
Increased EGFRvIII Epitope Accessibility after Tyrosine Kinase Inhibitor Treatment of Glioblastoma Cells Creates More Opportunities for Immunotherapy

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
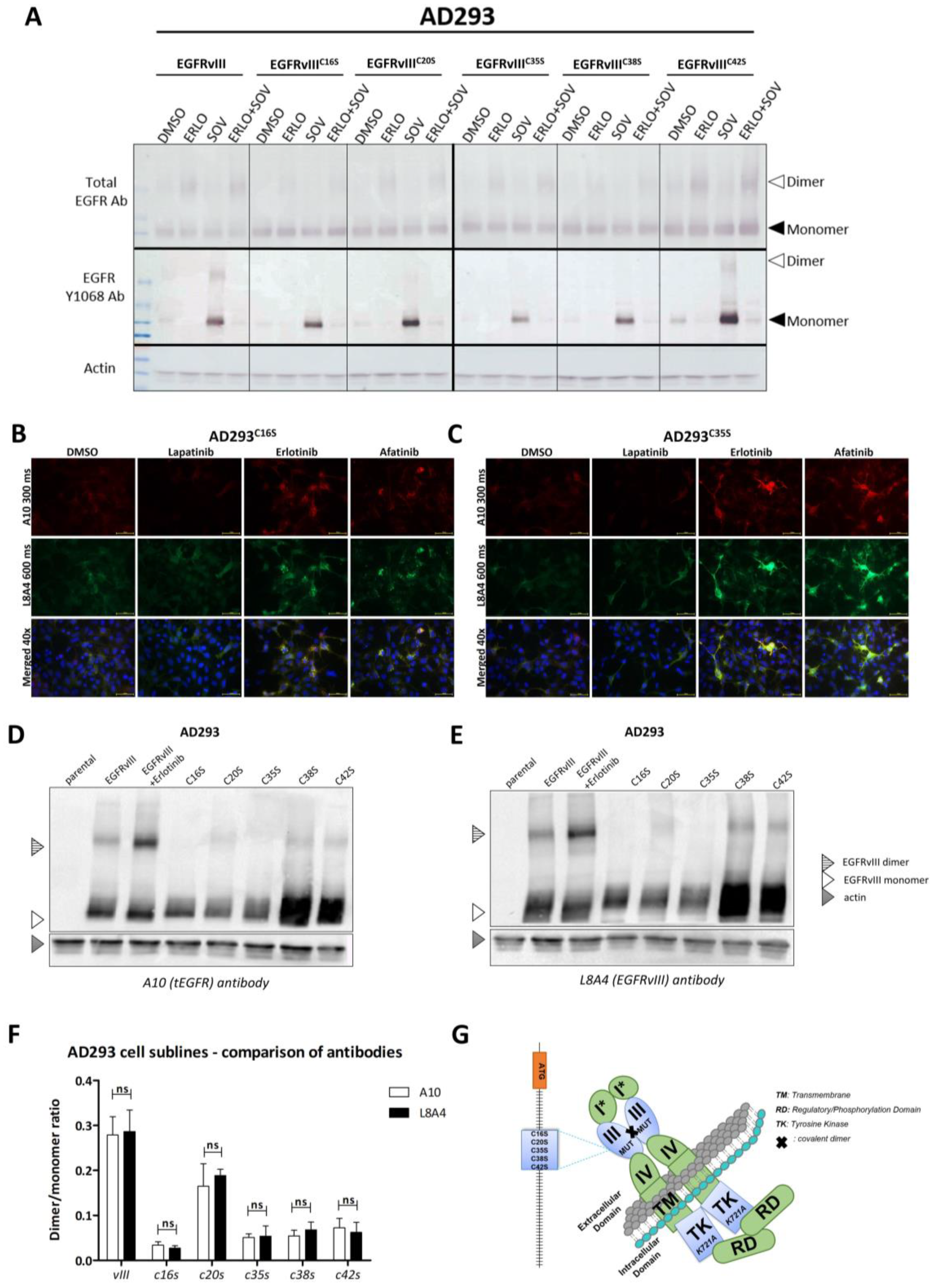
2.1. Characterization of Research Model According to the Different EGFRvIII Expression Profiles
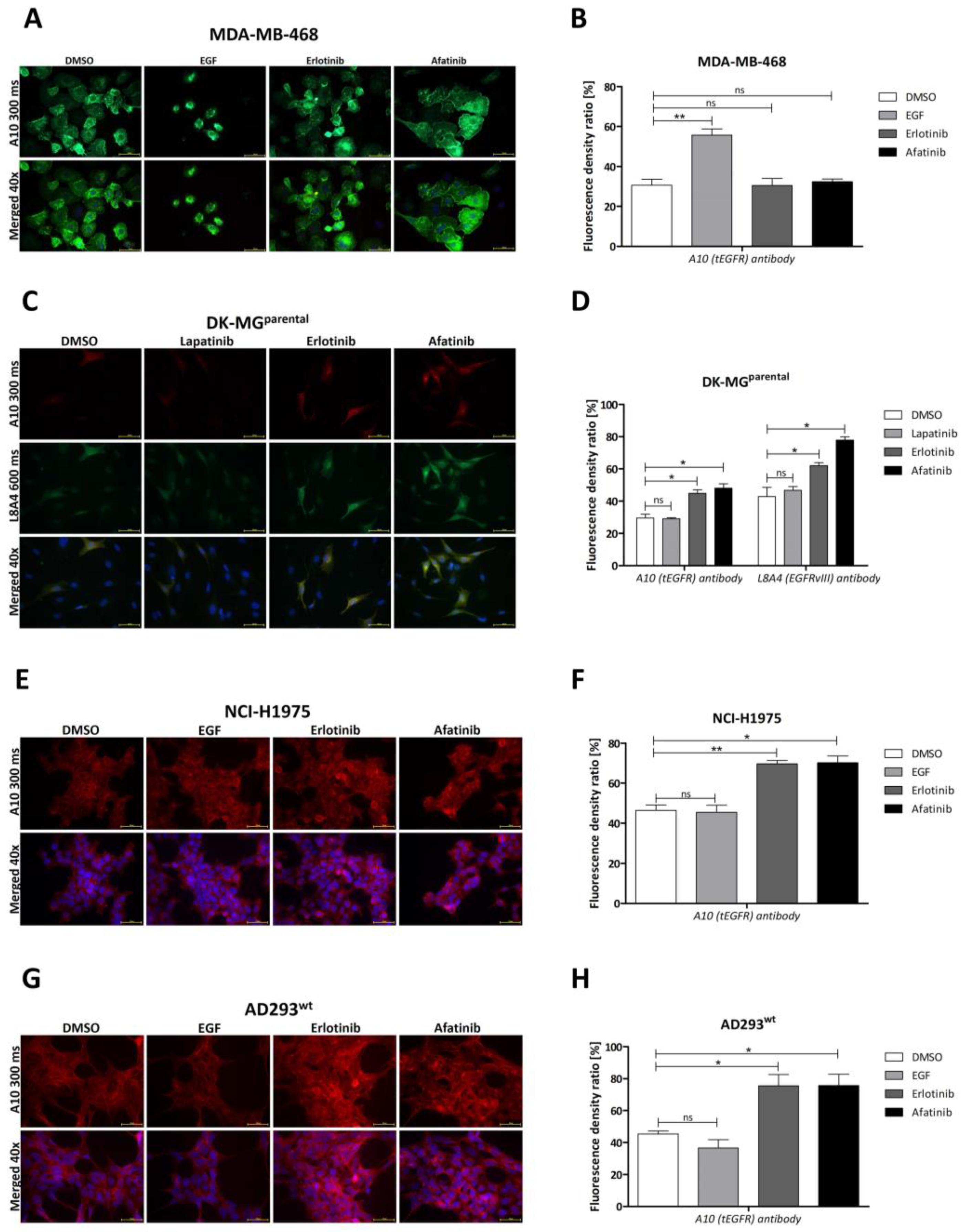
2.2. Influence of Particular TKIs
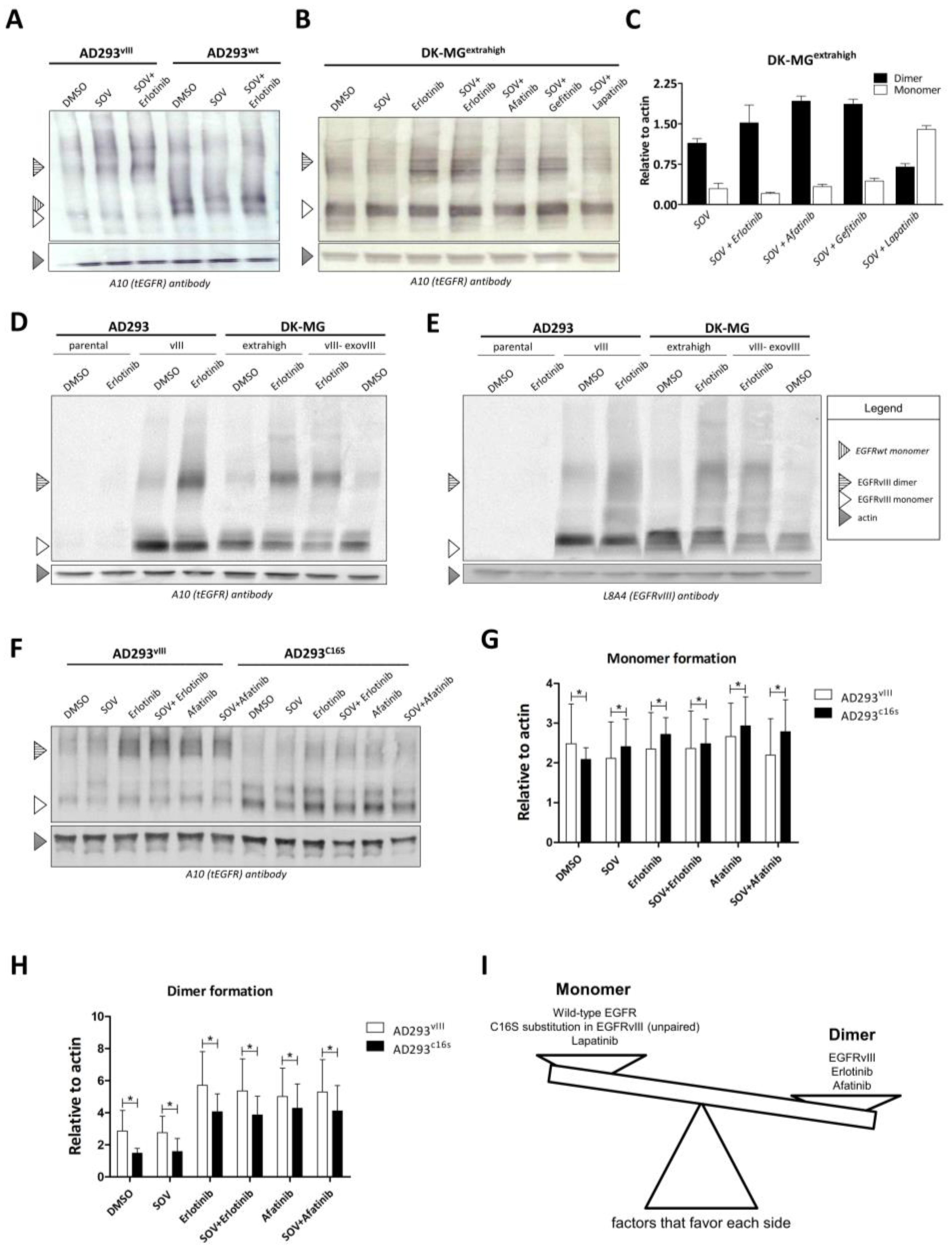
2.3. Dimer Formation and EGFR Variant
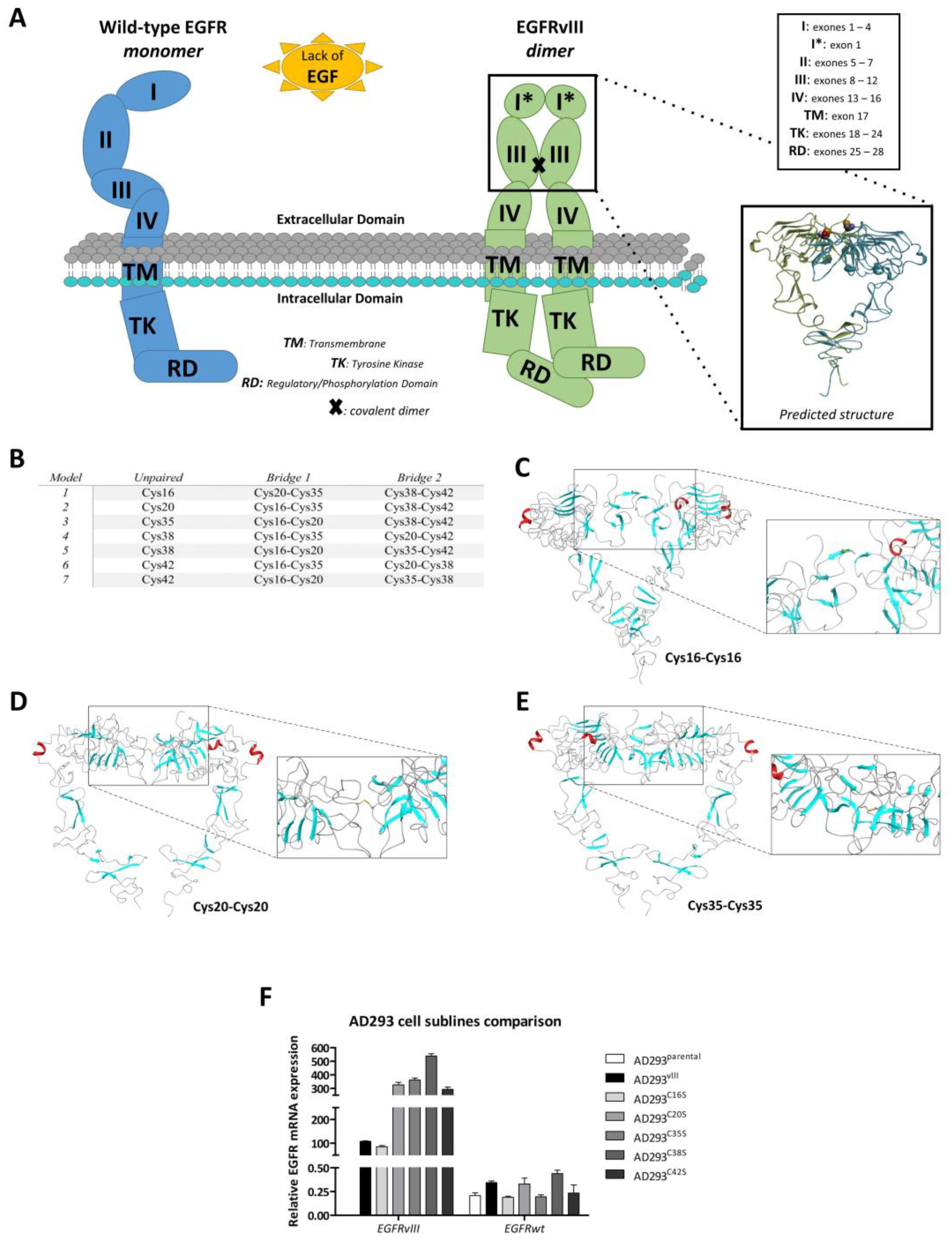
2.4. In Silico Analysis of Dimer Formation
2.5. Validation of In Silico Model
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construction of Plasmids and Site-Directed Mutagenesis in EGFRvIII Transgene
4.3. Preparation of Genetic Content Delivery Vehicle and Establishment of Cell Lines
4.4. Reverse-Transcription Real-Time PCR
4.5. Compounds
4.6. Western Blotting
4.7. Isolation of Protein Fractions
4.8. Immunofluorescence Analyses
4.9. Computational Modeling
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAR-T cells | chimeric antigen receptor T cells |
| DMSO | dimethyl sulfoxide |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| EGFRvIII | epidermal growth factor receptor variant III |
| EGFRwt | wild-type epidermal growth factor receptor |
| GB | glioblastoma |
| mAbs | monoclonal antibodies |
| SOV | sodium orthovanadate |
| TKI | tyrosine kinase inhibitor |
References
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Kasenda, B.; König, D.; Manni, M.; Ritschard, R.; Duthaler, U.; Bartoszek, E.; Bärenwaldt, A.; deuster, S.; Hutter, G.; Cordier, D.; et al. Targeting immunoliposomes to EGFR-positive glioblastoma. ESMO Open 2022, 7, 100365. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.W.; Weiss, W.A. Epidermal growth factor receptor (EGFR) and EGFRvIII in glioblastoma (GBM): Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- William, D.; Mokri, P.; Lamp, N.; Linnebacher, M.; Classen, C.F.; Erbersdobler, A.; Schneider, B. Amplification of the EGFR gene can be maintained and modulated by variation of EGF concentrations in in vitro models of glioblastoma multiforme. PLoS ONE 2017, 12, e01852082017. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, H.; Ohba, Y.; Tamaoki, N.; Shibuya, M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn. J. Cancer Res. 1990, 81, 773–779. [Google Scholar] [CrossRef]
- Huang, H.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.D.; Huang, C.M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Ramnarain, D.B.; Park, S.; Lee, D.Y.; Hatanpaa, K.J.; Scoggin, S.O.; Otu, H.; Libermann, T.A.; Raisanen, J.M.; Ashfaq, R.; Wong, E.T.; et al. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006, 66, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowska, A.; Stoczyńska-Fidelus, E.; Janik, K.; Włodarczyk, A.; Rieske, P. EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR phosphorylates tumor-derived EGFRvIII driving STAT3/5 and progression in glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Kancha, R.K.; von Bubnoff, N.; Duyster, J. Asymmetric kinase dimer formation is crucial for the activation of oncogenic EGFRvIII but not for ERBB3 phosphorylation. Cell Commun. Signal. 2013, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Greenall, S.A.; Donoghue, J.F.; Van Sinderen, M.; Dubljevic, V.; Budiman, S.; Devlin, M.; Street, I.; Adams, T.E.; Johns, T.G. EGFRvIII-mediated transactivation of receptor tyrosine kinases in glioma: Mechanism and therapeutic implications. Oncogene 2015, 34, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Stec, W.; Rosiak, K.; Treda, C.; Smolarz, M.; Peciak, J.; Pacholczyk, M.; Lenart, A.; Grzela, D.; Stoczynska-Fidelus, E.; Rieske, P. Cyclic trans-phosphorylation in a homodimer as the predominant mechanism of EGFRvIII action and regulation. Oncotarget 2018, 9, 8560–8572. [Google Scholar] [CrossRef] [Green Version]
- Ymer, S.I.; Greenall, S.A.; Cvrljevic, A.; Cao, D.X.; Donoghue, J.F.; Epa, V.C.; Scott, A.M.; Adams, T.E.; Johns, T.G. Glioma Specific Extracellular Missense Mutations in the First Cysteine Rich Region of Epidermal Growth Factor Receptor (EGFR) Initiate Ligand Independent Activation. Cancers 2011, 3, 2032–2049. [Google Scholar] [CrossRef] [Green Version]
- de Wit, M.; Gao, Y.; Mercieca, D.; de Heer, I.; Valkenburg, B.; van Royen, M.E.; Aerts, J.; Smitt, P.S.; French, P. Mutation and drug-specific intracellular accumulation of EGFR predict clinical responses to tyrosine kinase inhibitors. EBioMedicine 2020, 56, 102796. [Google Scholar] [CrossRef]
- Cavazzoni, A.; Alfieri, R.; Cretella, D.; Saccani, F.; Ampollini, L.; Galetti, M.; Quaini, F.; Graiani, G.; Madeddu, D.; Mozzoni, P.; et al. Combined use of anti-ErbB monoclonal antibodies and erlotinib enhances antibody-dependent cellular cytotoxicity of wild-type erlotinib-sensitive NSCLC cell lines. Mol. Cancer 2012, 11, 91. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Mi, L.Z.; Schürpf, T.; Walz, T.; Springer, T.A. Mechanisms for Kinase-mediated Dimerization of the Epidermal Growth Factor Receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef] [Green Version]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosc. 2021, 15, 662064. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Knackmuss, S.; Weichel, M.; Gantke, T.; Molkenthin, V.; Zhukovsky, E.A.; Tesar, M.; Treder, M. Highly Specific and Effective Targeting of EGFRvIII-Positive Tumors with TandAb Antibodies. Front. Oncol. 2017, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hu, Y.; Yang, J.; Li, W.; Zhang, M.; Wang, Q.; Zhang, L.; Wei, G.; Tian, Y.; Zhao, K.; et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature 2022, 609, 369–374. [Google Scholar] [CrossRef]
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and safety of CD19-directed CAR-T cell therapies in patients with relapsed/refractory aggressive B-cell lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND trials. Am. J. Hematol. 2021, 96, 1295–1312. [Google Scholar] [CrossRef]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Barth, R.F.; Wu, G.; Ciesielski, M.J.; Fenstermaker, R.A.; Moffat, B.A.; Ross, B.D.; Wikstrand, C.J. Development of a Syngeneic Rat Brain Tumor Model Expressing EGFRvIII and Its Use for Molecular Targeting Studies with Monoclonal Antibody L8A4. Clin. Cancer Res. 2005, 11, 341–350. [Google Scholar] [CrossRef]
- Chen, M.; Sun, R.; Shi, B.; Wang, Y.; Di, S.; Luo, H.; Sun, J.; Li, Z.; Zhou, M.; Jiang, H. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed. Pharmacother. 2019, 113, 108734. [Google Scholar] [CrossRef]
- Peciak, J.; Stec, W.J.; Treda, C.; Ksiazkiewicz, M.; Janik, K.; Popeda, M.; Smolarz, M.; Rosiak, K.; Hulas-Bigoszewska, K.; Och, W.; et al. Low Incidence along with Low mRNA Levels of EGFRvIII in Prostate and Colorectal Cancers Compared to Glioblastoma. J. Cancer 2017, 8, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Włodarczyk, A.; Tręda, C.; Rutkowska, A.; Grot, D.; Dobrewa, W.; Kierasińska, A.; Węgierska, M.; Wasiak, T.; Strózik, T.; Rieske, P.; et al. Phenotypical Flexibility of the EGFRvIII-Positive Glioblastoma Cell Line and the Multidirectional Influence of TGFβ and EGF on These Cells-EGFRvIII Appears as a Weak Oncogene. Int. J. Mol. Sci. 2022, 23, 12129. [Google Scholar] [CrossRef]
- Emlet, D.R.; Gupta, P.; Holgado-Madruga, M.; Del Vecchio, C.A.; Mitra, S.S.; Han, S.Y.; Li, G.; Jensen, K.C.; Vogel, H.; Xu, L.W.; et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Stockhausen, M.T.; Kristoffersen, K.; Stobbe, L.; Poulsen, H.S. Differentiation of glioblastoma multiforme stem-like cells leads to downregulation of EGFR and EGFRvIII and decreased tumorigenic and stem-like cell potential. Cancer Biol. Ther. 2014, 15, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.C.; Magge, R.S. Mechanisms of EGFR Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 8471. [Google Scholar] [CrossRef]
- Saleem, H.; Kulsoom Abdul, U.; Küçükosmanoglu, A.; Houweling, M.; Cornelissen, F.M.G.; Heiland, D.H.; Hegi, M.E.; Kouwenhoven, M.C.M.; Bailey, D.; Würdinger, T.; et al. The TICking clock of EGFR therapy resistance in glioblastoma: Target Independence or target Compensation. Drug Resist. Updates 2019, 43, 29–37. [Google Scholar] [CrossRef]
- Stec, W.J.; Rosiak, K.; Siejka, P.; Peciak, J.; Popeda, M.; Banaszczyk, M.; Pawlowska, R.; Treda, C.; Hulas-Bigoszewska, K.; Piaskowski, S.; et al. Cell line with endogenous EGFRvIII expression is a suitable model for research and drug development purposes. Oncotarget 2016, 7, 31907–31925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, C.A.; Giacomini, C.P.; Vogel, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.B. The Basics of Thiols and Cysteines in Redox Biology and Chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, R.H.; Siddiqui, M.K.; Salahuddin, P. Protein Disulfide Isomerase: Structure, Mechanism of Oxidative Protein Folding and Multiple Functional Roles. J. Biochem. Mol. Biol. Res. 2016, 2, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Ferrè, F.; Clote, P. DiANNA: A web server for disulfide connectivity prediction. Nucleic. Acids Res. 2005, 33 (Suppl. 2), W230–W232. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic. Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.H.; Tseng, L.Y. DBCP: A web server for disulfide bonding connectivity pattern prediction without the prior knowledge of the bonding state of cysteines. Nucleic. Acids Res. 2010, 38 (Suppl. 2), W503–W507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; He, B.J.; Jang, R.; Zhang, Y.; Shen, H.B. Accurate disulfide-bonding network predictions improve ab initio structure prediction of cysteine-rich proteins. Bioinformatics 2015, 31, 3773–3781. [Google Scholar] [CrossRef] [Green Version]
- Ceroni, A.; Passerini, A.; Vullo, A.; Frasconi, P. DISULFIND: A disulfide bonding state and cysteine connectivity prediction server. Nucleic. Acids Res. 2006, 34 (Suppl. 2), W177–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janik, K.; Treda, C.; Wlodarczyk, A.; Peciak, J.; Rosiak, K.; Zieba, J.; Grot, D.; Rutkowska, A.; Pawlowska, R.; Och, W.; et al. A way to understand idiopathic senescence and apoptosis in primary glioblastoma cells–possible approaches to circumvent these phenomena. BMC Cancer 2019, 19, 1–16. [Google Scholar] [CrossRef]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448 Pt 3, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Björkelund, H.; Gedda, L.; Malmqvist, M.; Andersson, K. Resolving the EGF-EGFR interaction characteristics through a multiple-temperature, multiple-inhibitor, real-time interaction analysis approach. Mol. Clin. Oncol. 2013, 1, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Sheff, D.R.; Daro, E.A.; Hull, M.; Mellman, I. The Receptor Recycling Pathway Contains Two Distinct Populations of Early Endosomes with Different Sorting Functions. J. Cell Biol. 1999, 145, 123–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X. EGFR Primary T790M and L858R Double Mutation Confers Clinical Benefit to Erlotinib and Resistance to Osimertinib in One Lung Adenocarcinoma Patient: A Case Report. J. Cancer Sci. Ther. 2018, 10, 366–370. [Google Scholar] [CrossRef]
- Haubrich, J.; Zwier, J.M.; Charrier-Savournin, F.; Prézeau, L.; Pin, J.P. Different conformations of EGF-induced receptor dimers involved in signaling and internalization. bioRxiv 2022, 19, 488777. [Google Scholar] [CrossRef]
- Sahin, A.; Sanchez, C.; Bullain, S.; Waterman, P.; Weissleder, R.; Carter, B.S. Development of third generation anti-EGFRvIII chimeric T cells and EGFRvIII-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS ONE 2018, 13, e01994142018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra22. [Google Scholar] [CrossRef] [Green Version]
- Ohno, M.; Ohkuri, T.; Kosaka, A.; Tanahashi, K.; June, C.H.; Natsume, A.; Okada, H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 2013, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.-T.; Song, H.; et al. Recognition of Glioma Stem Cells by Genetically Modified T Cells Targeting EGFRvIII and Development of Adoptive Cell Therapy for Glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.D.; Suryadevara, C.M.; Gedeon, P.C.; Herndon, J.E., 2nd; Sanchez-Perez, L.; Bigner, D.D.; Sampson, J.H. Intracerebral delivery of a third gener-ation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J. Clin. Neurosci. 2014, 21, 189–190. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Gao, H.; Kong, J.; Song, B.; Wang, P.; Shi, B.; Wang, H.; Li, Z. Selective Targeting of Glioblastoma with EGFRvIII/EGFR Bitargeted Chimeric Antigen Receptor T Cell. Cancer Immunol. Res. 2018, 6, 1314–1326. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-CAR T Cells Induce Remissions in CD19-CAR Naïve and Resistant B-ALL. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treda, C.; Popeda, M.; Ksiazkiewicz, M.; Grzela, D.P.; Walczak, M.P.; Banaszczyk, M.; Peciek, J.; Stoczynska-Fidelus, E.; Rieske, P. EGFR Activation Leads to Cell Death Independent of PI3K/AKT/mTOR in an AD293 Cell Line. PLoS ONE 2016, 11, e01552302016. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tręda, C.; Włodarczyk, A.; Pacholczyk, M.; Rutkowska, A.; Stoczyńska-Fidelus, E.; Kierasińska, A.; Rieske, P. Increased EGFRvIII Epitope Accessibility after Tyrosine Kinase Inhibitor Treatment of Glioblastoma Cells Creates More Opportunities for Immunotherapy. Int. J. Mol. Sci. 2023, 24, 4350. https://doi.org/10.3390/ijms24054350
Tręda C, Włodarczyk A, Pacholczyk M, Rutkowska A, Stoczyńska-Fidelus E, Kierasińska A, Rieske P. Increased EGFRvIII Epitope Accessibility after Tyrosine Kinase Inhibitor Treatment of Glioblastoma Cells Creates More Opportunities for Immunotherapy. International Journal of Molecular Sciences. 2023; 24(5):4350. https://doi.org/10.3390/ijms24054350
Chicago/Turabian StyleTręda, Cezary, Aneta Włodarczyk, Marcin Pacholczyk, Adrianna Rutkowska, Ewelina Stoczyńska-Fidelus, Amelia Kierasińska, and Piotr Rieske. 2023. "Increased EGFRvIII Epitope Accessibility after Tyrosine Kinase Inhibitor Treatment of Glioblastoma Cells Creates More Opportunities for Immunotherapy" International Journal of Molecular Sciences 24, no. 5: 4350. https://doi.org/10.3390/ijms24054350
APA StyleTręda, C., Włodarczyk, A., Pacholczyk, M., Rutkowska, A., Stoczyńska-Fidelus, E., Kierasińska, A., & Rieske, P. (2023). Increased EGFRvIII Epitope Accessibility after Tyrosine Kinase Inhibitor Treatment of Glioblastoma Cells Creates More Opportunities for Immunotherapy. International Journal of Molecular Sciences, 24(5), 4350. https://doi.org/10.3390/ijms24054350