
(S)-2-(Cyclobutylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)isonicotinamide Attenuates RANKL-Induced Osteoclast Differentiation by Inhibiting NF-κB Nuclear Translocation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
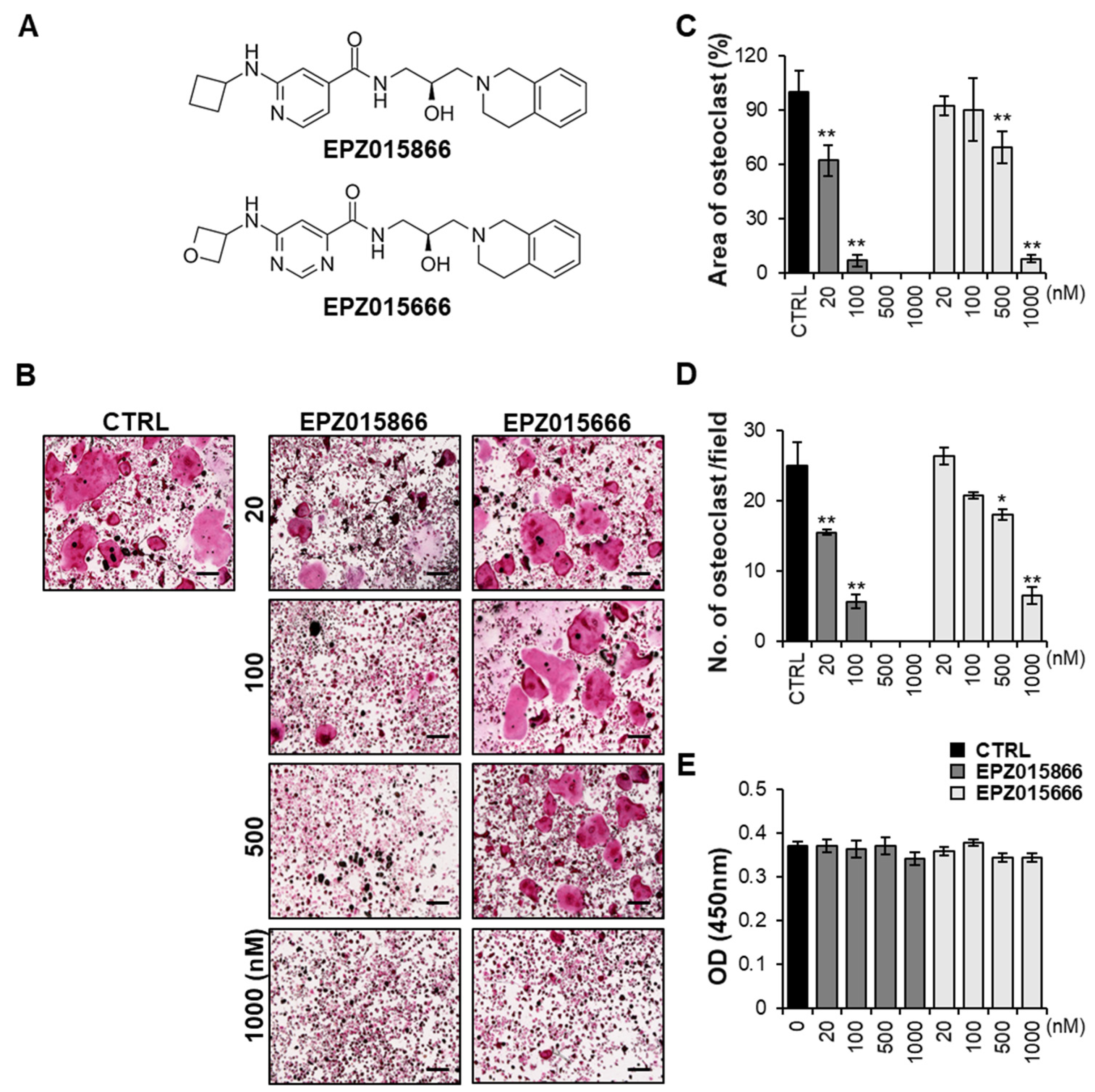
2.1. EPZ Compounds Attenuates RANKL-Induced Osteoclast Differentiation In Vitro
2.2. EPZ Compounds Suppress F-Actin Ring Formation and Bone Resorption
2.3. EPZ Compounds Inhibit the Expression of Osteoclast-Specific Genes
2.4. EPZ Compounds Decrease the Expression of the Transcription Factors PU.1 and NFATc1
2.5. EPZ Compounds Downregulate PRMT5 and H3R8me2s/H4R3me2s but Do Not Impact mRNA Levels of Prmt5 during Osteoclastogenesis
2.6. EPZ Compounds Reduce NF-κB Nuclear Translocation by Blocking the Demethylation of p65
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Osteoclast Cell Culture and Viability Assay
4.3. Tartrate-Resistant Acid Phosphatase (TRAP) Staining Assay
4.4. Osteoblast Cell Culture and In Vitro Differentiation
4.5. Phalloidin Staining and Immunofluorescence (IF) Staining
4.6. Bone Resorption Assay
4.7. Western Blot Assay
4.8. Real-Time PCR Assay
4.9. Nuclear and Cytoplasmic Extraction
4.10. Cell Transfection and Immunoprecipitation (IP)
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, X.; McDonald, J.M. Disorders of Bone Remodeling. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjidakis, D.J.; Androulakis, I.I. Bone Remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Dontas, I.A.; Yiannakopoulos, C.K. Risk Factors and Prevention of Osteoporosis-Related Fractures. J. Musculoskelet. Neuronal Interact. 2007, 7, 268–272. [Google Scholar] [PubMed]
- Coughlan, T.; Dockery, F. Osteoporosis and Fracture Risk in Older People. Clin. Med. 2014, 14, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Giusti, A.; Bianchi, G. Treatment of Primary Osteoporosis in Men. Clin. Interv. Aging 2015, 10, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasilakis, A.D.; Polyzos, S.A.; Makras, P.; Aubry-Rozier, B.; Kaouri, S.; Lamy, O. Clinical Features of 24 Patients With Rebound-Associated Vertebral Fractures After Denosumab Discontinuation: Systematic Review and Additional Cases. J. Bone Miner. Res. 2017, 32, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.R.; Ferrari, S.; Eastell, R.; Gilchrist, N.; Jensen, J.-E.B.; McClung, M.; Roux, C.; Törring, O.; Valter, I.; Wang, A.T.; et al. Vertebral Fractures After Discontinuation of Denosumab: A Post Hoc Analysis of the Randomized Placebo-Controlled FREEDOM Trial and Its Extension. J. Bone Miner. Res. 2018, 33, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Khosla, S.; Bilezikian, J.P.; Dempster, D.W.; Lewiecki, E.M.; Miller, P.D.; Neer, R.M.; Recker, R.R.; Shane, E.; Shoback, D.; Potts, J.T. Benefits and Risks of Bisphosphonate Therapy for Osteoporosis. J. Clin. Endocrinol. Metab. 2012, 97, 2272–2282. [Google Scholar] [CrossRef] [Green Version]
- Teitelbaum, S.L. Bone Resorption by Osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Otero, K.; Turnbull, I.R.; Poliani, P.L.; Vermi, W.; Cerutti, E.; Aoshi, T.; Tassi, I.; Takai, T.; Stanley, S.L.; Miller, M.; et al. Macrophage Colony-Stimulating Factor Induces the Proliferation and Survival of Macrophages via a Pathway Involving DAP12 and Beta-Catenin. Nat. Immunol. 2009, 10, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of Osteoclast Differentiation and Function by the New Members of the Tumor Necrosis Factor Receptor and Ligand Families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 Expression Determines Its Essential Role in Bone Homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential Role of P38 Mitogen-Activated Protein Kinase in Cathepsin K Gene Expression during Osteoclastogenesis through Association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jing, L.; Li, M.; He, L.; Guo, Z. Regulation of Histone Arginine Methylation/Demethylation by Methylase and Demethylase (Review). Mol. Med. Rep. 2019, 19, 3963–3971. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, W.-G. Advances in histone methyltransferases and histone demethylases. Ai Zheng Aizheng Chin. J. Cancer 2008, 27, 1018–1025. [Google Scholar]
- Zhao, L.; Duan, Y.-T.; Lu, P.; Zhang, Z.-J.; Zheng, X.-K.; Wang, J.-L.; Feng, W.-S. Epigenetic Targets and Their Inhibitors in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 2395–2419. [Google Scholar] [CrossRef]
- Wei, X.; Yi, X.; Zhu, X.-H.; Jiang, D.-S. Histone Methylation and Vascular Biology. Clin. Epigenetics 2020, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.F.; Levine, R.L. Genetic and Epigenetic Determinants of AML Pathogenesis. Semin. Hematol. 2019, 56, 84–89. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Sun, H.-L.; Liang, H.; Li, K.; Fan, Q.-M.; Zhao, Q.-H. Dynamic and Distinct Histone Modifications of Osteogenic Genes during Osteogenic Differentiation. J. Biochem. 2015, 158, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, A.; Bedford, M.T. Histone Arginine Methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, Y.; Cáceres, T.; May, K.; Hevel, J.M. Biochemistry and Regulation of the Protein Arginine Methyltransferases (PRMTs). Arch. Biochem. Biophys. 2016, 590, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Rui, L. PRMT5 in Gene Regulation and Hematologic Malignancies. Genes Dis. 2019, 6, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wen, C.; Jiang, H.; Ma, S.; Liu, X. Protein Arginine Methyltransferase 5 Functions via Interacting Proteins. Front. Cell Dev. Biol. 2021, 9, 725301. [Google Scholar] [CrossRef]
- Dong, Y.; Song, C.; Wang, Y.; Lei, Z.; Xu, F.; Guan, H.; Chen, A.; Li, F. Inhibition of PRMT5 Suppresses Osteoclast Differentiation and Partially Protects against Ovariectomy-Induced Bone Loss through Downregulation of CXCL10 and RSAD2. Cell. Signal. 2017, 34, 55–65. [Google Scholar] [CrossRef]
- Yan, Y.; Zhao, P.; Wang, Z.; Liu, Z.; Wang, Z.; Zhang, J.; Ding, Y.; Hua, X.; Yu, L. PRMT5 Regulates Colorectal Cancer Cell Growth and EMT via EGFR/Akt/GSK3β Signaling Cascades. Aging 2021, 13, 4468–4481. [Google Scholar] [CrossRef]
- Abu-Amer, Y. NF-ΚB Signaling and Bone Resorption. Osteoporos. Int. 2013, 24, 2377–2386. [Google Scholar] [CrossRef] [Green Version]
- Alles, N.; Soysa, N.S.; Hayashi, J.; Khan, M.; Shimoda, A.; Shimokawa, H.; Ritzeler, O.; Akiyoshi, K.; Aoki, K.; Ohya, K. Suppression of NF-KappaB Increases Bone Formation and Ameliorates Osteopenia in Ovariectomized Mice. Endocrinology 2010, 151, 4626–4634. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Stark, G.R. NF-ΚB: Regulation by Methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Yang, M.; Huang, D.-B.; Wei, H.; Ozer, G.H.; Ghosh, G.; Stark, G.R. Role of Lysine Methylation of NF-ΚB in Differential Gene Regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 13510–13515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Huang, X.; Qi, Y.; Qian, Z.; Ni, S.; Zhong, Z.; Zhang, X.; Li, D.; Yu, B. Juglanin Inhibits Osteoclastogenesis in Ovariectomized Mice via the Suppression of NF-ΚB Signaling Pathways. Front. Pharmacol. 2021, 11, 596230. [Google Scholar] [CrossRef]
- Ea, C.-K.; Baltimore, D. Regulation of NF-KappaB Activity through Lysine Monomethylation of P65. Proc. Natl. Acad. Sci. USA 2009, 106, 18972–18977. [Google Scholar] [CrossRef] [Green Version]
- Reintjes, A.; Fuchs, J.E.; Kremser, L.; Lindner, H.H.; Liedl, K.R.; Huber, L.A.; Valovka, T. Asymmetric Arginine Dimethylation of RelA Provides a Repressive Mark to Modulate TNFα/NF-ΚB Response. Proc. Natl. Acad. Sci. USA 2016, 113, 4326–4331. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, B.; Miyagi, M.; She, Y.; Gopalan, B.; Huang, D.-B.; Ghosh, G.; Stark, G.R.; Lu, T. PRMT5 Dimethylates R30 of the P65 Subunit to Activate NF-ΚB. Proc. Natl. Acad. Sci. USA 2013, 110, 13516–13521. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.P.; Bandyopadhyay, S.; Maxwell, T.J.; Willard, B.; DiCorleto, P.E. Tumor Necrosis Factor (TNF)-α Induction of CXCL10 in Endothelial Cells Requires Protein Arginine Methyltransferase 5 (PRMT5)-Mediated Nuclear Factor (NF)-ΚB P65 Methylation. J. Biol. Chem. 2014, 289, 15328–15339. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Nakayamada, S.; Okada, Y. Osteoblasts and Osteoclasts in Bone Remodeling and Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 325–328. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xiu, Y.; Li, J.; Xing, L.; Yao, Z. NF-ΚB-Mediated Regulation of Osteoclastogenesis. Endocrinol. Metab. 2015, 30, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Zuo, J.; Holliday, L.S. Specialized Roles for Actin in Osteoclasts: Unanswered Questions and Therapeutic Opportunities. Biomolecules 2019, 9, E17. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, T.; Myoui, A.; Ikeda, F.; Hata, K.; Yoshikawa, H.; Nishimura, R.; Yoneda, T. Critical Role of Cortactin in Actin Ring Formation and Osteoclastic Bone Resorption. J. Bone Miner. Metab. 2006, 24, 368–372. [Google Scholar] [CrossRef]
- Kwon, O.H.; Lee, C.-K.; Lee, Y.I.; Paik, S.-G.; Lee, H.-J. The Hematopoietic Transcription Factor PU.1 Regulates RANK Gene Expression in Myeloid Progenitors. Biochem. Biophys. Res. Commun. 2005, 335, 437–446. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.Q.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear Factor of Activated T-Cells (NFAT) Rescues Osteoclastogenesis in Precursors Lacking c-Fos. J. Biol. Chem. 2004, 279, 26475–26480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, H.; Rho, J.; Jeong, D.; Park, R.; Fisher, D.E.; Ostrowski, M.C.; Choi, Y.; Kim, N. Microphthalmia Transcription Factor and PU.1 Synergistically Induce the Leukocyte Receptor Osteoclast-Associated Receptor Gene Expression. J. Biol. Chem. 2003, 278, 24209–24216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Katagiri, H.; Kanegae, Y.; Takayanagi, H.; Sawada, Y.; Yamamoto, A.; Pando, M.P.; Asano, T.; Verma, I.M.; Oda, H.; et al. Reciprocal Role of ERK and NF-KappaB Pathways in Survival and Activation of Osteoclasts. J. Cell Biol. 2000, 148, 332–342. [Google Scholar] [CrossRef]
- Ishiyama, K.; Yashiro, T.; Nakano, N.; Kasakura, K.; Miura, R.; Hara, M.; Kawai, F.; Maeda, K.; Tamura, N.; Okumura, K.; et al. Involvement of PU.1 in NFATc1 Promoter Function in Osteoclast Development. Allergol. Int. 2015, 64, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Jimi, E.; Takakura, N.; Hiura, F.; Nakamura, I.; Hirata-Tsuchiya, S. The Role of NF-ΚB in Physiological Bone Development and Inflammatory Bone Diseases: Is NF-ΚB Inhibition “Killing Two Birds with One Stone”? Cells 2019, 8, 1636. [Google Scholar] [CrossRef] [Green Version]
- Iotsova, V.; Caamaño, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in Mice Lacking NF-KappaB1 and NF-KappaB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-KappaB in Osteoclast and B-Cell Development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [Green Version]
- Novack, D.V. Role of NF-ΚB in the Skeleton. Cell Res. 2011, 21, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Yang, X.-D.; Lamb, A.; Chen, L.-F. Posttranslational Modifications of NF-KappaB: Another Layer of Regulation for NF-KappaB Signaling Pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D.; Gilmore, T.D. Good Cop, Bad Cop: The Different Faces of NF-KappaB. Cell Death Differ. 2006, 13, 759–772. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. NF-KappaB RelA Phosphorylation Regulates RelA Acetylation. Mol. Cell. Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [Green Version]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-KappaB Signaling by Pin1-Dependent Prolyl Isomerization and Ubiquitin-Mediated Proteolysis of P65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Levy, D.; Kuo, A.J.; Chang, Y.; Schaefer, U.; Kitson, C.; Cheung, P.; Espejo, A.; Zee, B.M.; Liu, C.L.; Tangsombatvisit, S.; et al. Lysine Methylation of the NF-ΚB Subunit RelA by SETD6 Couples Activity of the Histone Methyltransferase GLP at Chromatin to Tonic Repression of NF-ΚB Signaling. Nat. Immunol. 2011, 12, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zhang, X.-O.; Verdejo-Torres, O.; Wigglesworth, K.; Sun, X.; Sallis, B.; Moon, D.; Huang, T.; Rozen, E.; Wang, G.; et al. Protein Arginine Methyltransferase 5 Promotes Metastasis via Enhancing EGFR Transcription and Modulating AKT1 Activation by Methylation. arXiv 2020. [Google Scholar] [CrossRef]
- Harris, D.P.; Chandrasekharan, U.M.; Bandyopadhyay, S.; Willard, B.; DiCorleto, P.E. PRMT5-Mediated Methylation of NF-ΚB P65 at Arg174 Is Required for Endothelial CXCL11 Gene Induction in Response to TNF-α and IFN-γ Costimulation. PLoS ONE 2016, 11, e0148905. [Google Scholar] [CrossRef] [Green Version]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.; Lee, J.-K.; Lee, J.-Y.; Chen, Z.; Ahn, S.-H.; Kim, N.D.; Kook, M.-S.; Min, S.H.; Park, B.-J.; Lee, T.-H. BCPA {N,N′-1,4-Butanediylbis[3-(2-Chlorophenyl)Acrylamide]} Inhibits Osteoclast Differentiation through Increased Retention of Peptidyl-Prolyl Cis-Trans Isomerase Never in Mitosis A-Interacting 1. Int. J. Mol. Sci. 2018, 19, 3436. [Google Scholar] [CrossRef] [Green Version]
- Weigert, J.; Neumeier, M.; Wanninger, J.; Schober, F.; Sporrer, D.; Weber, M.; Schramm, A.; Wurm, S.; Stögbauer, F.; Filarsky, M.; et al. Adiponectin Upregulates Monocytic Activin A but Systemic Levels Are Not Altered in Obesity or Type 2 Diabetes. Cytokine 2009, 45, 86–91. [Google Scholar] [CrossRef]
- Cho, E.; Chen, Z.; Ding, M.; Seong, J.; Lee, S.; Min, S.H.; Choi, D.K.; Lee, T.-H. PMSA Prevents Osteoclastogenesis and Estrogen-Dependent Bone Loss in Mice. Bone 2021, 142, 115707. [Google Scholar] [CrossRef]
- Chen, Z.; Ding, M.; Cho, E.; Seong, J.; Lee, S.; Lee, T.-H. 2-NPPA Mitigates Osteoclastogenesis via Reducing TRAF6-Mediated c-Fos Expression. Front. Pharmacol. 2021, 11, 599081. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, M.; Cho, E.; Chen, Z.; Park, S.-W.; Lee, T.-H. (S)-2-(Cyclobutylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)isonicotinamide Attenuates RANKL-Induced Osteoclast Differentiation by Inhibiting NF-κB Nuclear Translocation. Int. J. Mol. Sci. 2023, 24, 4327. https://doi.org/10.3390/ijms24054327
Ding M, Cho E, Chen Z, Park S-W, Lee T-H. (S)-2-(Cyclobutylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)isonicotinamide Attenuates RANKL-Induced Osteoclast Differentiation by Inhibiting NF-κB Nuclear Translocation. International Journal of Molecular Sciences. 2023; 24(5):4327. https://doi.org/10.3390/ijms24054327
Chicago/Turabian StyleDing, Mina, Eunjin Cho, Zhihao Chen, Sang-Wook Park, and Tae-Hoon Lee. 2023. "(S)-2-(Cyclobutylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)isonicotinamide Attenuates RANKL-Induced Osteoclast Differentiation by Inhibiting NF-κB Nuclear Translocation" International Journal of Molecular Sciences 24, no. 5: 4327. https://doi.org/10.3390/ijms24054327
APA StyleDing, M., Cho, E., Chen, Z., Park, S.-W., & Lee, T.-H. (2023). (S)-2-(Cyclobutylamino)-N-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl)isonicotinamide Attenuates RANKL-Induced Osteoclast Differentiation by Inhibiting NF-κB Nuclear Translocation. International Journal of Molecular Sciences, 24(5), 4327. https://doi.org/10.3390/ijms24054327