
TCF-1 Is Required for CD4 T Cell Persistence Functions during AlloImmunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TCF-1 Regulates CD4 T Cell Phenotype and Memory Formation
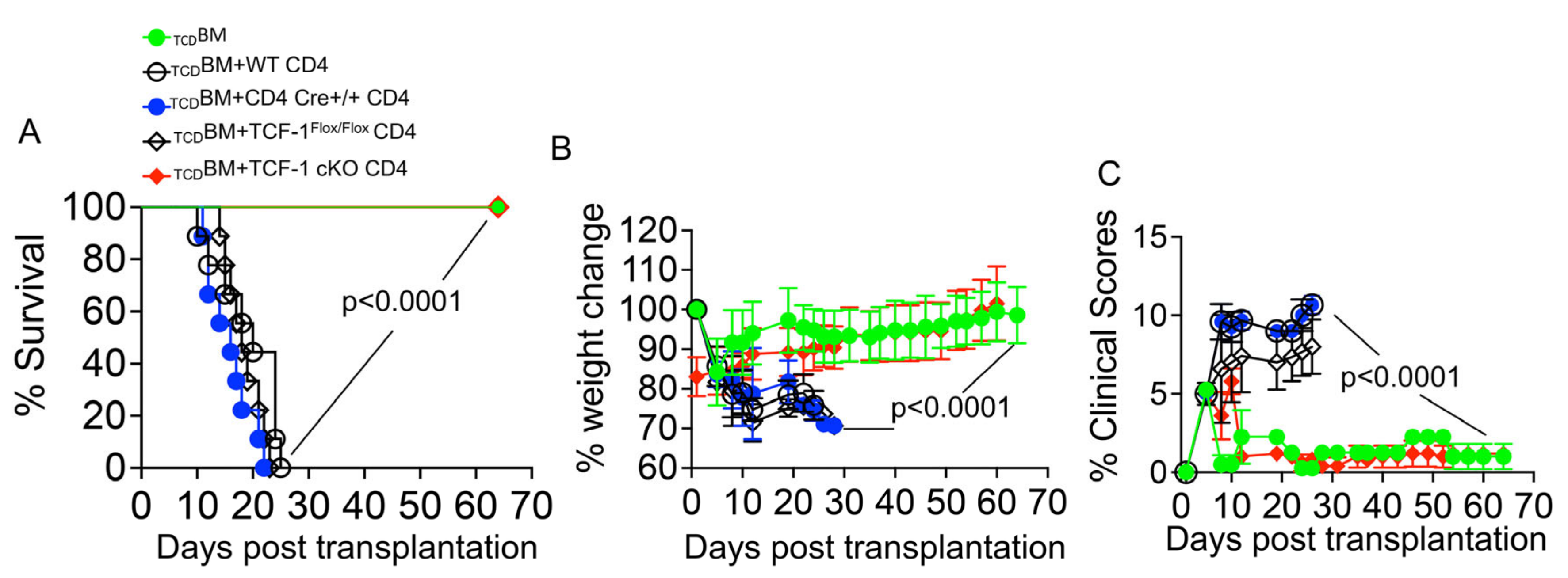
2.2. Loss of TCF-1 in Donor CD4 T Cells Reduces Severity and Persistence of GvHD Symptoms
2.3. TCF-1 Regulates Chemokine/Chemokine Receptor Expression in Mature CD4 T Cells during Allo-Activation
2.4. TCF-1 Regulates CD4 T Cell Damage to GvHD Target Organs
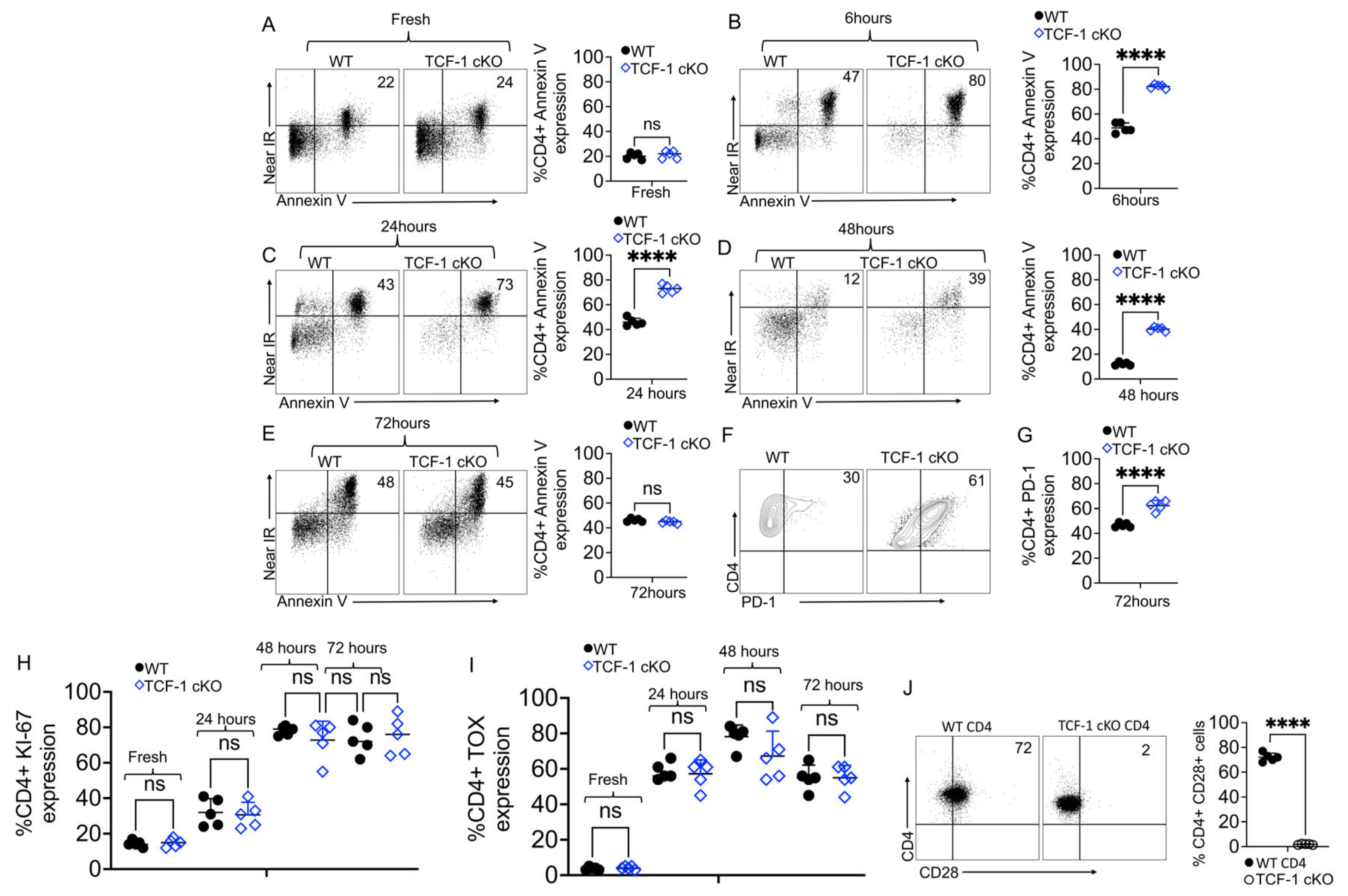
2.5. TCF-1 Regulates CD4 T Cell Survival and Persistence
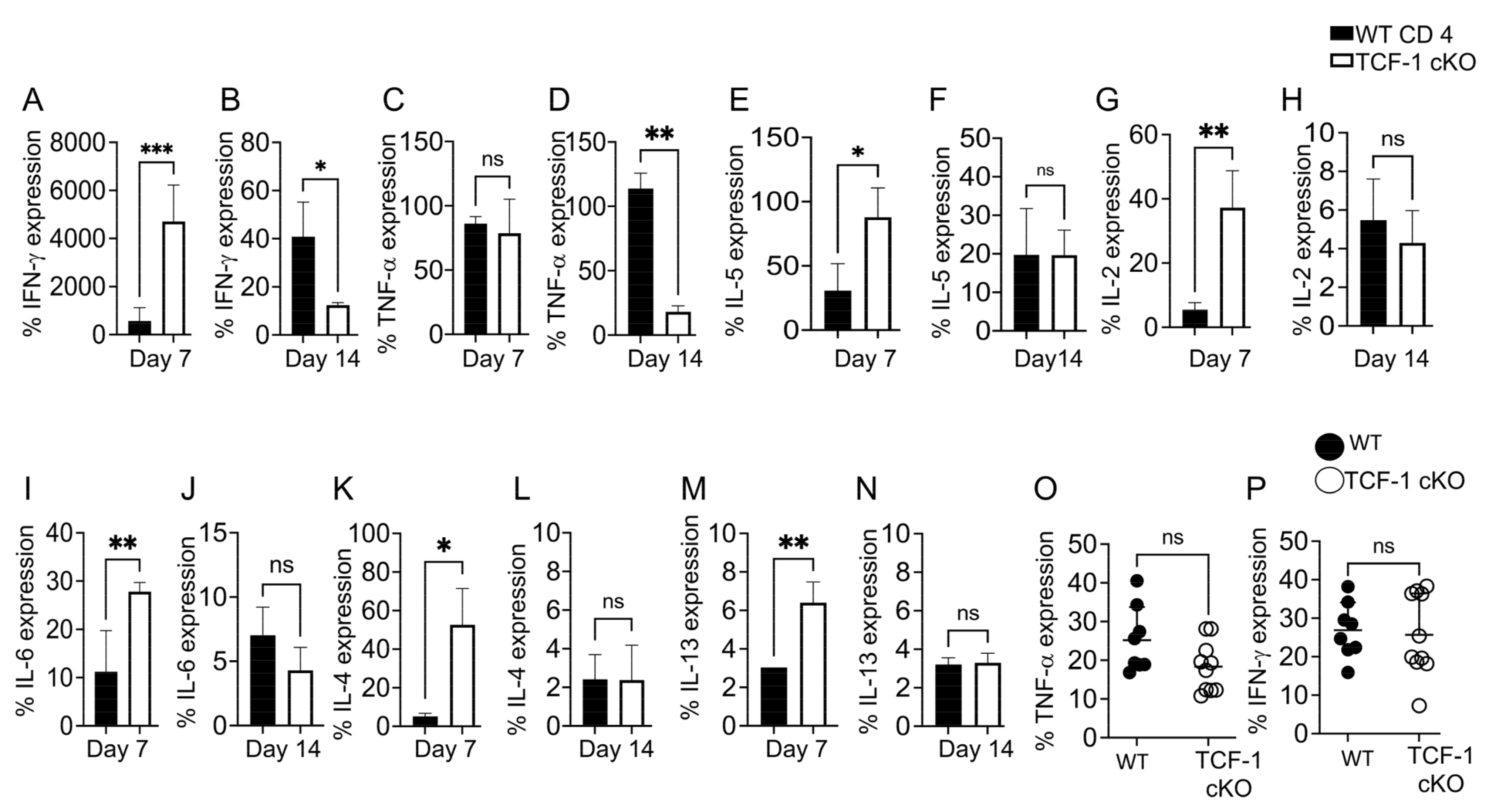
2.6. TCF-1 Regulates Serum Levels of Cytokines during Alloimmunity
2.7. TCF-1 Regulates Key Signaling Pathways in Donor CD4 T Cells
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. DNA Extraction and PCR
4.3. Flow Cytometry, Sorting, and Phenotyping
4.4. Bone Marrow Transplants
4.4.1. Allo-HSCT and GVHD Studies
4.4.2. Allo-HSCT Sort Term Experiments
4.4.3. Bone Marrow Chimera Model
4.5. qPCR Analysis
4.6. Histopathological Evaluation
4.7. Isolation of Lymphocytes from the Liver and Small Intestine
4.8. Cellular Level Cytokine Production Assay
4.9. Serum Level Cytokine Production Assay
4.10. Cell Death Assay
4.10.1. In Vitro
4.10.2. In Vivo
4.11. RNA Sequencing
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weber, B.N.; Chi, A.W.; Chavez, A.; Yashiro-Ohtani, Y.; Yang, Q.; Shestova, O.; Bhandoola, A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 2011, 476, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5, eabb9726. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 1996, 156, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Sullivan, B.; Szabo, S.J.; Sobel, R.A.; Glimcher, L.H.; Kuchroo, V.K. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004, 200, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Oestreich, K.J.; Huang, A.C.; Weinmann, A.S. The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J. Exp. Med. 2011, 208, 1001–1013. [Google Scholar] [CrossRef]
- Nish, S.A.; Zens, K.D.; Kratchmarov, R.; Lin, W.W.; Adams, W.C.; Chen, Y.H.; Yen, B.; Rothman, N.J.; Bhandoola, A.; Xue, H.H.; et al. CD4+ T cell effector commitment coupled to self-renewal by asymmetric cell divisions. J. Exp. Med. 2017, 214, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.B.; Hosokawa, H.; Hasegawa, A.; Watarai, H.; Taniguchi, M.; Yamashita, M.; Nakayama, T. Lymphoid enhancer factor interacts with GATA-3 and controls its function in T helper type 2 cells. Immunology 2008, 125, 377–386. [Google Scholar] [CrossRef]
- Xu, L.; Cao, Y.; Xie, Z.; Huang, Q.; Bai, Q.; Yang, X.; He, R.; Hao, Y.; Wang, H.; Zhao, T.; et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat. Immunol. 2015, 16, 991–999. [Google Scholar] [CrossRef]
- Donnarumma, T.; Young, G.R.; Merkenschlager, J.; Eksmond, U.; Bongard, N.; Nutt, S.L.; Boyer, C.; Dittmer, U.; Le-Trilling, V.T.; Trilling, M.; et al. Opposing Development of Cytotoxic and Follicular Helper CD4 T Cells Controlled by the TCF-1-Bcl6 Nexus. Cell Rep. 2016, 17, 1571–1583. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Gullicksrud, J.A.; Xing, S.; Zeng, Z.; Shan, Q.; Li, F.; Love, P.E.; Peng, W.; Xue, H.H.; Crotty, S. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat. Immunol. 2015, 16, 980–990. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; He, Z.; Sen, S.; Wang, F.; Zhang, Q.; Sun, Z. TCF-1 Inhibits IL-17 Gene Expression To Restrain Th17 Immunity in a Stage-Specific Manner. J. Immunol. 2018, 200, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Arber, C.; Brenner, M.K.; Reddy, P. Mouse models in bone marrow transplantation and adoptive cellular therapy. Semin. Hematol. 2013, 50, 131–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paczesny, S.; Hanauer, D.; Sun, Y.; Reddy, P. New perspectives on the biology of acute GVHD. Bone Marrow Transpl. 2010, 45, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Xing, C.; Zhang, W.; Chen, C.; Xu, J.; Meng, A.; Pan, Y. Coordinate involvement of Nodal-dependent inhibition and Wnt-dependent activation in the maintenance of organizer-specific bmp2b in zebrafish. Int. J. Dev. Biol. 2016, 60, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.; Mammadli, M.; Hiner, S.; Suo, L.; Yang, Q.; Sen, J.M.; Karimi, M. TCF-1 regulates NKG2D expression on CD8 T cells during anti-tumor responses. Cancer Immunol. Immunother. 2022, 1–21. [Google Scholar] [CrossRef]
- Chen, Z.; Ji, Z.; Ngiow, S.F.; Manne, S.; Cai, Z.; Huang, A.C.; Johnson, J.; Staupe, R.P.; Bengsch, B.; Xu, C.; et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity 2019, 51, 840–855.e845. [Google Scholar] [CrossRef]
- Issuree, P.D.; Day, K.; Au, C.; Raviram, R.; Zappile, P.; Skok, J.A.; Xue, H.H.; Myers, R.M.; Littman, D.R. Stage-specific epigenetic regulation of CD4 expression by coordinated enhancer elements during T cell development. Nat. Commun. 2018, 9, 3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinos, D.A.; Soskic, B.; Williams, C.; Kennedy, A.; Jostins, L.; Sansom, D.M.; Trynka, G. Genomic profiling of T-cell activation suggests increased sensitivity of memory T cells to CD28 costimulation. Genes Immun. 2020, 21, 390–408. [Google Scholar] [CrossRef] [PubMed]
- Linterman, M.A.; Denton, A.E.; Divekar, D.P.; Zvetkova, I.; Kane, L.; Ferreira, C.; Veldhoen, M.; Clare, S.; Dougan, G.; Espeli, M.; et al. CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. elife 2014, 3, e03180. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Blaise, D.; Faucher, C.; Vey, N.; Bouabdallah, R.; Stoppa, A.M.; Viret, F.; Gravis, G.; Olive, D.; Gaugler, B. Inflammatory cytokines and acute graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation. Blood 2005, 106, 4407–4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohty, M.; Gaugler, B. Inflammatory cytokines and dendritic cells in acute graft-versus-host disease after allogeneic stem cell transplantation. Cytokine Growth Factor Rev. 2008, 19, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Henden, A.S.; Hill, G.R. Cytokines in Graft-versus-Host Disease. J. Immunol. 2015, 194, 4604–4612. [Google Scholar] [CrossRef] [Green Version]
- Lynch Kelly, D.; Lyon, D.E.; Ameringer, S.A.; Elswick, R.K. Symptoms, Cytokines, and Quality of Life in Patients Diagnosed with Chronic Graft-Versus-Host Disease Following Allogeneic Hematopoietic Stem Cell Transplantation. Oncol. Nurs. Forum. 2015, 42, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Jin, J.; Weyand, C.M.; Goronzy, J.J. The Transcription Factor TCF1 in T Cell Differentiation and Aging. Int. J. Mol. Sci. 2020, 21, 6497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berga-Bolanos, R.; Zhu, W.S.; Steinke, F.C.; Xue, H.H.; Sen, J.M. Cell-autonomous requirement for TCF1 and LEF1 in the development of Natural Killer T cells. Mol. Immunol. 2015, 68, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulze-Koops, H. The role of the T cell in autoimmune inflammation. Arthritis Res. Ther. 2005, 7, S4–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, D.M.; Burma, A.M.; Kitakule, M.M.; Luo, Y.; Mehta, N.N. T Cells in Autoimmunity-Associated Cardiovascular Diseases. Front. Immunol. 2020, 11, 588776. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Joern, R.R.; Forsthuber, T.G. Memory CD4(+) T Cells in Immunity and Autoimmune Diseases. Cells 2020, 9, 531. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Dong, C. A complex issue on CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 5–11. [Google Scholar] [CrossRef]
- Jiang, H.; Fu, D.; Bidgoli, A.; Paczesny, S. T Cell Subsets in Graft Versus Host Disease and Graft Versus Tumor. Front. Immunol. 2021, 12, 761448. [Google Scholar] [CrossRef] [PubMed]
- Mammadli, M.; Huang, W.; Harris, R.; Xiong, H.; Weeks, S.; May, A.; Gentile, T.; Henty-Ridilla, J.; Waickman, A.T.; August, A.; et al. Targeting SLP76:ITK interaction separates GVHD from GVL in allo-HSCT. iScience 2021, 24, 102286. [Google Scholar] [CrossRef]
- Papaioannou, V.E. The T-box gene family: Emerging roles in development, stem cells and cancer. Development 2014, 141, 3819–3833. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, D.; Yu, Y.; Heinrichs, J.; Wu, Y.; Schutt, S.; Kaosaard, K.; Liu, C.; Haarberg, K.; Bastian, D.; et al. T-bet is critical for the development of acute graft-versus-host disease through controlling T cell differentiation and function. J. Immunol. 2015, 194, 388–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, J.L.; Zhang, Q.; Ndonye, R.; Richardson, S.K.; Howell, A.R.; Gapin, L. T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells. Blood 2006, 107, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Yang, Q.; Zhu, Y.; Wang, H.R.; Chen, X.; Zhang, X.; Lu, B. T-Bet and Eomes Regulate the Balance between the Effector/Central Memory T Cells versus Memory Stem Like T Cells. PLoS ONE 2013, 8, e67401. [Google Scholar] [CrossRef] [Green Version]
- Mbitikon-Kobo, F.M.; Vocanson, M.; Michallet, M.C.; Tomkowiak, M.; Cottalorda, A.; Angelov, G.S.; Coupet, C.A.; Djebali, S.; Marcais, A.; Dubois, B.; et al. Characterization of a CD44/CD122int memory CD8 T cell subset generated under sterile inflammatory conditions. J. Immunol. 2009, 182, 3846–3854. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Dong, Y.; Tsurushita, N.; Tso, J.Y.; Fu, W. CD122 blockade restores immunological tolerance in autoimmune type 1 diabetes via multiple mechanisms. JCI Insight 2018, 3, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Heng, A.H.S.; Han, C.W.; Abbott, C.; McColl, S.R.; Comerford, I. Chemokine-Driven Migration of Pro-Inflammatory CD4(+) T Cells in CNS Autoimmune Disease. Front. Immunol. 2022, 13, 817473. [Google Scholar] [CrossRef]
- Chen, B.J.; Deoliveira, D.; Cui, X.; Le, N.T.; Son, J.; Whitesides, J.F.; Chao, N.J. Inability of memory T cells to induce graft-versus-host disease is a result of an abortive alloresponse. Blood 2007, 109, 3115–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.Z.; Liang, Y.; Nurieva, R.I.; Guo, F.; Anasetti, C.; Dong, C. Opposing effects of ICOS on graft-versus-host disease mediated by CD4 and CD8 T cells. J. Immunol. 2006, 176, 7394–7401. [Google Scholar] [CrossRef] [Green Version]
- Amatore, F.; Gorvel, L.; Olive, D. Role of Inducible Co-Stimulator (ICOS) in cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 141–150. [Google Scholar] [CrossRef]
- Mahajan, S.; Cervera, A.; MacLeod, M.; Fillatreau, S.; Perona-Wright, G.; Meek, S.; Smith, A.; MacDonald, A.; Gray, D. The role of ICOS in the development of CD4 T cell help and the reactivation of memory T cells. Eur. J. Immunol. 2007, 37, 1796–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhume, K.; Finn, C.M.; Strutt, T.M.; Sell, S.; McKinstry, K.K. T-bet optimizes CD4 T-cell responses against influenza through CXCR3-dependent lung trafficking but not functional programming. Mucosal. Immunol. 2019, 12, 1220–1230. [Google Scholar] [CrossRef]
- Duffner, U.; Lu, B.; Hildebrandt, G.C.; Teshima, T.; Williams, D.L.; Reddy, P.; Ordemann, R.; Clouthier, S.G.; Lowler, K.; Liu, C.; et al. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 2003, 31, 897–902. [Google Scholar] [CrossRef]
- Wu, J.Q.; Seay, M.; Schulz, V.P.; Hariharan, M.; Tuck, D.; Lian, J.; Du, J.; Shi, M.; Ye, Z.; Gerstein, M.; et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet. 2012, 8, e1002565. [Google Scholar] [CrossRef] [Green Version]
- Harly, C.; Kenney, D.; Wang, Y.; Ding, Y.; Zhao, Y.; Awasthi, P.; Bhandoola, A. A Shared Regulatory Element Controls the Initiation of Tcf7 Expression During Early T Cell and Innate Lymphoid Cell Developments. Front. Immunol. 2020, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Decman, V.; Laidlaw, B.J.; Dimenna, L.J.; Abdulla, S.; Mozdzanowska, K.; Erikson, J.; Ertl, H.C.; Wherry, E.J. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol. 2010, 184, 5151–5159. [Google Scholar] [CrossRef] [Green Version]
- Mammadli, M.; Huang, W.; Harris, R.; Sultana, A.; Cheng, Y.; Tong, W.; Pu, J.; Gentile, T.; Dsouza, S.; Yang, Q.; et al. Targeting Interleukin-2-Inducible T-Cell Kinase (ITK) Differentiates GVL and GVHD in Allo-HSCT. Front. Immunol. 2020, 11, 593863. [Google Scholar] [CrossRef]
- Mammadli, M.; Harris, R.; Suo, L.; May, A.; Gentile, T.; Waickman, A.T.; Bah, A.; August, A.; Nurmemmedov, E.; Karimi, M. Interleukin-2-inducible T-cell kinase (Itk) signaling regulates potent noncanonical regulatory T cells. Clin. Transl. Med. 2021, 11, e625. [Google Scholar] [CrossRef]
- Mammadli, M.; Harris, R.; Mahmudlu, S.; Verma, A.; May, A.; Dhawan, R.; Waickman, A.T.; Sen, J.M.; August, A.; Karimi, M. Human Wnt/beta-Catenin Regulates Alloimmune Signaling during Allogeneic Transplantation. Cancers 2021, 13, 3798. [Google Scholar] [CrossRef]
- Cooke, K.R.; Kobzik, L.; Martin, T.R.; Brewer, J.; Delmonte, J., Jr.; Crawford, J.M.; Ferrara, J.L. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 1996, 88, 3230–3239. [Google Scholar] [CrossRef] [Green Version]
- Peter, C.; Kielstein, J.T.; Clarke-Katzenberg, R.; Adams, M.C.; Pitsiouni, M.; Kambham, N.; Karimi, M.A.; Kengatharan, K.M.; Cooke, J.P. A novel bioluminescent tumor model of human renal cancer cell lines: An in vitro and in vivo characterization. J. Urol. 2007, 177, 2342–2346. [Google Scholar] [CrossRef]
- Nishimura, R.; Baker, J.; Beilhack, A.; Zeiser, R.; Olson, J.A.; Sega, E.I.; Karimi, M.; Negrin, R.S. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood 2008, 112, 2563–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breems, D.A.; Lowenberg, B. Autologous stem cell transplantation in the treatment of adults with acute myeloid leukaemia. Br. J. Haematol. 2005, 130, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Hale, G.; Waldmann, H. Control of graft-versus-host disease and graft rejection by T cell depletion of donor and recipient with Campath-1 antibodies. Results of matched sibling transplants for malignant diseases. Bone Marrow Transpl. 1994, 13, 597–611. [Google Scholar]
- Mony, J.T.; Khorooshi, R.; Owens, T. Chemokine receptor expression by inflammatory T cells in EAE. Front. Cell. Neurosci. 2014, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, S.; Allavena, P.; Albanesi, C.; Nasorri, F.; Bianchi, G.; Traidl, C.; Sozzani, S.; Girolomoni, G.; Cavani, A. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J. Immunol. 2001, 166, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonsdorf, A.S.; Hwang, S.T.; Enk, A.H. Chemokine receptors in T-cell-mediated diseases of the skin. J. Investig. Dermatol. 2009, 129, 2552–2566. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Scarponi, C.; Pallotta, S.; Daniele, R.; Bosisio, D.; Madonna, S.; Fortugno, P.; Gonzalvo-Feo, S.; Franssen, J.D.; Parmentier, M.; et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J. Exp. Med. 2009, 206, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelot, N.; Enot, D.P.; Flament, C.; Vimond, N.; Blattner, C.; Pitt, J.M.; Yamazaki, T.; Roberti, M.P.; Daillere, R.; Vetizou, M.; et al. Chemokine receptor patterns in lymphocytes mirror metastatic spreading in melanoma. J. Clin. Investig. 2016, 126, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shang, H.; Jiang, Y. Chemokines and Chemokine Receptors: Accomplices for Human Immunodeficiency Virus Infection and Latency. Front. Immunol. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Jagasia, M.; Storer, B.; Chai, X.; Pidala, J.; Cutler, C.; Arora, M.; Weisdorf, D.J.; Flowers, M.E.; Martin, P.J.; et al. Global and organ-specific chronic graft-versus-host disease severity according to the 2005 NIH Consensus Criteria. Blood 2011, 118, 4242–4249. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, K.; Eckert, F.; Welker, C.; Muller, H.; Muller, F.; Zips, D.; Sipos, B.; Klein, R.; Blank, G.; Feuchtinger, T.; et al. Chronic graft-versus-host-disease in CD34(+)-humanized NSG mice is associated with human susceptibility HLA haplotypes for autoimmune disease. J. Autoimmun. 2015, 62, 55–66. [Google Scholar] [CrossRef]
- Beilhack, A.; Schulz, S.; Baker, J.; Beilhack, G.F.; Wieland, C.B.; Herman, E.I.; Baker, E.M.; Cao, Y.A.; Contag, C.H.; Negrin, R.S. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood 2005, 106, 1113–1122. [Google Scholar] [CrossRef]
- Schreiner, D.; King, C.G. CD4+ Memory T Cells at Home in the Tissue: Mechanisms for Health and Disease. Front. Immunol. 2018, 9, 2394. [Google Scholar] [CrossRef] [Green Version]
- Zaph, C.; Rook, K.A.; Goldschmidt, M.; Mohrs, M.; Scott, P.; Artis, D. Persistence and function of central and effector memory CD4+ T cells following infection with a gastrointestinal helminth. J. Immunol. 2006, 177, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Rosetti, F.; Madera-Salcedo, I.K.; Rodriguez-Rodriguez, N.; Crispin, J.C. Regulation of activated T cell survival in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2022, 18, 232–244. [Google Scholar] [CrossRef]
- Oh, D.Y.; Fong, L. Cytotoxic CD4(+) T cells in cancer: Expanding the immune effector toolbox. Immunity 2021, 54, 2701–2711. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Utzschneider, D.T.; Gabriel, S.S.; Chisanga, D.; Gloury, R.; Gubser, P.M.; Vasanthakumar, A.; Shi, W.; Kallies, A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat. Immunol. 2020, 21, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, J.; Li, Y.; Xiao, M.; Wang, H.; Tian, Q.; Li, Z.; Tang, J.; Hu, L.; Tan, Y.; et al. The Transcription Factor TCF1 Preserves the Effector Function of Exhausted CD8 T Cells During Chronic Viral Infection. Front. Immunol. 2019, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, G.; Romano, M.; Nova-Lamperti, E.; Werner Sunderland, M.; Nerviani, A.; Scotta, C.; Bombardieri, M.; Quezada, S.A.; Sacks, S.H.; Noelle, R.J.; et al. PD-L1 signaling on human memory CD4+ T cells induces a regulatory phenotype. PLoS Biol. 2021, 19, e3001199. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.C.; Dundar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef]
- Seo, H.; Chen, J.; Gonzalez-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; Lopez-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais Ferreira, D.; Silva, J.G.; Wyss, T.; Fuertes Marraco, S.A.; Scarpellino, L.; Charmoy, M.; Maas, R.; Siddiqui, I.; Tang, L.; Joyce, J.A.; et al. Central memory CD8(+) T cells derive from stem-like Tcf7(hi) effector cells in the absence of cytotoxic differentiation. Immunity 2020, 53, 985–1000.e1011. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hathcock, K.S.; Klug, D.; Harada, Y.; Choudhury, B.; Allison, J.P.; Abe, R.; Hodes, R.J. Regulated costimulation in the thymus is critical for T cell development: Dysregulated CD28 costimulation can bypass the pre-TCR checkpoint. J. Immunol. 2005, 175, 4199–4207. [Google Scholar] [CrossRef] [Green Version]
- Teshima, T. Th1 and Th17 join forces for acute GVHD. Blood 2011, 118, 4765–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Tugues, S.; Amorim, A.; Spath, S.; Martin-Blondel, G.; Schreiner, B.; De Feo, D.; Lutz, M.; Guscetti, F.; Apostolova, P.; Haftmann, C.; et al. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Sci. Transl. Med. 2018, 10, eaat8410. [Google Scholar] [CrossRef] [Green Version]
- Bastien, J.P.; Roy, J.; Roy, D.C. Selective T-cell depletion for haplotype-mismatched allogeneic stem cell transplantation. Semin. Oncol. 2012, 39, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Bleakley, M.; Turtle, C.J.; Riddell, S.R. Augmentation of anti-tumor immunity by adoptive T-cell transfer after allogeneic hematopoietic stem cell transplantation. Expert Rev. Hematol. 2012, 5, 409–425. [Google Scholar] [CrossRef]
- Ferrara, J.L. Blood and Marrow Transplant Clinical Trials Network: Progress since the State of the Science Symposium 2007. Biol. Blood Marrow Transpl. 2014, 20, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, Y.G. The complex and central role of interferon-gamma in graft-versus-host disease and graft-versus-tumor activity. Immunol. Rev. 2014, 258, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherwinski, H.M.; Schumacher, J.H.; Brown, K.D.; Mosmann, T.R. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J. Exp. Med. 1987, 166, 1229–1244. [Google Scholar] [CrossRef] [Green Version]
- Abraham, K.M.; Teale, J.M. The contribution of parasite-specific T cells to isotype restriction in Mesocestoides corti-infected mice. J. Immunol. 1987, 139, 2530–2537. [Google Scholar] [CrossRef]
- Coghill, J.M.; Sarantopoulos, S.; Moran, T.P.; Murphy, W.J.; Blazar, B.R.; Serody, J.S. Effector CD4+ T cells, the cytokines they generate, and GVHD: Something old and something new. Blood 2011, 117, 3268–3276. [Google Scholar] [CrossRef]
- Germar, K.; Dose, M.; Konstantinou, T.; Zhang, J.; Wang, H.; Lobry, C.; Arnett, K.L.; Blacklow, S.C.; Aifantis, I.; Aster, J.C.; et al. T-cell factor 1 is a gatekeeper for T-cell specification in response to Notch signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 20060–20065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gounari, F.; Khazaie, K. TCF-1: A maverick in T cell development and function. Nat. Immunol. 2022, 23, 671–678. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Huang, F.; Kannan, A.K.; Hu, J.; August, A. ITK tunes IL-4-induced development of innate memory CD8+ T cells in a gammadelta T and invariant NKT cell-independent manner. J. Leukoc. Biol. 2014, 96, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.E.; McNiff, J.; Yan, J.; Doyle, H.; Mamula, M.; Shlomchik, M.J.; Shlomchik, W.D. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Investig. 2003, 112, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Wu, Y.; Bastian, D.; Iamsawat, S.; Chang, J.; Daenthanasanmak, A.; Nguyen, H.D.; Schutt, S.; Dai, M.; Chen, F.; et al. Inducible T-Cell Co-Stimulator Impacts Chronic Graft-Versus-Host Disease by Regulating Both Pathogenic and Regulatory T Cells. Front. Immunol. 2018, 9, 1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.X.; Holland, A.M.; Na, I.K.; Terwey, T.H.; Alpdogan, O.; Bautista, J.L.; Smith, O.M.; Suh, D.; King, C.; Kochman, A.; et al. Absence of P-selectin in recipients of allogeneic bone marrow transplantation ameliorates experimental graft-versus-host disease. J. Immunol. 2010, 185, 1912–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, C.A.; Burkett, S.B.; Panoskaltsis-Mortari, A.; Kirby, S.L.; Luster, A.D.; McKinnon, K.; Blazar, B.R.; Serody, J.S. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J. Immunol. 2004, 173, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, P.M. The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 1994, 12, 593–633. [Google Scholar] [CrossRef] [PubMed]
- Galeano Nino, J.L.; Pageon, S.V.; Tay, S.S.; Colakoglu, F.; Kempe, D.; Hywood, J.; Mazalo, J.K.; Cremasco, J.; Govendir, M.A.; Dagley, L.F.; et al. Cytotoxic T cells swarm by homotypic chemokine signalling. elife 2020, 9, e56554. [Google Scholar] [CrossRef]
- Verbeek, S.; Izon, D.; Hofhuis, F.; Robanus-Maandag, E.; te Riele, H.; van de Wetering, M.; Oosterwegel, M.; Wilson, A.; MacDonald, H.R.; Clevers, H. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 1995, 374, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 2020, 11, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro-Garcia, M.A.; Alonso-Arias, R.; Lopez-Larrea, C. When Aging Reaches CD4+ T-Cells: Phenotypic and Functional Changes. Front. Immunol. 2013, 4, 107. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, J.; Zhou, H.; Zhou, J.; Deng, Z.; Lu, Y.; Rao, J.; Ji, G.; Gu, J.; Yang, X.; et al. Inhibition of Glycogen Synthase Kinase 3beta Increases the Proportion and Suppressive Function of CD19(+)CD24(hi)CD27(+) Breg Cells. Front. Immunol. 2020, 11, 603288. [Google Scholar] [CrossRef]
- Steinke, F.C.; Yu, S.; Zhou, X.; He, B.; Yang, W.; Zhou, B.; Kawamoto, H.; Zhu, J.; Tan, K.; Xue, H.-H. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat. Immunol. 2014, 15, 646–656. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mammadli, M.; Suo, L.; Sen, J.M.; Karimi, M. TCF-1 Is Required for CD4 T Cell Persistence Functions during AlloImmunity. Int. J. Mol. Sci. 2023, 24, 4326. https://doi.org/10.3390/ijms24054326
Mammadli M, Suo L, Sen JM, Karimi M. TCF-1 Is Required for CD4 T Cell Persistence Functions during AlloImmunity. International Journal of Molecular Sciences. 2023; 24(5):4326. https://doi.org/10.3390/ijms24054326
Chicago/Turabian StyleMammadli, Mahinbanu, Liye Suo, Jyoti Misra Sen, and Mobin Karimi. 2023. "TCF-1 Is Required for CD4 T Cell Persistence Functions during AlloImmunity" International Journal of Molecular Sciences 24, no. 5: 4326. https://doi.org/10.3390/ijms24054326