancer Res. 2013, 11, 780–792. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Spyrou, I.; Ploumidis, A.; Papalampros, A.E.; Kyriakakis, M.; Delakas, D.; Spandidos, D.A.; Tsatsakis, A.M. Expression profile of CYP1A1 and CYP1B1 enzymes in colon and bladder tumors. PLoS ONE 2013, 8, e82487. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Liu, X.-L.; Liu, H.-S.; Luo, X.-Y.; Yuan, Y.; Ji, Y.-M.; Liu, T.; Guo, J.-L.; Zhang, J. The risk model based on the three oxidative stress-related genes evaluates the prognosis of LAC patients. Oxidative Med. Cell. Longev. 2022, 2022, 4022896. [Google Scholar] [CrossRef]
- Sun, C.; Guo, E.; Zhou, B.; Shan, W.; Huang, J.; Weng, D.; Wu, P.; Wang, C.; Wang, S.; Zhang, W.; et al. A reactive oxygen species scoring system predicts cisplatin sensitivity and prognosis in ovarian cancer patients. BMC Cancer 2019, 19, 1061. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jee, B.A.; Kwon, S.M.; Yoon, Y.; Xu, W.G.; Wang, H.; Wang, X.W.; Thorgeirsson, S.S.; Lee, J.; Woo, H.G.; et al. Identification of a mitochondrial defect gene signature reveals NUPR1 as a key regulator of liver cancer progression. Hepatology 2015, 62, 1174–1189. [Google Scholar] [CrossRef]
- Patop, I.L.; Kadener, S. circRNAs in cancer. Curr. Opin. Genet. Dev. 2018, 48, 121–127. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Wan, Y.; Zhao, Y.; Wen, Q.; Tang, X.; Shen, J.; Wu, X.; Li, M.; Li, X.; et al. Circular RNAs in the regulation of oxidative stress. Front. Pharmacol. 2021, 12, 1906. [Google Scholar] [CrossRef]
- Ding, C.; Wu, Z.; You, H.; Ge, H.; Zheng, S.; Lin, Y.; Wu, X.; Lin, Z.; Kang, D. CircNFIX promotes progression of glioma through regulating miR-378e/RPN2 axis. J. Exp. Clin. Cancer Res. 2019, 38, 506. [Google Scholar] [CrossRef]
- Lu, J.; Zhu, Y.; Qin, Y.; Chen, Y. CircNFIX Acts as a miR-212-3p sponge to enhance the malignant progression of non-small cell lung cancer by up-regulating ADAM10. Cancer Manag. Res. 2020, 12, 9577–9587. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. A review of the molecular mechanisms of hyperglycemia-induced free radical generation leading to oxidative stress. J. Cell. Physiol. 2019, 234, 1300–1312. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Zhang, W.; Ke, Z.; Lv, Y.; Zhang, B.; Liao, Z. Ribophorin II promotes the epithelial–mesenchymal transition and aerobic glycolysis of laryngeal squamous cell carcinoma via regulating reactive oxygen species-mediated Phosphatidylinositol-3-kinase/protein kinase B activation. Bioengineered 2022, 13, 5141–5151. [Google Scholar] [CrossRef]
- Crawford, H.; Dempsey, P.; Brown, G.; Adam, L.; Moss, M. ADAM10 as a therapeutic target for cancer and inflammation. Curr. Pharm. Des. 2009, 15, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-C.; Ni, J.-J.; Cui, W.-Y.; Wang, B.-Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Wang, X.; Zhou, J.; Xu, M.; Yan, Y.; Huang, L.; Kuang, Y.; Liu, Y.; Li, P.; Zheng, W.; Liu, H.; et al. A 15-lncRNA signature predicts survival and functions as a ceRNA in patients with colorectal cancer. Cancer Manag. Res. 2018, 10, 5799. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, P.; Pardieck, J.; Saulsberry, A.C.; Nandakumar, S.K.; Finkelstein, D.; Gray, J.T.; Persons, D.A.; McKinney-Freeman, S. Nfix is a novel regulator of murine hematopoietic stem and progenitor cell survival. Blood 2013, 122, 2987–2996. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.P.; Castresana, J.S.; Shahi, M.H. Role of circular RNA in brain tumor development. Cells 2022, 11, 2130. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, C.; Liu, J.; Jiang, H.; Jiang, X.; Liu, Z. A novel tumor-promoting role for nuclear factor IX in glioblastoma is mediated through transcriptional activation of GINS1. Mol. Cancer Res. 2022, 2022, OF1–OF10. [Google Scholar] [CrossRef]
- Walker, M.; Li, Y.; Morales-Hernandez, A.; Qi, Q.; Parupalli, C.; Brown, S.A.; Christian, C.; Clements, W.K.; Cheng, Y.; Freeman, S.L.M. An NFIX-mediated regulatory network governs the balance of hematopoietic stem and progenitor cells during hematopoiesis. Blood Adv. 2022. [Google Scholar] [CrossRef]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef]
- Akbar, H.; Duan, X.; Saleem, S.; Davis, A.K.; Zheng, Y. RhoA and Rac1 GTPases differentially regulate agonist-receptor mediated reactive oxygen species generation in platelets. PLoS ONE 2016, 11, e0163227. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kim, J.-G.; Jeon, C.-Y.; Won, H.-Y.; Moon, M.-Y.; Seo, J.-Y.; Kim, J.-I.; Kim, J.; Lee, J.-Y.; Choi, S.-Y.; et al. Downstream components of RhoA required for signal pathway of superoxide formation during phagocytosis of serum opsonized zymosans in macrophages. Exp. Mol. Med. 2005, 37, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G.; Caloca, M.J. The rac GTPase in cancer: From old concepts to new paradigms. Cancer Res. 2017, 77, 5445–5451. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Yin, C.; Huang, X.; Huang, Y.; Ding, L.; Jin, M.; Wang, Z.; Wei, J.; Li, X. ROS/TGF-β signal mediated accumulation of SOX4 in OA-FLS promotes cell senescence. Exp. Gerontol. 2021, 156, 111616. [Google Scholar] [CrossRef] [PubMed]
- Wendler, W.M.F.; Kremmer, E.; Förster, R.; Winnacker, E.-L. Identification of pirin, a novel highly conserved nuclear protein. J. Biol. Chem. 1997, 272, 8482–8489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Knatko, E.V.; Higgins, M.; Naidu, S.D.; Smith, G.; Honda, T.; de la Vega, L.; Dinkova-Kostova, A.T. Pirin, an Nrf2-regulated protein, is overexpressed in human colorectal tumors. Antioxidants 2022, 11, 262. [Google Scholar] [CrossRef]
- Brzóska, K.; Stępkowski, T.M.; Kruszewski, M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol. Cell. Biochem. 2014, 389, 99–111. [Google Scholar] [CrossRef]
- Perez-Dominguez, F.; Carrillo-Beltrán, D.; Blanco, R.; Muñoz, J.; León-Cruz, G.; Corvalan, A.; Urzúa, U.; Calaf, G.; Aguayo, F. Role of pirin, an oxidative stress sensor protein, in epithelial carcinogenesis. Biology 2021, 10, 116. [Google Scholar] [CrossRef]
- Miyazaki, I.; Simizu, S.; Okumura, H.; Takagi, S.; Osada, H. A small-molecule inhibitor shows that pirin regulates migration of melanoma cells. Nat. Chem. Biol. 2010, 6, 667–673. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. Non-coding RNAs are involved in the response to oxidative stress. Biomed. Pharmacother. 2020, 127, 110228. [Google Scholar] [CrossRef]
- Wang, X.; Shen, C.; Zhu, J.; Shen, G.; Li, Z.; Dong, J. Long noncoding RNAs in the regulation of oxidative stress. Oxidative Med. Cell. Longev. 2019, 2019, 1318795. [Google Scholar] [CrossRef]
- Banerjee, J.; Khanna, S.; Bhattacharya, A. MicroRNA regulation of oxidative stress. Oxidative Med. Cell. Longev. 2017, 2017, 2872156. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.O.; Reiisi, S.; Shareef, S. miRNAs, oxidative stress, and cancer: A comprehensive and updated review. J. Cell. Physiol. 2020, 235, 8812–8825. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Majd, H.M.; Rahnama, R.; Heshmati, J.; Morvaridzadeh, M.; Agah, S.; Amini, S.M.; Masoodi, M. Cross-talk between oxidative stress signaling and microRNA regulatory systems in carcinogenesis: Focused on gastrointestinal cancers. Biomed. Pharmacother. 2020, 131, 110729. [Google Scholar] [CrossRef]
- Wu, L.; Du, Q.; Wu, C. CircLPAR1/miR-212-3p/ZNF217 feedback loop promotes amyloid β-induced neuronal injury in Alzheimer’s Disease. Brain Res. 2021, 1770, 147622. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Zhou, R.; Sun, J.; Zhang, F.; Tang, X.; Chen, K.K.; Zhao, J.; Lan, X.; Lin, S.; Zhang, Z.; et al. LncRNA SNHG5 promotes the progression of osteosarcoma by sponging the miR-212-3p/SGK3 axis. Cancer Cell Int. 2018, 18, 141. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Carlos, A.R.; Weis, S.; Soares, M.P. Cross-talk between iron and glucose metabolism in the establishment of disease tolerance. Front. Immunol. 2018, 9, 2498. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Nakamura, A.; Miyoshi, H.; Yamauchi, Y.; Kawata, S.; Takahashi, K.; Kitao, N.; Nomoto, H.; Kameda, H.; Cho, K.Y.; et al. Glucokinase inactivation paradoxically ameliorates glucose intolerance by increasing β-cell mass in db/db Mice. Diabetes 2021, 70, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Dong, H.; Yang, Y.; Liu, B.; Zheng, M.; Cheng, Q.; Peng, L.; Li, J. NFIX downregulation independently predicts poor prognosis in lung adenocarcinoma, but not in squamous cell carcinoma. Futur. Oncol. 2018, 14, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).