

Construction of Two Independent RAB Family-Based Scoring Systems Based on Machine Learning Algorithms and Definition of RAB13 as a Novel Therapeutic Target for Hepatocellular Carcinoma
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
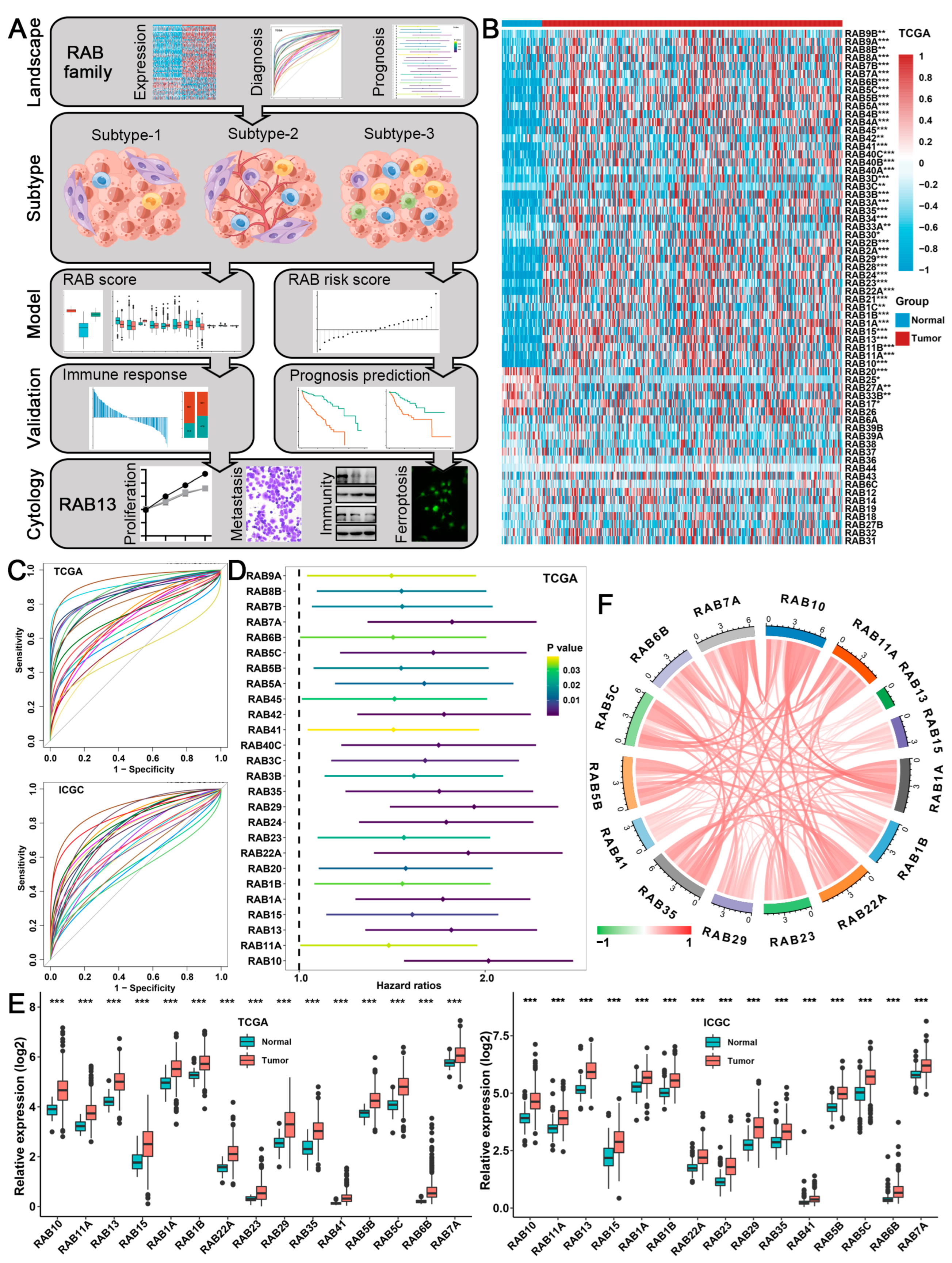
2.1. Expression, Diagnosis, and Prognosis of the RAB Family in HCC
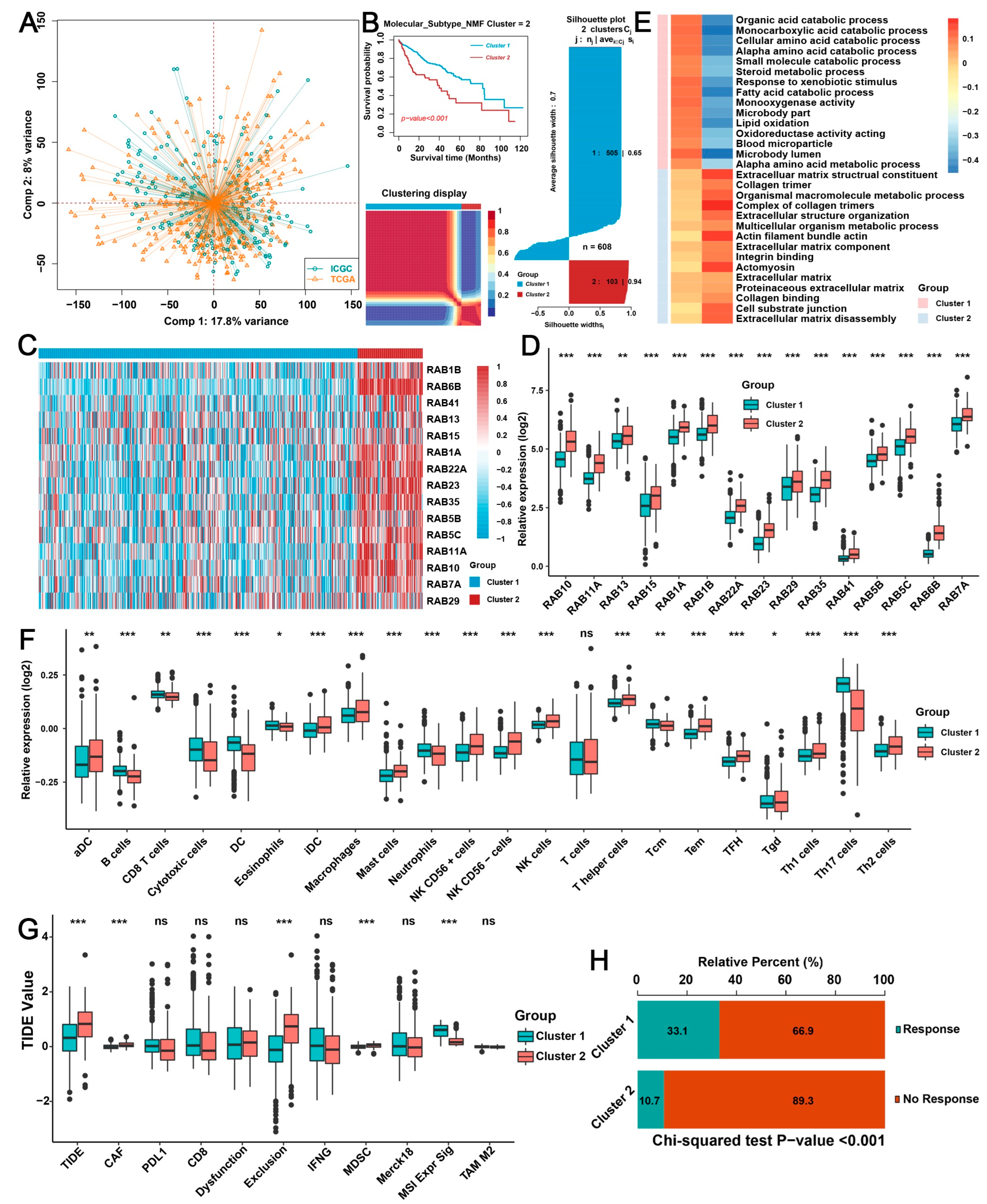
2.2. Biological Characteristics of Distinct RAB Clusters in HCC
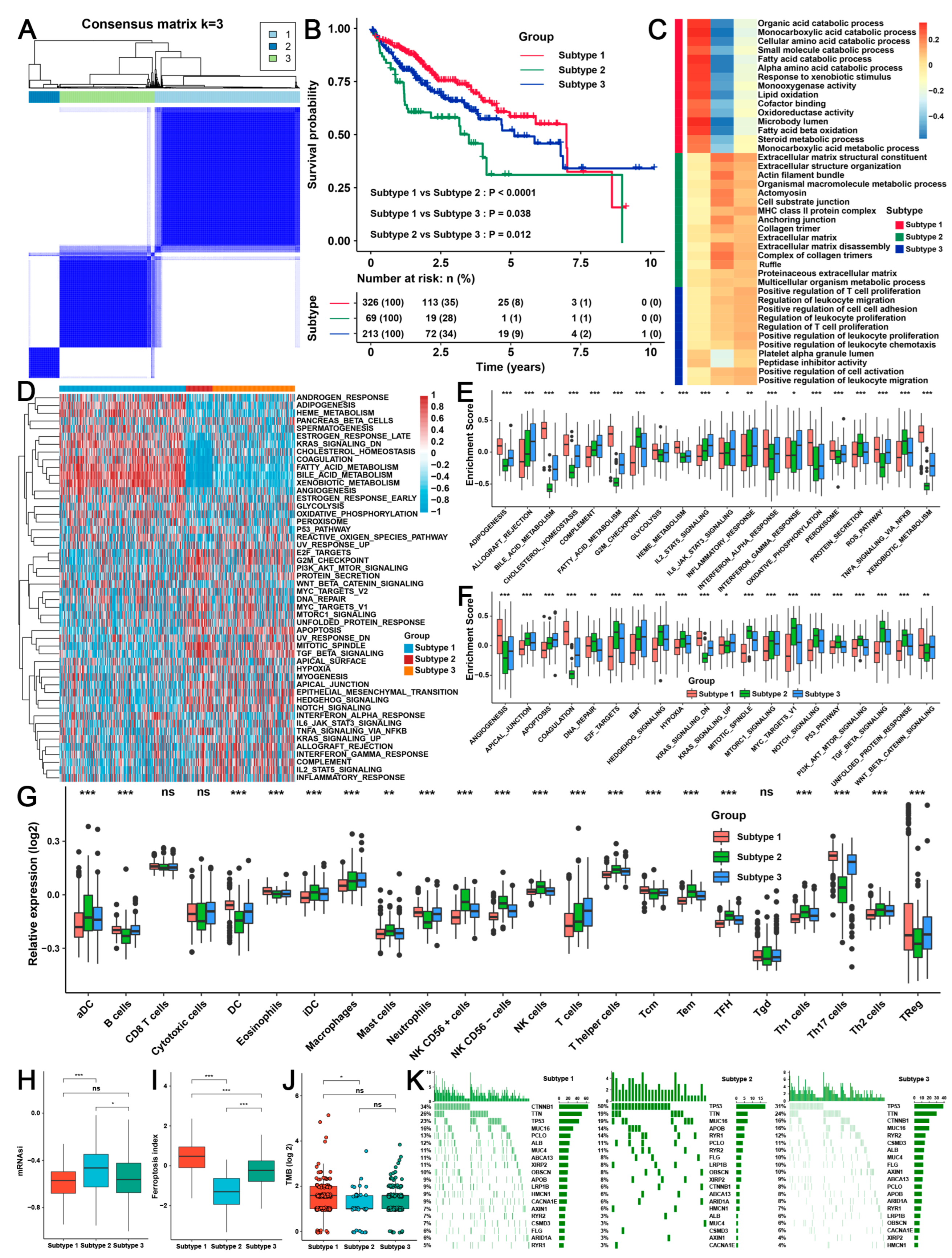
2.3. Three TME Subtypes Were Revealed by Unsupervised Clustering Analysis of the RAB-Associated Signatures in HCC
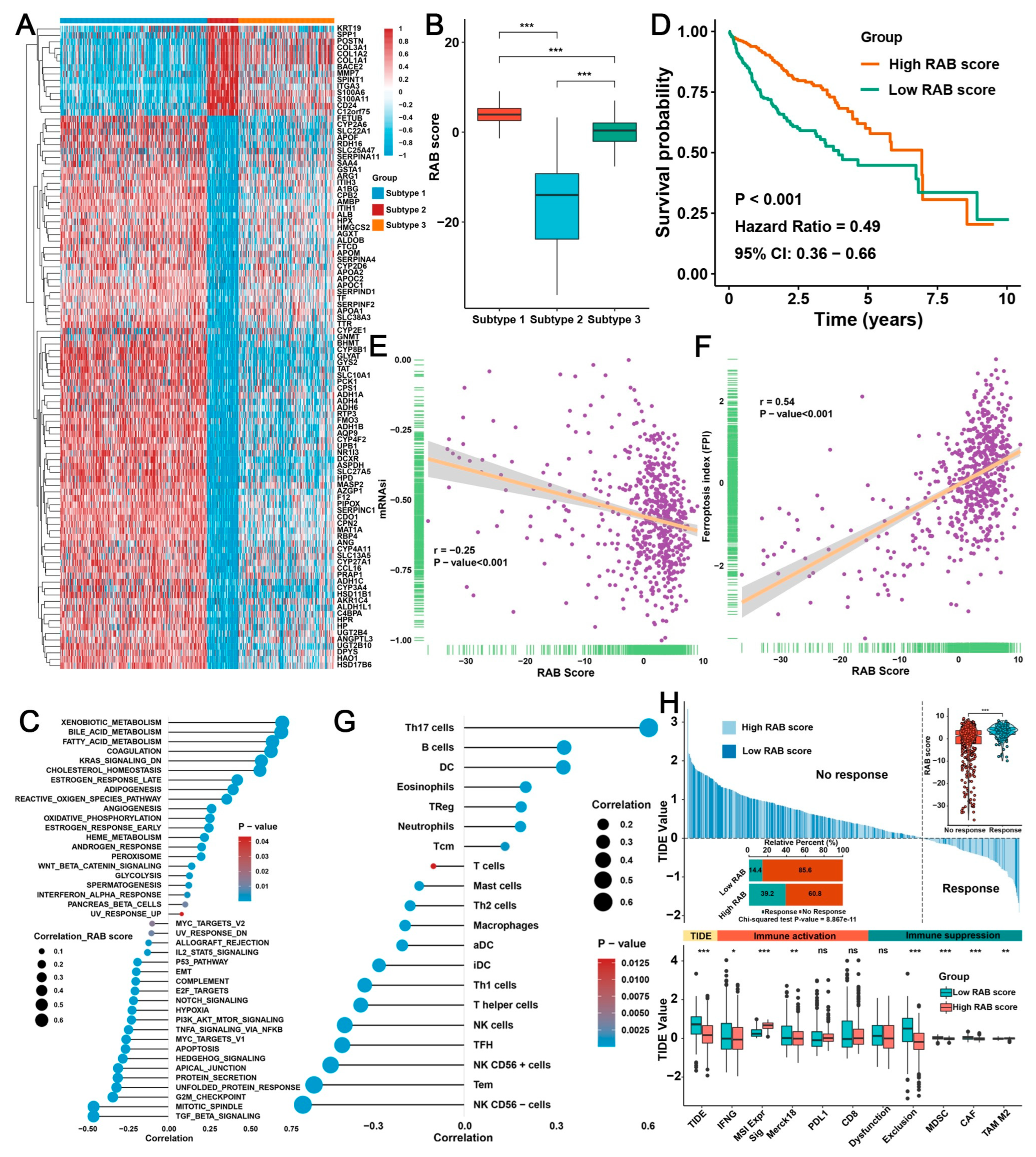
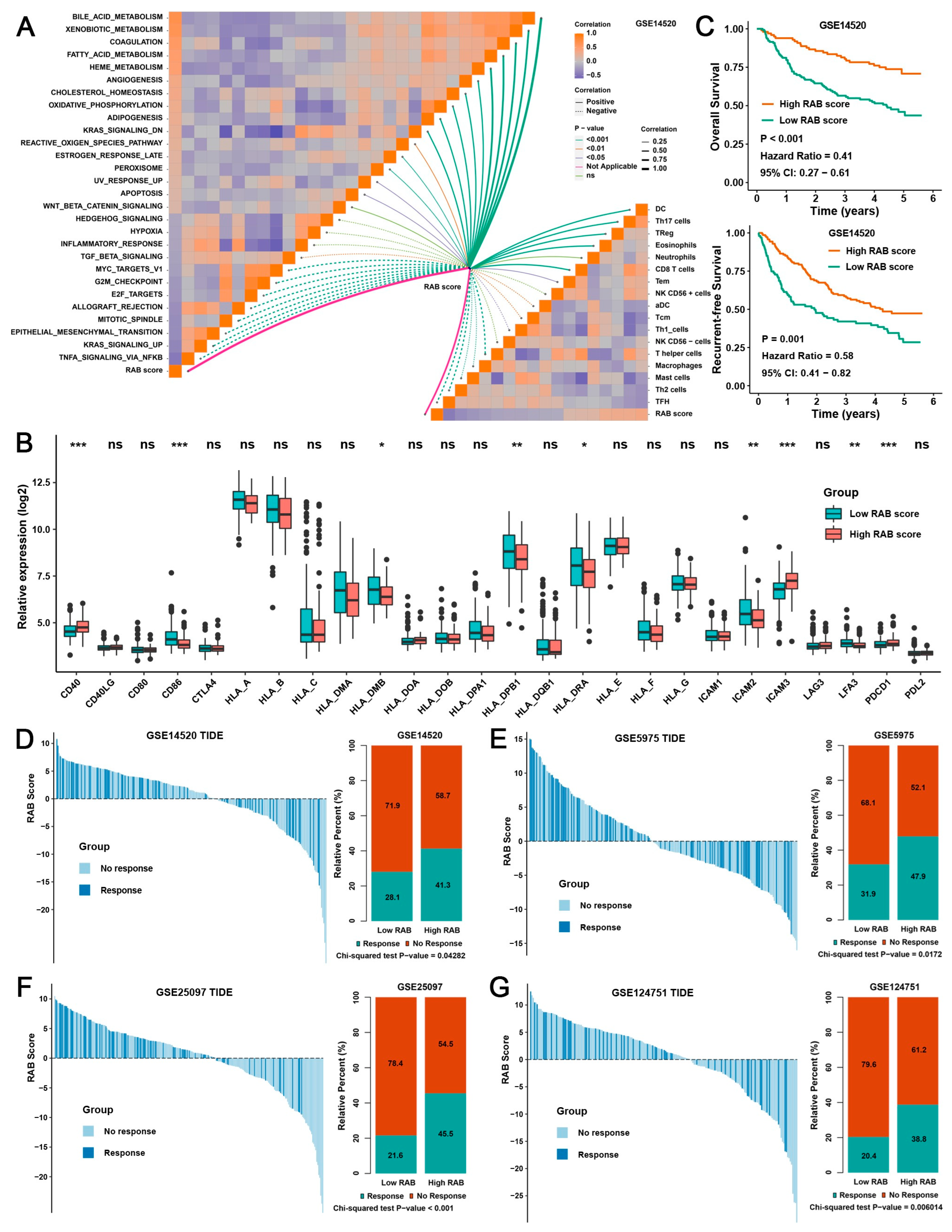
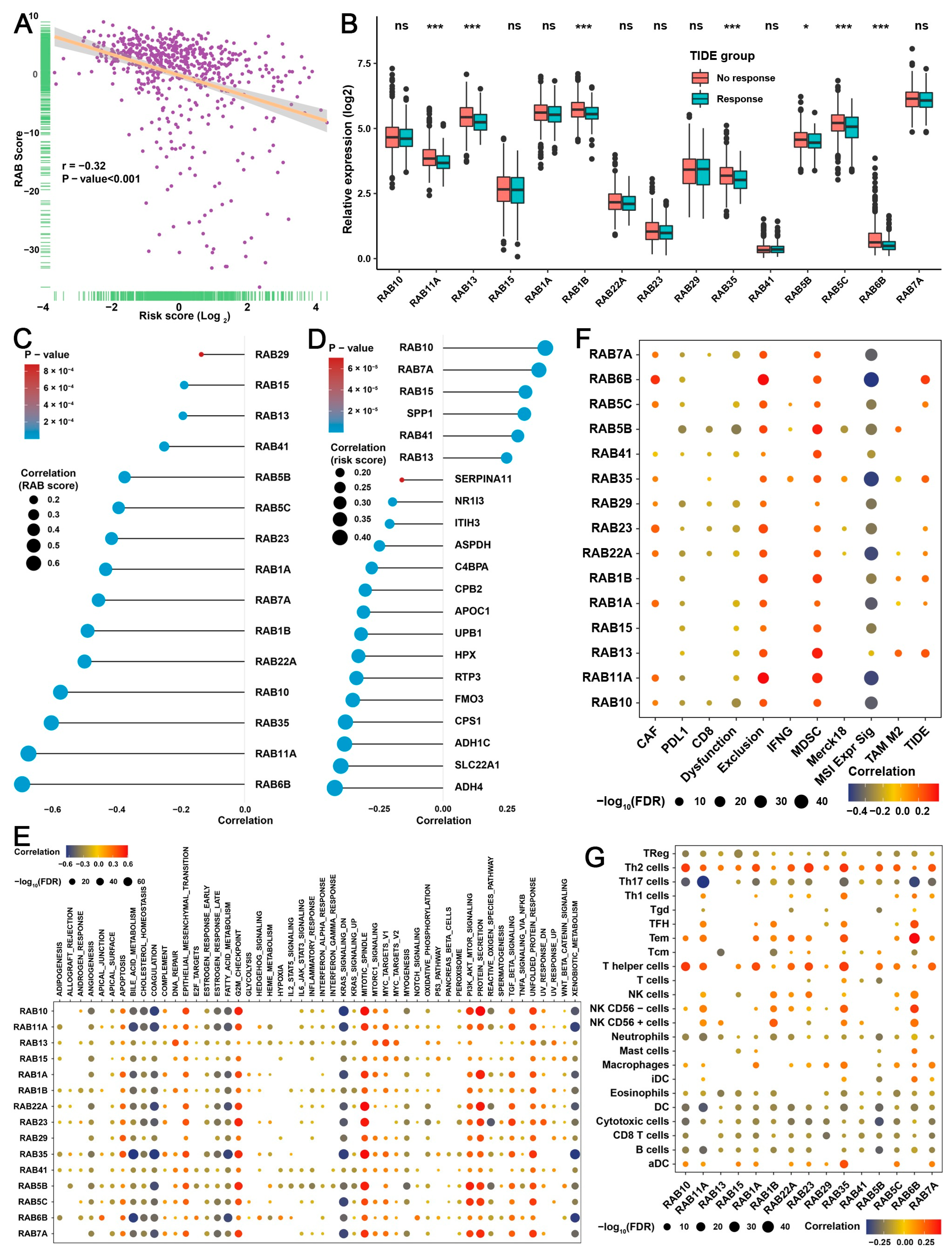
2.4. Construction of a RAB Score and Evaluation of Its Predictive Ability in the Pooled HCC Cohort
2.5. Validation of the RAB Score in Response to Immunotherapy
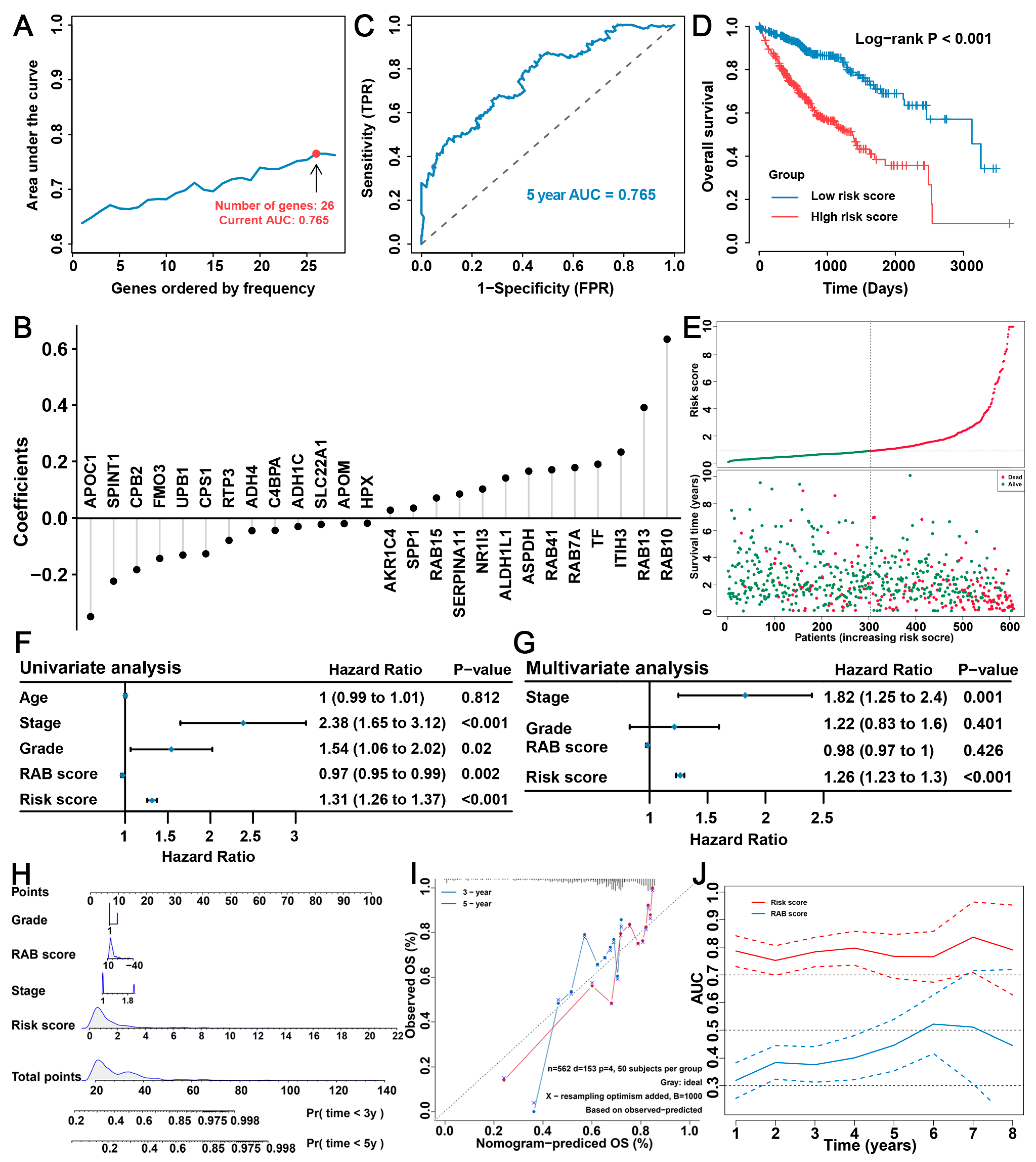
2.6. Construction of the RAB Risk Score to Better Predict the Prognosis of HCC Patients
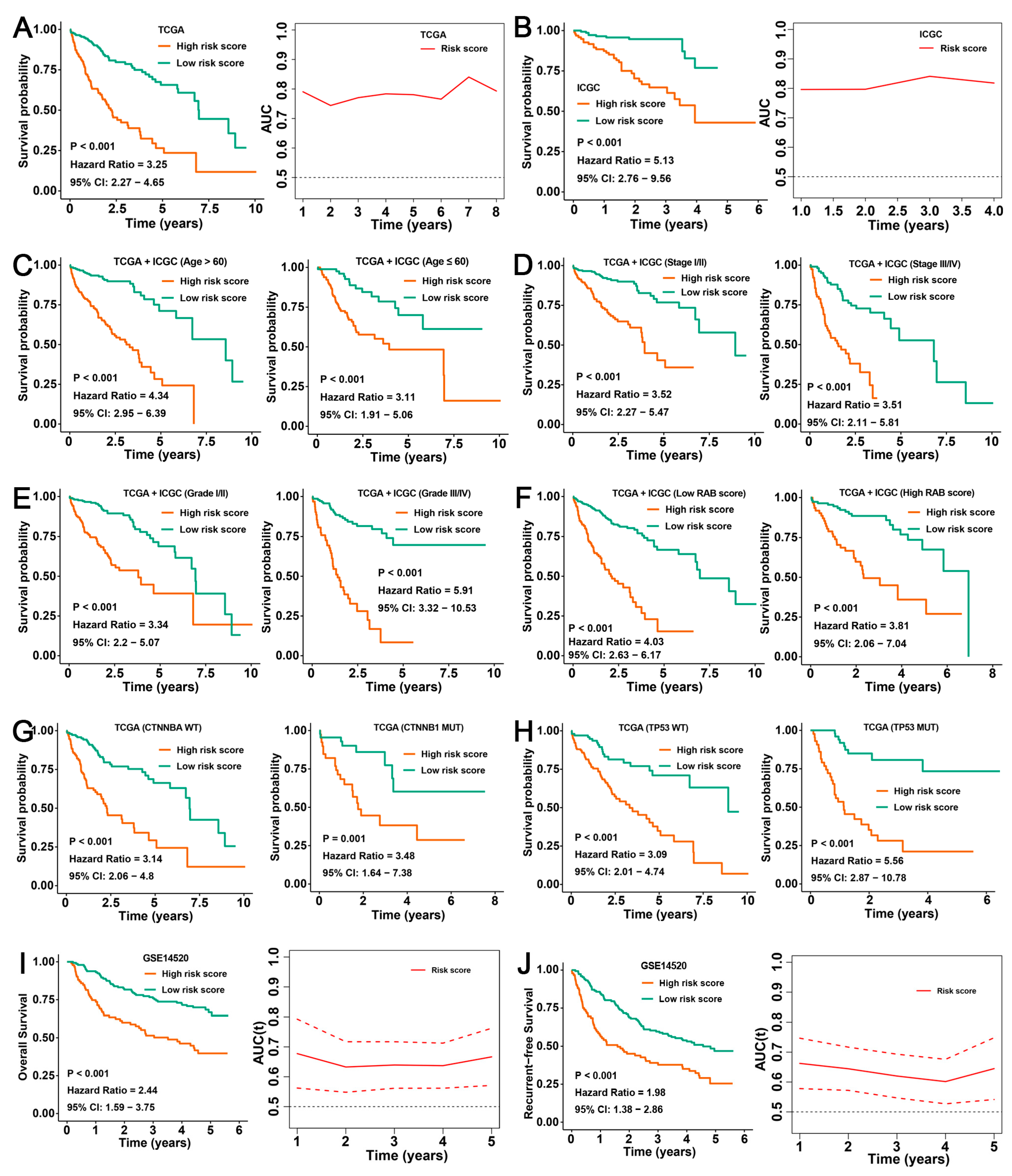
2.7. Validation of the Prognostic Predictive Ability of the RAB Risk Score
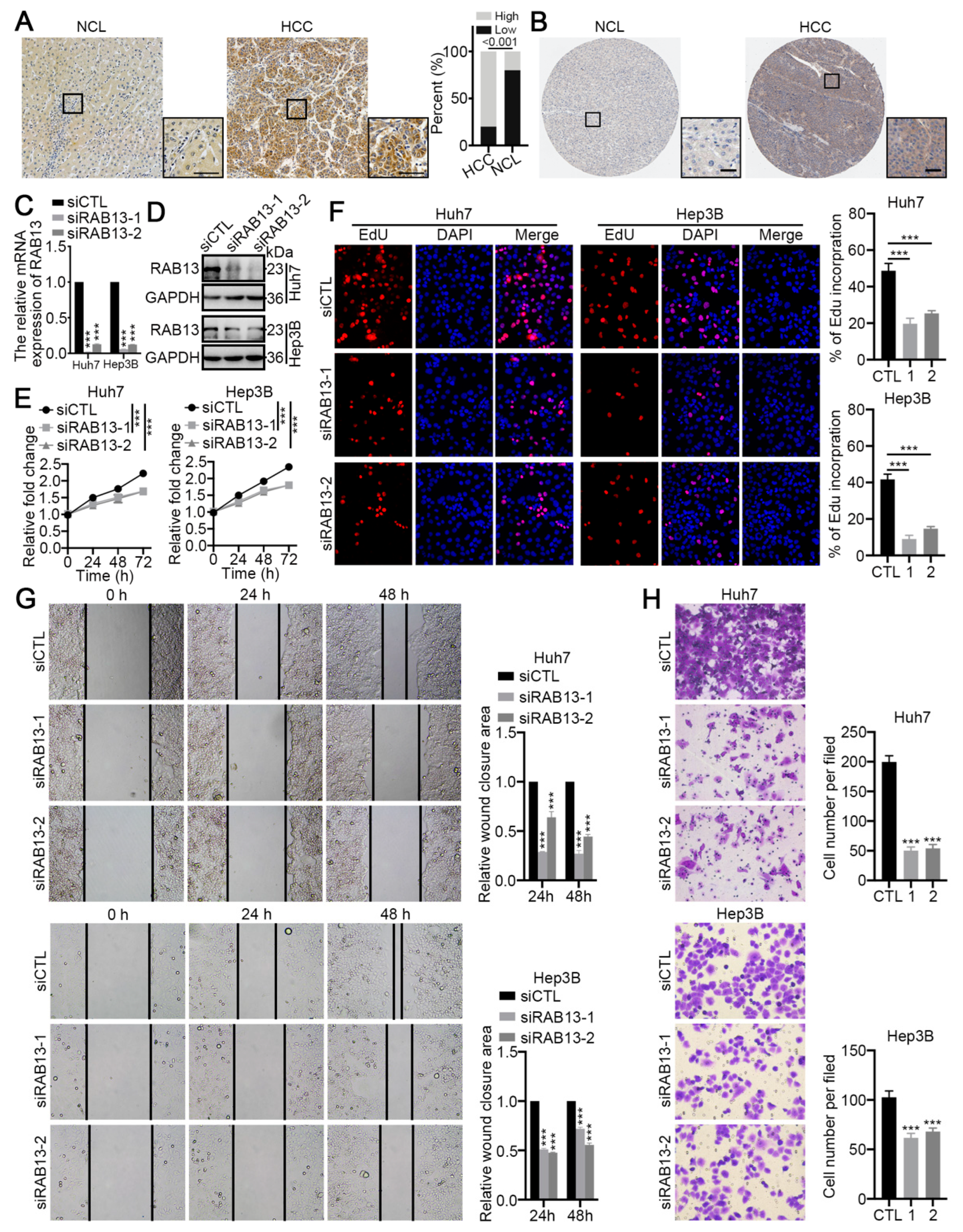
2.8. RAB13 Is Essential for the Malignant Biological Behaviors of HCC Cells
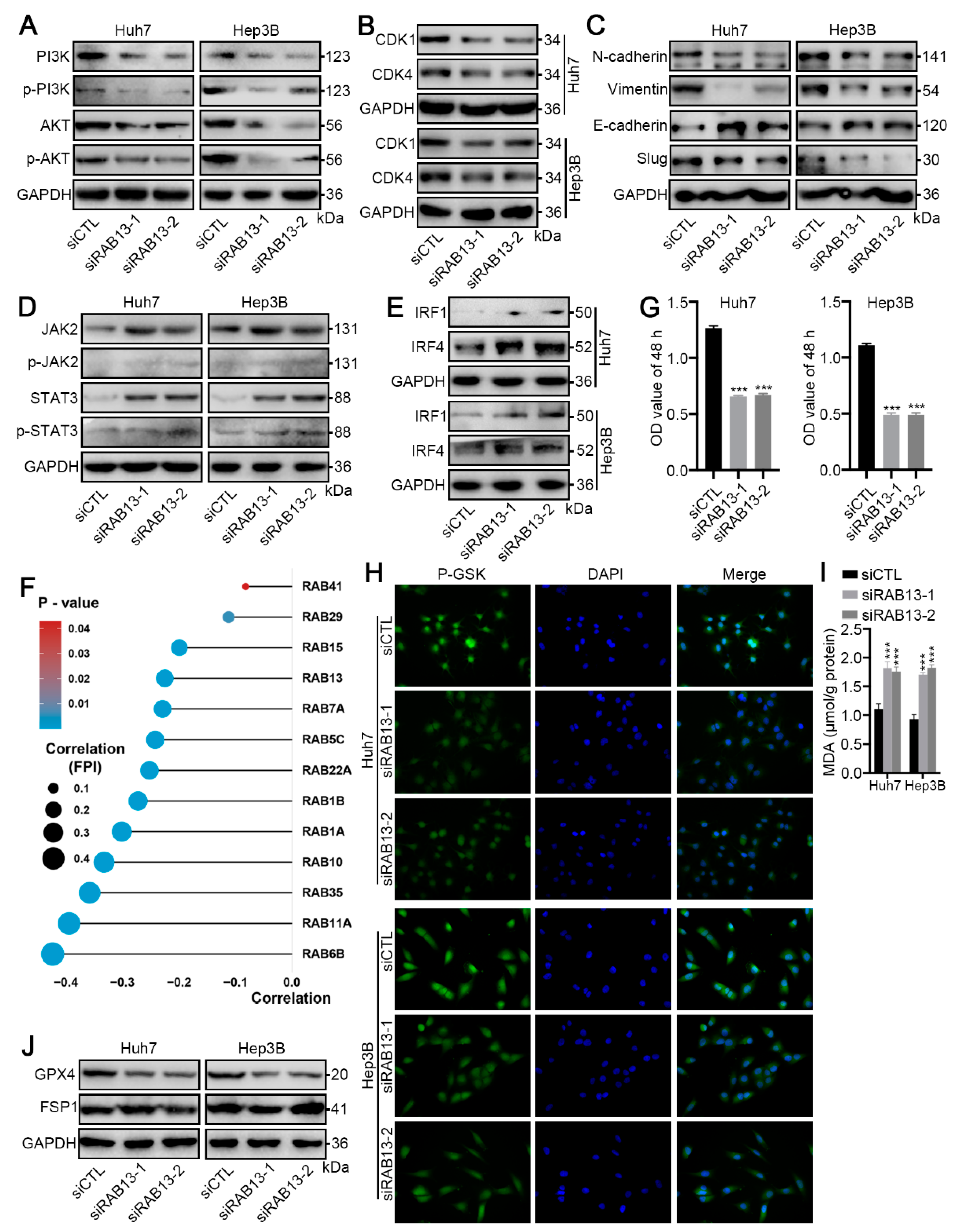
2.9. RAB13 Knockdown Promotes GPX4-Dependent Ferroptosis Vulnerability in HCC Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Samples and Immunohistochemistry
4.2. Cell Culture and Reagents
4.3. Transfection
4.4. Quantitative Real-Time Polymerase Chain Reaction (qRT–PCR) and Western Blot Analysis
4.5. Wound Healing and Transwell Assays
4.6. Cell Counting Kit-8
4.7. EdU Assays
4.8. Ferroptosis Detection
4.9. Data Sources and Preprocessing
4.10. Pathway Enrichment Analysis
4.11. Consensus Clustering with Nonnegative Matrix Factorization
4.12. Unsupervised Clustering for RAB-Associated Gene Signatures
4.13. Construction of the Risk Models
4.14. Immune Response Prediction and Immune Microenvironment Assessment
4.15. mRNA-Based Stemness Index (mRNAsi) and Ferroptosis Potential Index (FPI)
4.16. Development and Validation of the Prognostic Nomogram
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Chen, H.-D.; Yu, Y.-W.; Li, N.; Chen, W.-Q. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef]
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Gairing, S.J.; Müller, L.; Galle, P.R. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J. Hepatol. 2022, 76, 446–457. [Google Scholar] [CrossRef]
- Fulgenzi, C.A.M.; Talbot, T.; Murray, S.M.; Silletta, M.; Vincenzi, B.; Cortellini, A.; Pinato, D.J. Immunotherapy in Hepatocellular Carcinoma. Curr. Treat. Options Oncol. 2021, 22, 87. [Google Scholar] [CrossRef]
- Ioannou, G.N. HCC surveillance after SVR in patients with F3/F4 fibrosis. J. Hepatol. 2021, 74, 458–465. [Google Scholar] [CrossRef]
- Adeniji, N.; Dhanasekaran, R. Genomic Landscape of HCC. Curr. Hepatol. Rep. 2020, 19, 448–461. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Khemlina, G.; Ikeda, S.; Kurzrock, R. The biology of Hepatocellular carcinoma: Implications for genomic and immune therapies. Mol. Cancer 2017, 16, 149. [Google Scholar] [CrossRef]
- Xu, L.; Nagai, Y.; Kajihara, Y.; Ito, G.; Tomita, T. The Regulation of Rab GTPases by Phosphorylation. Biomolecules 2021, 11, 1340. [Google Scholar] [CrossRef]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef] [PubMed]
- Waschbüsch, D.; Khan, A.R. Phosphorylation of RabGTPasesin the regulation of membrane trafficking. Traffic 2020, 21, 712–719. [Google Scholar] [CrossRef]
- Shi, L.-W.; Zhao, Z.-B.; Zhong, L.; Gao, J.; Gong, J.-P.; Chen, H.; Min, Y.; Zhang, Y.-Y.; Li, Z. Overexpression of Rab40b Promotes Hepatocellular Carcinoma Cell Proliferation and Metastasis via PI3K/AKT Signaling Pathway. Cancer Manag. Res. 2020, 12, 10139–10150. [Google Scholar] [CrossRef]
- Zhang, Z.-Y.; Lu, M.; Liu, Z.-K.; Li, H.; Yong, Y.-L.; Zhang, R.-Y.; Chen, Z.-N.; Bian, H. Rab11a regulates MMP2 expression by activating the PI3K/AKT pathway in human hepatocellular carcinoma cells. Pathol. Res. Pr. 2020, 216, 153046. [Google Scholar] [CrossRef]
- You, X.; Liu, F.; Zhang, T.; Li, Y.; Ye, L.; Zhang, X. Hepatitis B virus X protein upregulates oncogene Rab18 to result in the dysregulation of lipogenesis and proliferation of hepatoma cells. Carcinogenesis 2013, 34, 1644–1652. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Zheng, X.; Zhao, D. Rab31 promoted hepatocellular carcinoma (HCC) progression via inhibition of cell apoptosis induced by PI3K/AKT/Bcl-2/BAX pathway. Tumor Biol. 2015, 36, 8661–8670. [Google Scholar] [CrossRef]
- Ben Salem, K.; Ben Abdelaziz, A. Principal Component Analysis (PCA). Tunis. Med. 2021, 99, 383–389. [Google Scholar] [PubMed]
- Zhao, Z.; Liu, H.; Zhou, X.; Fang, D.; Ou, X.; Ye, J.; Peng, J.; Xu, J. Necroptosis-Related lncRNAs: Predicting Prognosis and the Distinction between the Cold and Hot Tumors in Gastric Cancer. J. Oncol. 2021, 2021, 6718443. [Google Scholar] [CrossRef] [PubMed]
- Ruf, B.; Heinrich, B.; Greten, T.F. Immunobiology and immunotherapy of HCC: Spotlight on innate and innate-like immune cells. Cell. Mol. Immunol. 2021, 18, 112–127. [Google Scholar] [CrossRef]
- Zongyi, Y.; Xiaowu, L. Immunotherapy for hepatocellular carcinoma. Cancer Lett. 2020, 470, 8–17. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Orlando, D.A.; Lin, C.Y.; Bernard, A.; Wang, J.Y.; Socolar, J.E.S.; Iversen, E.S.; Hartemink, A.J.; Haase, S.B. Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature 2008, 453, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, H.; Chen, W.; Cheng, M.; Zou, L.; Yang, Q.; Chan, C.B.; Zhu, H.; Chen, C.; Nie, J.-Y.; et al. Rab13 Sustains Breast Cancer Stem Cells by Supporting Tumor–Stroma Cross-talk. Cancer Res. 2022, 82, 2124–2140. [Google Scholar] [CrossRef]
- Hinger, S.A.; Abner, J.J.; Franklin, J.L.; Jeppesen, D.K.; Coffey, R.J.; Patton, J.G. Rab13 regulates sEV secretion in mutant KRAS colorectal cancer cells. Sci. Rep. 2020, 10, 15804. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Gan, H.; Tang, Z.; Guo, Y.; Yao, S.; Liuyu, T.; Zhong, B.; Lin, D. RNF115 Inhibits the Post-ER Trafficking of TLRs and TLRs-Mediated Immune Responses by Catalyzing K11-Linked Ubiquitination of RAB1A and RAB13. Adv. Sci. 2022, 9, e2105391. [Google Scholar] [CrossRef]
- Yuan, J.; Yin, Z.; Tan, L.; Zhu, W.; Tao, K.; Wang, G.; Shi, W.; Gao, J. Interferon regulatory factor-1 reverses chemoresistance by downregulating the expression of P-glycoprotein in gastric cancer. Cancer Lett. 2019, 457, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Tan, L.; Yin, Z.; Zhu, W.; Tao, K.; Wang, G.; Shi, W.; Gao, J. MIR17HG-miR-18a/19a axis, regulated by interferon regulatory factor-1, promotes gastric cancer metastasis via Wnt/β-catenin signalling. Cell Death Dis. 2019, 10, 454. [Google Scholar] [CrossRef]
- Brunet, J.-P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Zhang, Q.; Wang, Y.; Zhang, L.; Zhao, H.; Chen, C.; Wang, Y.; Liu, S.; Lu, T.; Wang, F.; et al. Molecular classification and subtype-specific characterization of skin cutaneous melanoma by aggregating multiple genomic platform data. J. Cancer Res. Clin. Oncol. 2018, 144, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhao, Q.; Zuo, Z.-X.; Yuan, S.-Q.; Yu, K.; Zhang, Q.; Zhang, X.; Sheng, H.; Ju, H.-Q.; Cheng, H.; et al. Systematic Analysis of the Aberrances and Functional Implications of Ferroptosis in Cancer. iScience 2020, 23, 101302. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Liu, Z.; Yuan, J.; Wu, Z.; Kong, L.; Yang, J.; Lv, T. Construction of Two Independent RAB Family-Based Scoring Systems Based on Machine Learning Algorithms and Definition of RAB13 as a Novel Therapeutic Target for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 4335. https://doi.org/10.3390/ijms24054335
Jiang C, Liu Z, Yuan J, Wu Z, Kong L, Yang J, Lv T. Construction of Two Independent RAB Family-Based Scoring Systems Based on Machine Learning Algorithms and Definition of RAB13 as a Novel Therapeutic Target for Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2023; 24(5):4335. https://doi.org/10.3390/ijms24054335
Chicago/Turabian StyleJiang, Chenhao, Zijian Liu, Jingsheng Yuan, Zhenru Wu, Lingxiang Kong, Jiayin Yang, and Tao Lv. 2023. "Construction of Two Independent RAB Family-Based Scoring Systems Based on Machine Learning Algorithms and Definition of RAB13 as a Novel Therapeutic Target for Hepatocellular Carcinoma" International Journal of Molecular Sciences 24, no. 5: 4335. https://doi.org/10.3390/ijms24054335
APA StyleJiang, C., Liu, Z., Yuan, J., Wu, Z., Kong, L., Yang, J., & Lv, T. (2023). Construction of Two Independent RAB Family-Based Scoring Systems Based on Machine Learning Algorithms and Definition of RAB13 as a Novel Therapeutic Target for Hepatocellular Carcinoma. International Journal of Molecular Sciences, 24(5), 4335. https://doi.org/10.3390/ijms24054335