

Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease



Abstract
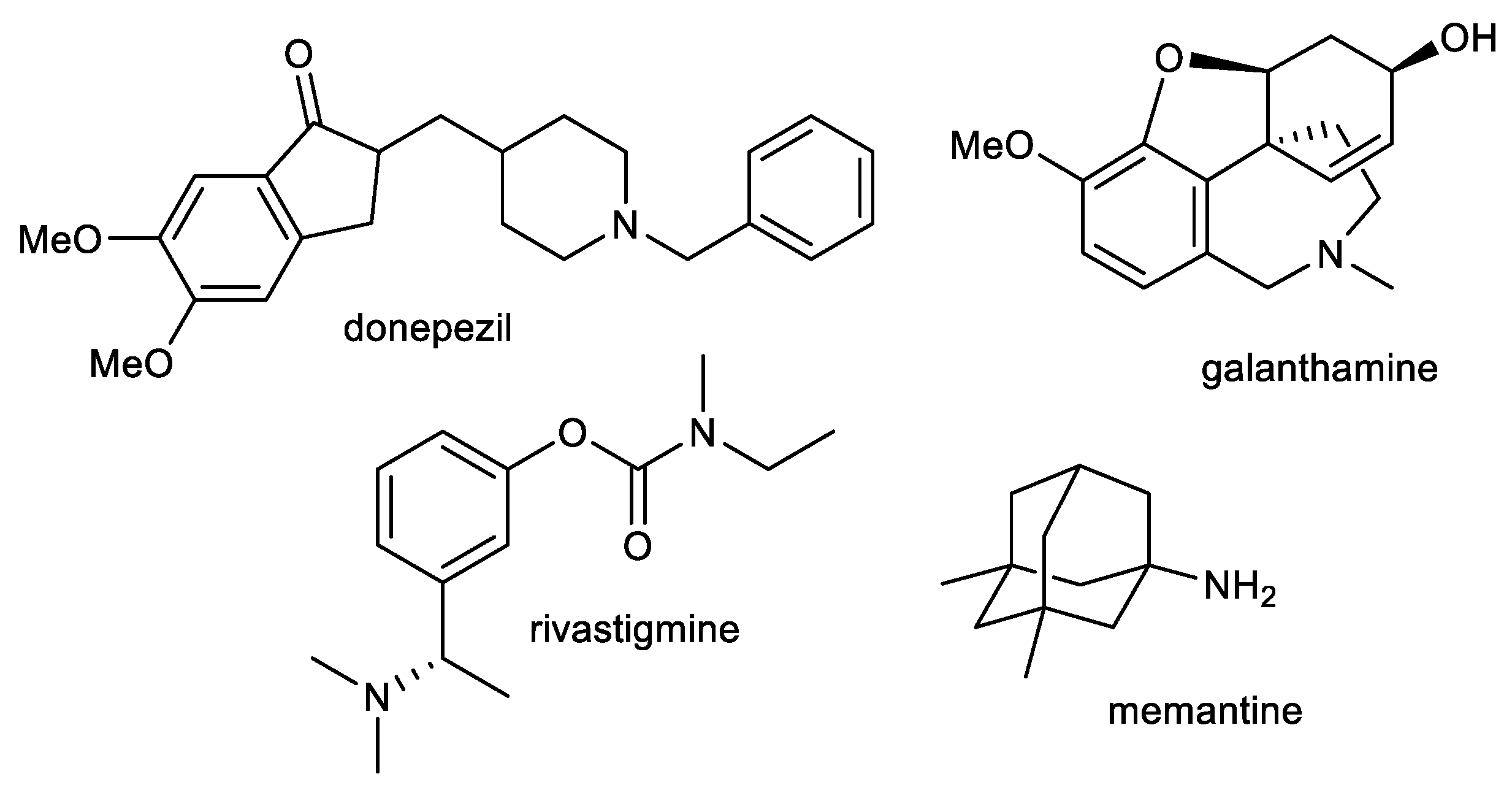
:1. Introduction
2. Plant Alkaloids as Multi-Target Compounds for the Treatment of AD
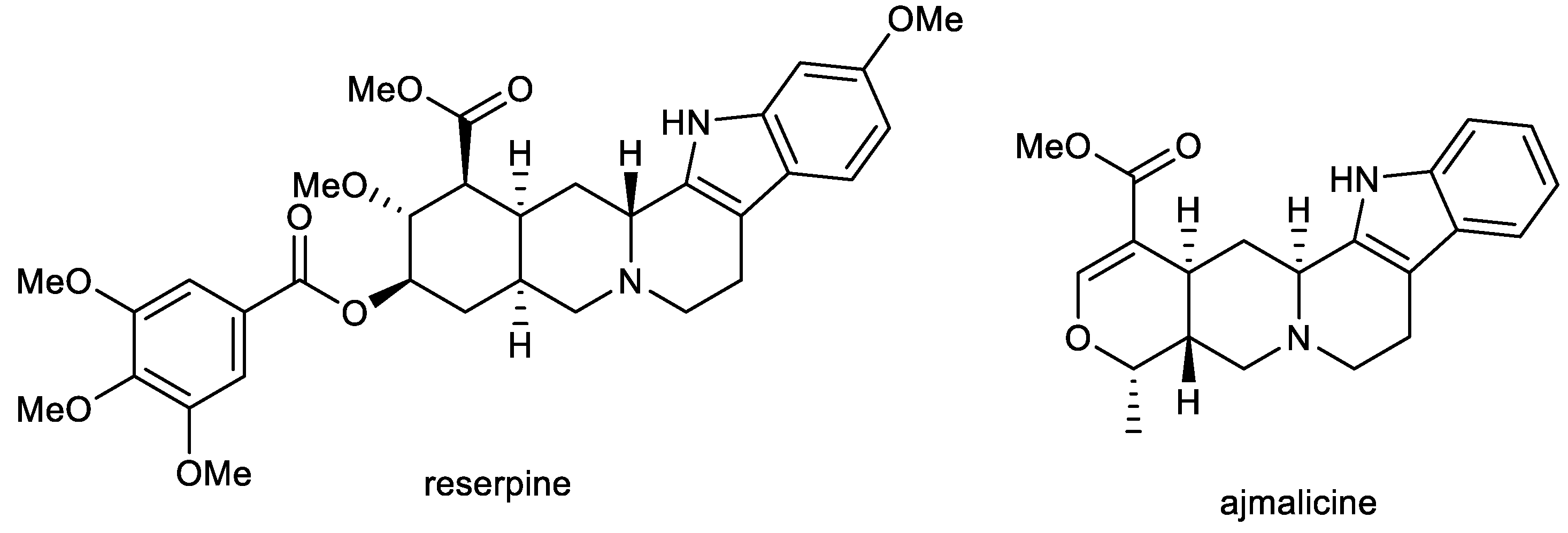
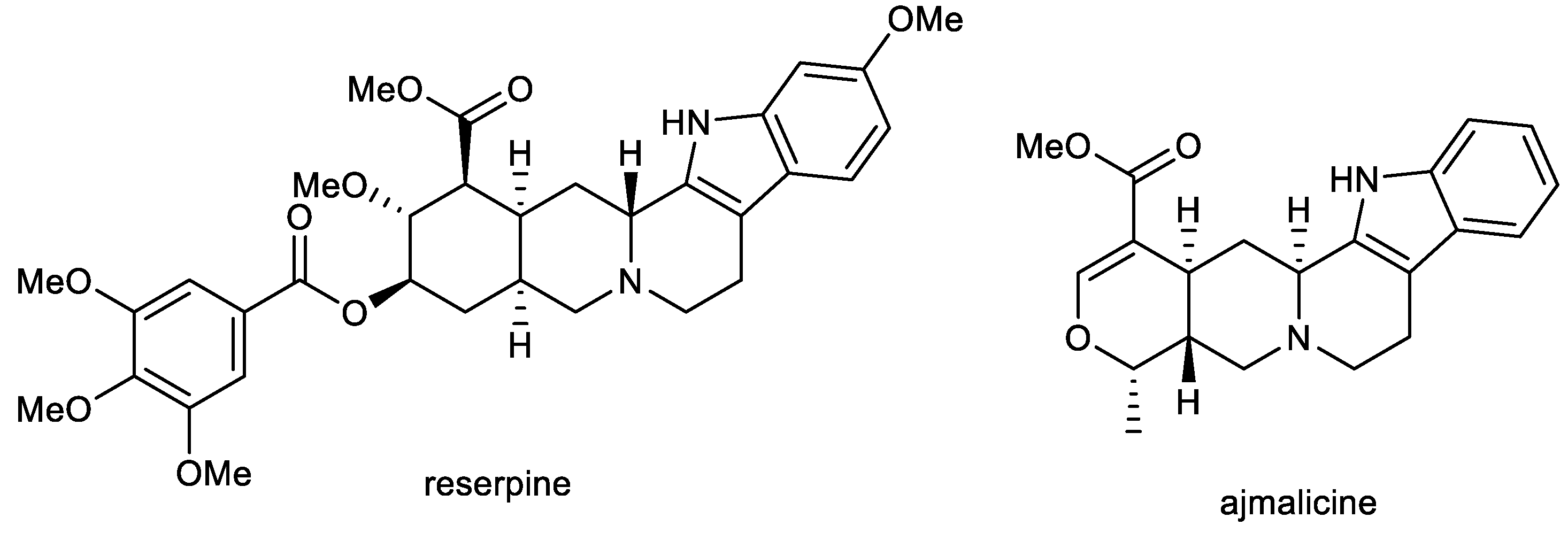
2.1. Indole Alkaloids: Ajmalicine and Reserpine
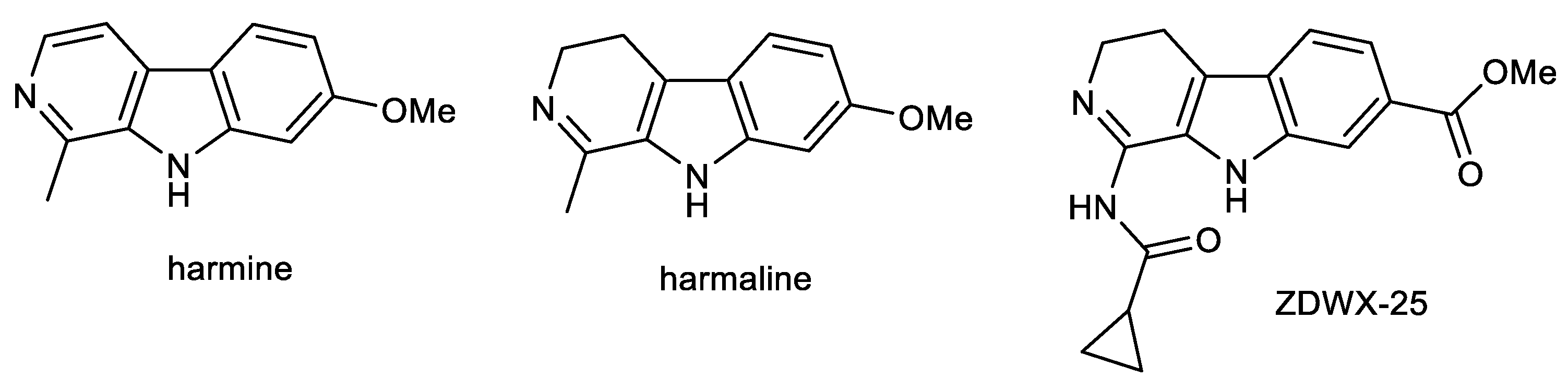
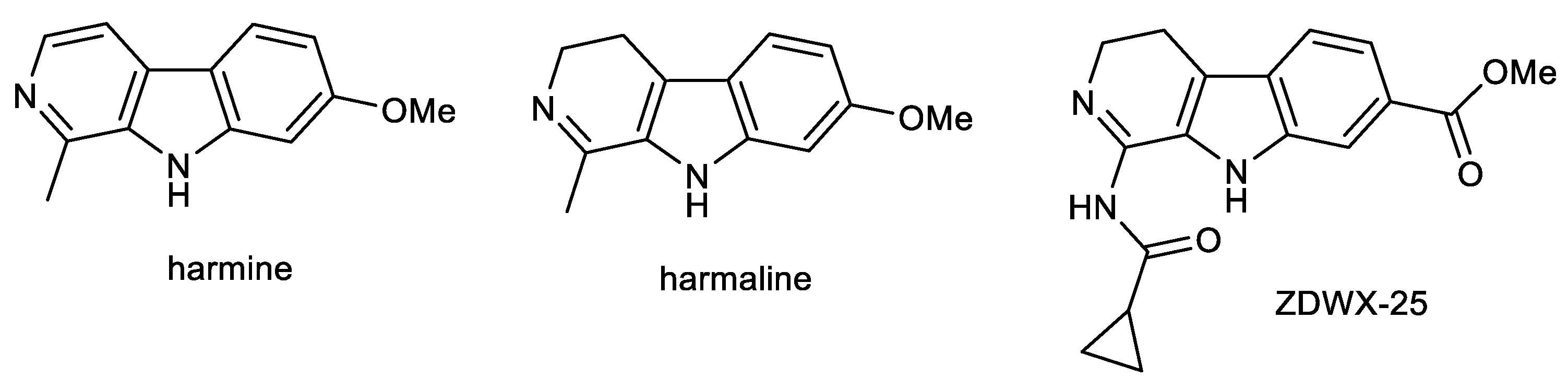
2.2. β-Carboline Alkaloids: Harmine and Harmaline
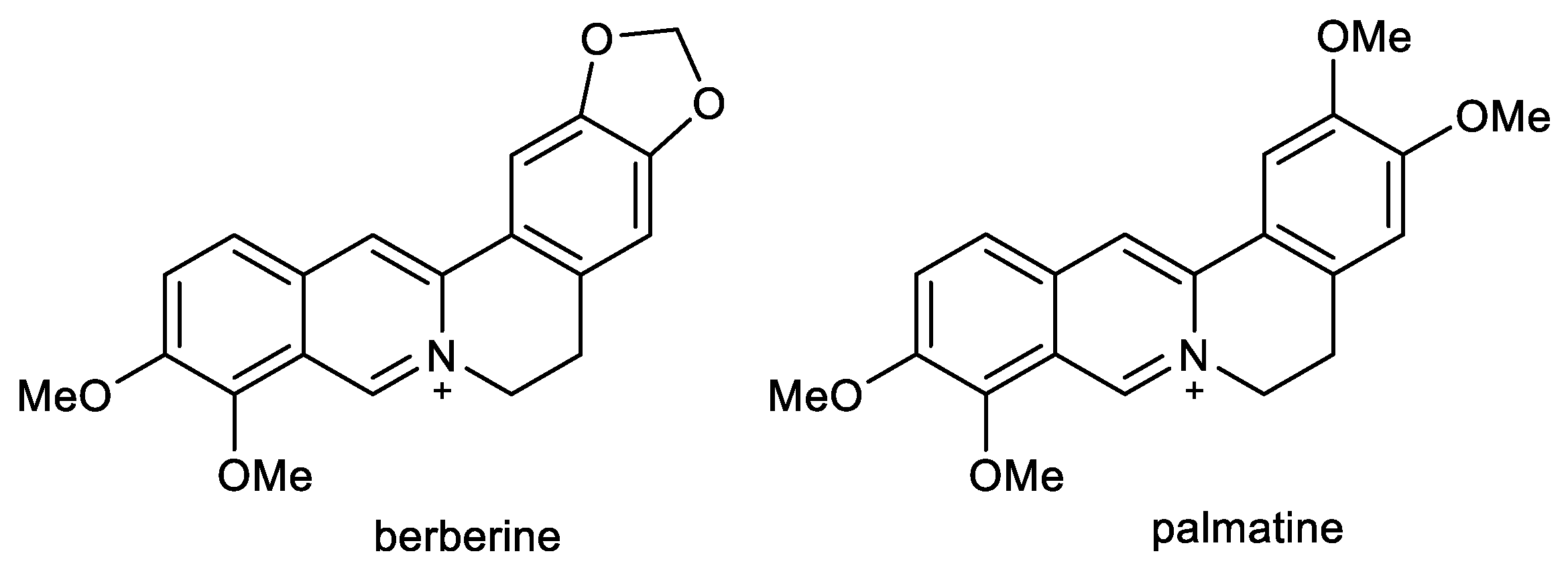
2.3. Protoberberine Alkaloids: Berberine and Palmatine
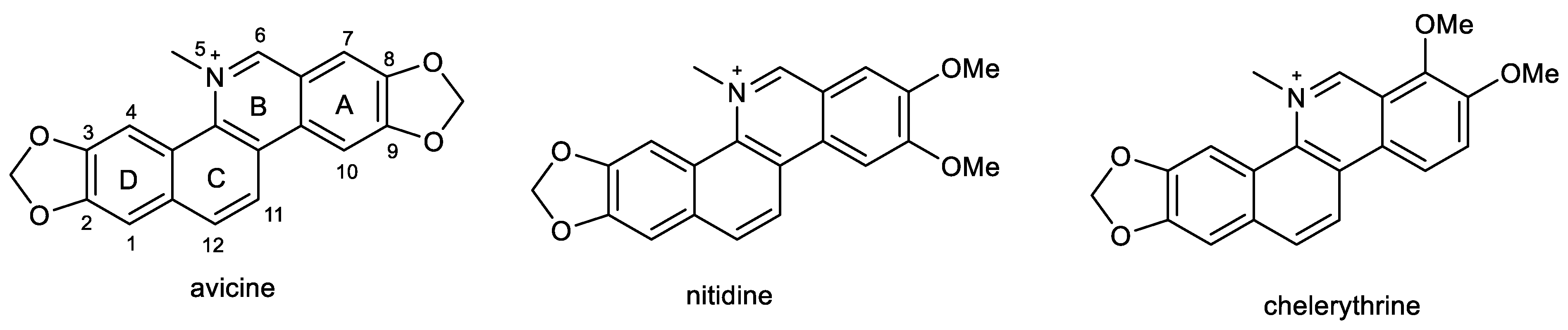
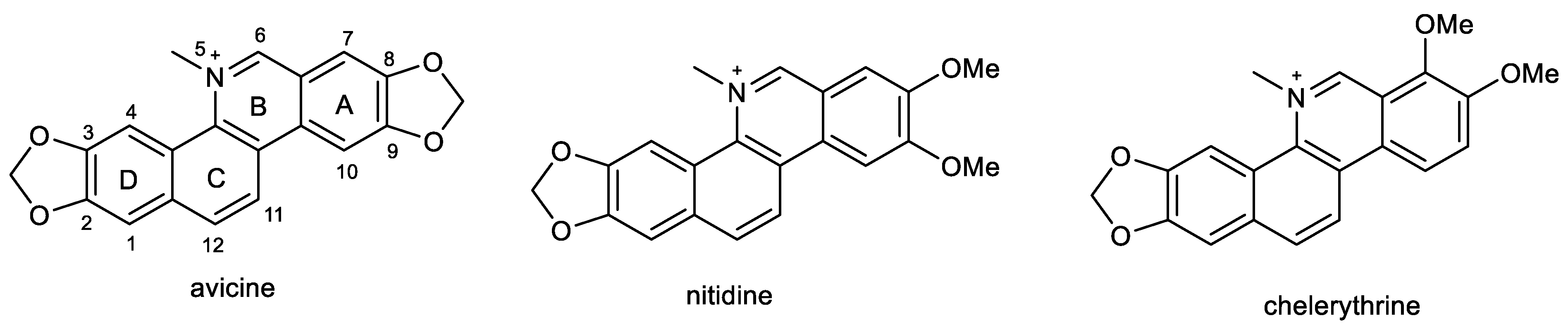
2.4. Benzophenathridine Alkaloids: Avicine, Nitidine, and Chelerythrine
3. Marine Alkaloids and Nitrogen Containing Compounds as Multi-Target Compounds for the Treatment of AD
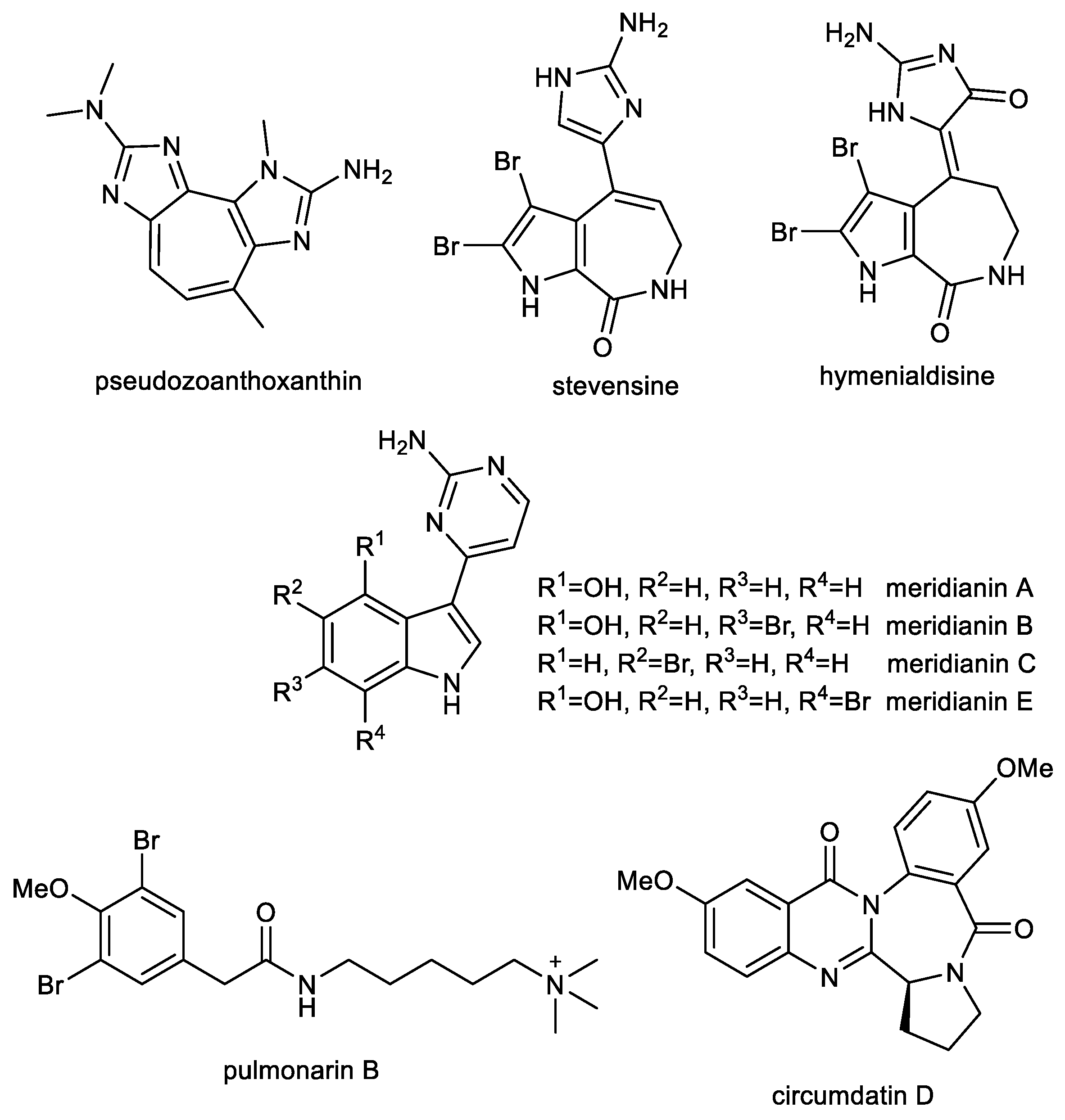
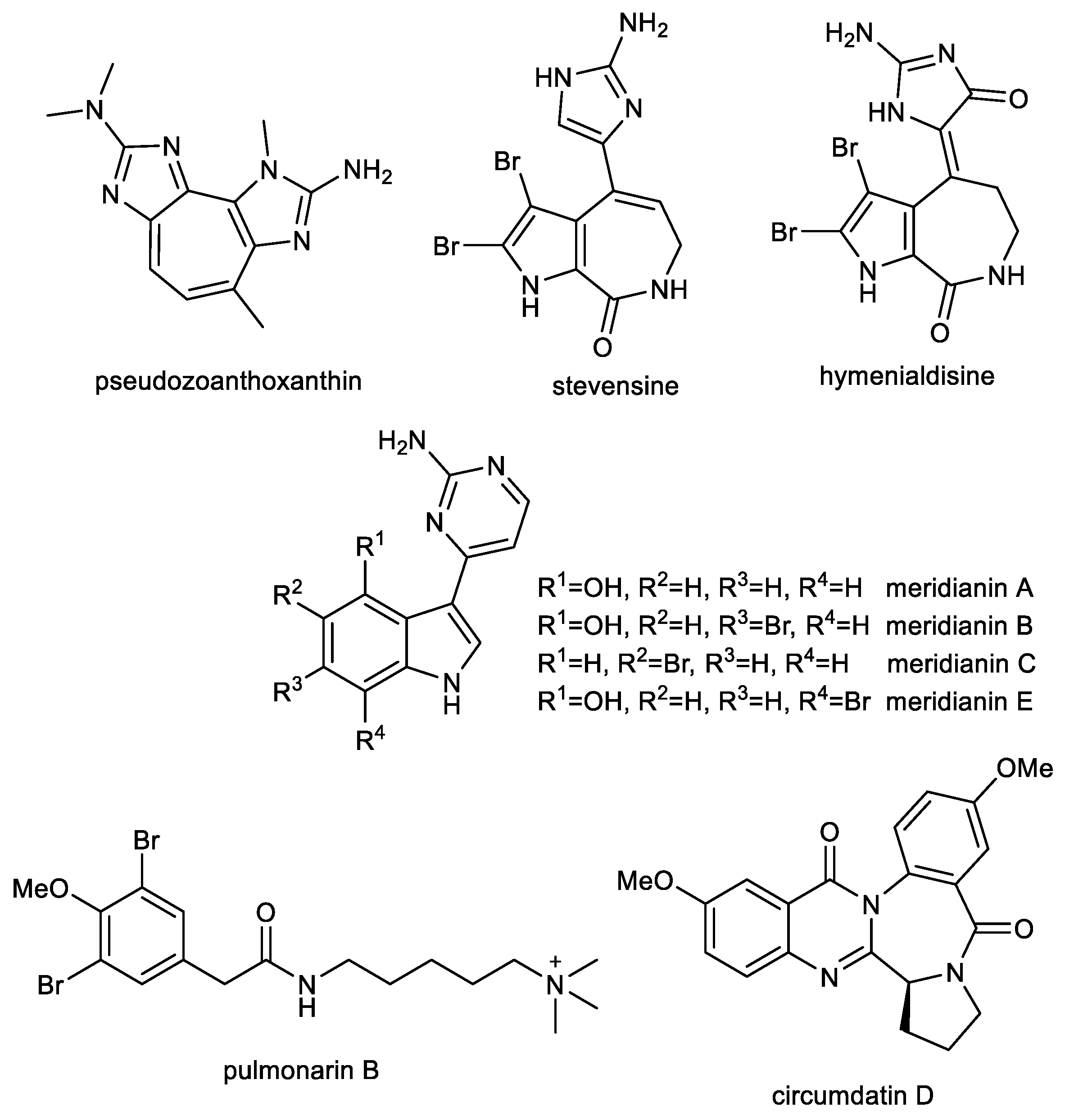
3.1. Imidazole Alkaloids: Pseudozoanthoxanthin and Stevensine
3.2. Indole Alkaloids: Meridianins
3.3. Further Nitrogen Containing Marine Compounds as Multi-Target Compounds for the Treatment of AD: Pulmonarin B
3.4. Quinazoline-Benzodiazepine Alkaloid: Circumdatin D
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Cheng, M.; Liu, P.; Cao, D.; Luo, J.; Wan, Y.; Fang, Y.; Jin, Y.; Xie, S.-S.; Liu, J. A multi-target directed ligands strategy for the treatment of Alzheimer’s disease: Dimethyl fumarate plus Tranilast modified Dithiocarbate as AChE inhibitor and Nrf2 activator. Eur. J. Med. Chem. 2022, 242, 114630. [Google Scholar] [CrossRef] [PubMed]
- Wattmo, C.; Wallin, Å.K. Early- versus late-onset Alzheimer’s disease in clinical practice: Cognitive and global outcomes over 3 years. Alzheimer’s Res. Ther. 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cahlíková, L.; Vrabec, R.; Pidaný, F.; Peřinová, R.; Maafi, N.; Mamun, A.A.; Ritomská, A.; Wijaya, V.; Blunden, G. Recent Progress on Biological Activity of Amaryllidaceae and Further Isoquinoline Alkaloids in Connection with Alzheimer’s Disease. Molecules 2021, 26, 5240. [Google Scholar] [CrossRef]
- Forest, K.H.; Nichols, R.A. Assessing Neuroprotective Agents for Aβ-Induced Neurotoxicity. Trends Mol. Med. 2019, 25, 685–695. [Google Scholar] [CrossRef]
- Cahlíková, L.; Macáková, K.; Benešová, N.; Chlebek, J.; Hošťálková, A.; Opletal, L. Chapter 6—Natural Compounds (Small Molecules) as Potential and Real Drugs of Alzheimer’s Disease: A Critical Review. In Studies in Natural Products Chemistry; Atta ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 42, pp. 153–194. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Hasselmo, M.E.; Anderson, B.P.; Bower, J.M. Cholinergic modulation of cortical associative memory function. J. Neurophysiol. 1992, 67, 1230–1246. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Silva, T.; Reis, J.; Teixeira, J.; Borges, F. Alzheimer’s disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes. Ageing Res. Rev. 2014, 15, 116–145. [Google Scholar] [CrossRef]
- de Almeida, J.P.; Saldanha, C. Nonneuronal cholinergic system in human erythrocytes: Biological role and clinical relevance. J. Membr. Biol. 2010, 234, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciro, A.; Park, J.; Burkhard, G.; Yan, N.; Geula, C. Biochemical differentiation of cholinesterases from normal and Alzheimer’s disease cortex. Curr. Alzheimer Res. 2012, 9, 138–143. [Google Scholar] [CrossRef]
- García-Ayllón, M.S.; Silveyra, M.X.; Sáez-Valero, J. Association between acetylcholinesterase and beta-amyloid peptide in Alzheimer’s cerebrospinal fluid. Chem. Biol. Interact. 2008, 175, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, G.R.; Nirmala, G.; Apparao, A.; Madhavi, A.S.; Sreelatha, S.; Rani, J.S.; Vijayalakshmi, P. Serum butyrylcholinesterase in type 2 diabetes mellitus: A biochemical and bioinformatics approach. Lipids Health Dis. 2005, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liston, D.R.; Nielsen, J.A.; Villalobos, A.; Chapin, D.; Jones, S.B.; Hubbard, S.T.; Shalaby, I.A.; Ramirez, A.; Nason, D.; White, W.F. Pharmacology of selective acetylcholinesterase inhibitors: Implications for use in Alzheimer’s disease. Eur. J. Pharmacol. 2004, 486, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Amat-ur-Rasool, H.; Ahmed, M.; Hasnain, S.; Carter, W.G. Anti-Cholinesterase Combination Drug Therapy as a Potential Treatment for Alzheimer’s Disease. Brain Sci. 2021, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Wu, H.-X.; Ye, S.-S.; Pan, L.; Qian, Y. Expression of recombinant human acetylcholinesterase and its application in screening its inhibitors. Acta Pharm. Sin. 2014, 49, 50–54. [Google Scholar]
- Wu, C.; Zhang, G.; Zhang, Z.-W.; Jiang, X.; Zhang, Z.; Li, H.; Qin, H.-L.; Tang, W. Structure–activity relationship, in vitro and in vivo evaluation of novel dienyl sulphonyl fluorides as selective BuChE inhibitors for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2021, 36, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Wang, M.; Shentu, J.; Huang, C.; Bai, Y.; Pan, H.; Zhang, D.; Yuan, Z.; Zhang, H.; Xiao, X.; et al. Inhibition of acetylcholinesterase activity and β-amyloid oligomer formation by 6-bromotryptamine A, a multi-target anti-Alzheimer’s molecule. Oncol. Lett. 2020, 19, 1593–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, C.; das Ros, L.U.; da Rocha, A.S.; Machado, L.S.; Bellaver, B.; Zimmer, E.R. The Glutamatergic System in Alzheimer’s Disease: A Systematic Review with Meta-Analysis. Alzheimer’s Dement. 2022, 18, e064821. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Pocernich, C.B. The Glutamatergic System and Alzheimer’s Disease. CNS Drugs 2003, 17, 641–652. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [Green Version]
- de Paula, V.J.R.; Guimarães, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dement. Neuropsychol. 2009, 3, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [Green Version]
- Agis-Torres, A.; Sölhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-Target-Directed Ligands and other Therapeutic Strategies in the Search of a Real Solution for Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Wang, H.H.; Liu, S.J.; Deng, Y.Q.; Zhang, Y.J.; Tian, Q.; Wang, X.C.; Chen, X.Q.; Yang, Y.; Zhang, J.Y.; et al. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 3591–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020, 163, 1599–1617. [Google Scholar] [CrossRef]
- Sabbagh, J.J.; Dickey, C.A. The Metamorphic Nature of the Tau Protein: Dynamic Flexibility Comes at a Cost. Front. Neurosci. 2016, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3β (GSK3β) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Plattner, F.; Angelo, M.; Giese, K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006, 281, 25457–25465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, A.; Perez, D.I. GSK-3 inhibitors: A ray of hope for the treatment of Alzheimer’s disease? J. Alzheimers Dis. 2008, 15, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hah, J.M. A Perspective on the Development of c-Jun N-terminal Kinase Inhibitors as Therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies. Biomedicines 2021, 9, 1431. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of Monoamine Oxidase Activity in Alzheimer’s Disease: An Insight into the Therapeutic Potential of Inhibitors. Molecules 2021, 26, 3724. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S.; et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimer’s Res. Ther. 2017, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, R.; Gottfries, C.G.; Oreland, L.; Wiberg, A.; Winblad, B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 1980, 27, 1029–1034. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinová, L.; Nepovimová, E.; Sepsova, V.; Korábečný, J.; Klimeš, J.; Kuča, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Woloshin, S.; Kesselheim, A.S. What to Know About the Alzheimer Drug Aducanumab (Aduhelm). JAMA Intern. Med. 2022, 182, 892. [Google Scholar] [CrossRef]
- Lalli, G.; Schott, J.M.; Hardy, J.; De Strooper, B. Aducanumab: A new phase in therapeutic development for Alzheimer’s disease? EMBO Mol. Med. 2021, 13, e14781. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Three FDA advisory panel members resign over approval of Alzheimer’s drug. BMJ 2021, 373, n1503. [Google Scholar] [CrossRef] [PubMed]
- Gujral, S.S.; Shakeri, A.; Hejazi, L.; Rao, P.P.N. Design, synthesis and structure-activity relationship studies of 3-phenylpyrazino [1,2-a]indol-1(2H)-ones as amyloid aggregation and cholinesterase inhibitors with antioxidant activity. Eur. J. Med. Chem. Rep. 2022, 6, 100075. [Google Scholar] [CrossRef]
- Reardon, S. FDA approves Alzheimer’s drug lecanemab amid safety concerns. Nature 2023, 613, 227–228. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.S.; Jhanji, N. Natural Products as Potential Anti-Alzheimer Agents. Curr. Med. Chem. 2020, 27, 5887–5917. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Khan, T.S.; Zargaham, M.K.; Khan, A.U.; Khan, S.; Hussain, A.; Uddin, J.; Khan, A.; Al-Harrasi, A. Potential therapeutic natural products against Alzheimer’s disease with Reference of Acetylcholinesterase. Biomed. Pharmacother. 2021, 139, 111609. [Google Scholar] [CrossRef]
- Kong, Y.R.; Tay, K.C.; Su, Y.X.; Wong, C.K.; Tan, W.N.; Khaw, K.Y. Potential of Naturally Derived Alkaloids as Multi-Targeted Therapeutic Agents for Neurodegenerative Diseases. Molecules 2021, 26, 728. [Google Scholar] [CrossRef]
- Hügel, H.M.; de Silva, N.H.; Siddiqui, A.; Blanch, E.; Lingham, A. Natural spirocyclic alkaloids and polyphenols as multi target dementia leads. Bioorg. Med. Chem. 2021, 43, 116270. [Google Scholar] [CrossRef]
- Roy, A. A review on the alkaloids an important therapeutic compound from plants. Int. J. Plant Biotechnol. 2017, 3, 1–9. [Google Scholar]
- Heinrich, M.; Mah, J.; Amirkia, V. Alkaloids Used as Medicines: Structural Phytochemistry Meets Biodiversity—An Update and Forward Look. Molecules 2021, 26, 1836. [Google Scholar] [CrossRef]
- Lima, J.A.; Hamerski, L. Chapter 8—Alkaloids as Potential Multi-Target Drugs to Treat Alzheimer’s Disease. In Studies in Natural Products Chemistry; Atta ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 61, pp. 301–334. [Google Scholar] [CrossRef]
- Mondal, A.; Gandhi, A.; Fimognari, C.; Atanasov, A.G.; Bishayee, A. Alkaloids for cancer prevention and therapy: Current progress and future perspectives. Eur. J. Pharmacol. 2019, 858, 172472. [Google Scholar] [CrossRef]
- Tilaoui, M.; Ait Mouse, H.; Zyad, A. Update and New Insights on Future Cancer Drug Candidates From Plant-Based Alkaloids. Front. Pharmacol. 2021, 12, 719694. [Google Scholar] [CrossRef]
- Yarnell, E.; Abascal, K. Treating Hypertension Botanically. Altern. Complement. Ther. 2001, 7, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Kundu, A.; Kumar, A.; Gupta, M.; Lee, B.; Bhakta, T.; Dash, S.; Kim, N.-H. Chapter 15—Analysis of alkaloids (indole alkaloids, isoquinoline alkaloids, tropane alkaloids). In Recent Advances in Natural Products Analysis; Silva, A.S., Nabavi, S.F., Saeedi, M., Nabavi, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 505–567. [Google Scholar] [CrossRef]
- Kashyap, P.; Kalaiselvan, V.; Kumar, R.; Kumar, S. Ajmalicine and Reserpine: Indole Alkaloids as Multi-Target Directed Ligands Towards Factors Implicated in Alzheimer’s Disease. Molecules 2020, 25, 1609. [Google Scholar] [CrossRef] [Green Version]
- Wiatrak, B.; Kubis-Kubiak, A.; Piwowar, A.; Barg, E. PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions. Cells 2020, 9, 958. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Meng, L.; Zou, N.; Wang, H.; Li, S.; Huang, L.; Cheng, X.; Wang, Z.; Chen, W.; Wang, C. Mechanism-based pharmacokinetics-pharmacodynamics studies of harmine and harmaline on neurotransmitters regulatory effects in healthy rats: Challenge on monoamine oxidase and acetylcholinesterase inhibition. Phytomedicine 2019, 62, 152967. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, X.; Tian, L.; Gao, Y.; Liu, W.; Chen, H.; Jiang, X.; Xu, Z.; Ding, H.; Zhao, Q. Design, synthesis and biological evaluation of harmine derivatives as potent GSK-3β/DYRK1A dual inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2021, 222, 113554. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ding, K.-M.; Zhang, L.; Cheng, X.-M.; Wang, C.-H.; Wang, Z.-T. Acetylcholinesterase and Butyrylcholinesterase Inhibitory Activities of β-Carboline and Quinoline Alkaloids Derivatives from the Plants of Genus Peganum. J. Chem. 2013, 2013, 717232. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shi, Z.-Q.; Zhang, M.; Xin, G.-Z.; Pang, T.; Zhou, P.; Chen, J.; Qi, L.-W.; Yang, H.; Xu, X.; et al. Camptothecin and its Analogs Reduce Amyloid-β Production and Amyloid-β42-Induced IL-1β Production. J. Alzheimer’s Dis. 2015, 43, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kesuma, D.; Wang, J.; Deng, Y.; Duan, J.; Wang, J.H.; Qi, R.Z. Specific inhibition of cyclin-dependent kinases and cell proliferation by harmine. Biochem. Biophys. Res. Commun. 2004, 317, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Vucicevic, J.; Nikolic, K.; Dobričić, V.; Agbaba, D. Prediction of blood–brain barrier permeation of α-adrenergic and imidazoline receptor ligands using PAMPA technique and quantitative-structure permeability relationship analysis. Eur. J. Pharm. Sci. 2015, 68, 94–105. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, X.; Liu, W.; Chou, G.; Wang, Z.; Wang, C. Potent AChE and BChE inhibitors isolated from seeds of Peganum harmala Linn by a bioassay-guided fractionation. J. Ethnopharmacol. 2015, 168, 279–286. [Google Scholar] [CrossRef]
- Song, Y.; Wang, J.; Teng, S.F.; Kesuma, D.; Deng, Y.; Duan, J.; Wang, J.H.; Qi, R.Z.; Sim, M.M. β-Carbolines as specific inhibitors of cyclin-Dependent kinases. Bioorg. Med. Chem. Lett. 2002, 12, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- GB2447791; Pharmaceutical Formulations and Compounds Such as Harmine, Harmaline, Harmane or Harmalol for Use in Alleviating Conditions Related to Down’s Syndrome. Intellectual Property Office: Newport, UK, 2008.
- Vijayalakshmi, V.; Lele, J.V.; Daginawala, H.F. Effect of reserpine on the monoamine oxidase (MAO) activity in rat liver and brain. Biochem. Pharmacol. 1978, 27, 1985–1986. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.; Luk, W.W.K.; Cui, W.; Hu, S.; Tsim, K.W.K.; Han, Y. Synergistic Inhibition on Acetylcholinesterase by the Combination of Berberine and Palmatine Originally Isolated from Chinese Medicinal Herbs. J. Mol. Neurosci. 2014, 53, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Min, B.-S.; Yokozawa, T.; Lee, J.-H.; Kim, Y.S.; Choi, J.S. Anti-Alzheimer and antioxidant activities of coptidis Rhizoma alkaloids. Biol. Pharm. Bull. 2009, 32, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Brunhofer, G.; Fallarero, A.; Karlsson, D.; Batista-Gonzalez, A.; Shinde, P.; Gopi Mohan, C.; Vuorela, P. Exploration of natural compounds as sources of new bifunctional scaffolds targeting cholinesterases and beta amyloid aggregation: The case of chelerythrine. Bioorg. Med. Chem. 2012, 20, 6669–6679. [Google Scholar] [CrossRef]
- Hošťálková, A.; Maříková, J.; Opletal, L.; Korábečný, J.; Hulcová, D.; Kuneš, J.; Nováková, L.; Perez, D.I.; Jun, D.; Kučera, T.; et al. Isoquinoline Alkaloids from Berberis vulgaris as Potential Lead Compounds for the Treatment of Alzheimer’s Disease. J. Nat. Prod. 2019, 82, 239–248. [Google Scholar] [CrossRef]
- Carradori, S.; D’Ascenzio, M.; Chimenti, P.; Secci, D.; Bolasco, A. Selective MAO-B inhibitors: A lesson from natural products. Mol. Divers. 2014, 18, 219–243. [Google Scholar] [CrossRef]
- Fischer, H.; Kansy, M.; Avdeef, A.; Senner, F. Permeation of permanently positive charged molecules through artificial membranes—Influence of physico-chemical properties. Eur. J. Pharm. Sci. 2007, 31, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Plazas, E.; Hagenow, S.; Avila Murillo, M.; Stark, H.; Cuca, L.E. Isoquinoline alkaloids from the roots of Zanthoxylum rigidum as multi-target inhibitors of cholinesterase, monoamine oxidase A and Aβ1-42 aggregation. Bioorg. Chem. 2020, 98, 103722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q.; Liu, R.; Zhou, H.; Crommen, J.; Moaddel, R.; Jiang, Z.; Zhang, T. Rapid screening and identification of monoamine oxidase-A inhibitors from Corydalis Rhizome using enzyme-immobilized magnetic beads based method. J. Chromatogr. A 2019, 1592, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.C.; Ryu, H.W.; Kang, M.G.; Lee, H.; Park, D.; Cho, M.L.; Oh, S.R.; Kim, H. Selective inhibition of monoamine oxidase A by chelerythrine, an isoquinoline alkaloid. Bioorg. Med. Chem. Lett. 2018, 28, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Arya, U.; Dwivedi, H.; Subramaniam, J.R. Reserpine ameliorates Aβ toxicity in the Alzheimer’s disease model in Caenorhabditis elegans. Exp. Gerontol. 2009, 44, 462–466. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [Green Version]
- Herraiz, T.; González, D.; Ancín-Azpilicueta, C.; Arán, V.J.; Guillén, H. beta-Carboline alkaloids in Peganum harmala and inhibition of human monoamine oxidase (MAO). Food Chem. Toxicol. 2010, 48, 839–845. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D.; Yu, S. Pharmacological effects of harmine and its derivatives: A review. Arch. Pharm. Res. 2020, 43, 1259–1275. [Google Scholar] [CrossRef]
- He, D.; Wu, H.; Wei, Y.; Liu, W.; Huang, F.; Shi, H.; Zhang, B.; Wu, X.; Wang, C. Effects of harmine, an acetylcholinesterase inhibitor, on spatial learning and memory of APP/PS1 transgenic mice and scopolamine-induced memory impairment mice. Eur. J. Pharmacol. 2015, 768, 96–107. [Google Scholar] [CrossRef]
- Li, S.-P.; Wang, Y.-W.; Qi, S.-L.; Zhang, Y.-P.; Deng, G.; Ding, W.-Z.; Ma, C.; Lin, Q.-Y.; Guan, H.-D.; Liu, W.; et al. Analogous β-Carboline Alkaloids Harmaline and Harmine Ameliorate Scopolamine-Induced Cognition Dysfunction by Attenuating Acetylcholinesterase Activity, Oxidative Stress, and Inflammation in Mice. Front. Pharmacol. 2018, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Ai, X.; Yu, P.; Peng, L.; Luo, L.; Liu, J.; Li, S.; Lai, X.; Luan, F.; Meng, X. Berberine: A Review of its Pharmacokinetics Properties and Therapeutic Potentials in Diverse Vascular Diseases. Front. Pharmacol. 2021, 12, 762654. [Google Scholar] [CrossRef] [PubMed]
- Neag, M.A.; Mocan, A.; Echeverría, J.; Pop, R.M.; Bocsan, C.I.; Crişan, G.; Buzoianu, A.D. Berberine: Botanical Occurrence, Traditional Uses, Extraction Methods, and Relevance in Cardiovascular, Metabolic, Hepatic, and Renal Disorders. Front. Pharmacol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imenshahidi, M.; Hosseinzadeh, H. Berberis vulgaris and Berberine: An Update Review. Phytother. Res. 2016, 30, 1745–1764. [Google Scholar] [CrossRef] [PubMed]
- Imenshahidi, M.; Hosseinzadeh, H. Berberine and barberry (Berberis vulgaris): A clinical review. Phytother. Res. 2019, 33, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.-F.; Yang, C.-J.; Morris-Natschke, S.L.; Li, J.-C.; Yin, X.-D.; Liu, Y.-Q.; Guo, X.; Peng, J.-W.; Goto, M.; Zhang, J.-Y.; et al. Biologically active isoquinoline alkaloids covering 2014–2018. Med. Res. Rev. 2020, 40, 2212–2289. [Google Scholar] [CrossRef]
- Cheng, Z.; Kang, C.; Che, S.; Su, J.; Sun, Q.; Ge, T.; Guo, Y.; Lv, J.; Sun, Z.; Yang, W.; et al. Berberine: A Promising Treatment for Neurodegenerative Diseases. Front. Pharmacol. 2022, 13, 845591. [Google Scholar] [CrossRef]
- Akbar, M.; Shabbir, A.; Rehman, K.; Akash, M.S.H.; Shah, M.A. Neuroprotective potential of berberine in modulating Alzheimer’s disease via multiple signaling pathways. J. Food Biochem. 2021, 45, e13936. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, C.; Yang, W. Role of berberine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016, 12, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Yuan, N.N.; Cai, C.Z.; Wu, M.Y.; Su, H.X.; Li, M.; Lu, J.H. Neuroprotective effects of berberine in animal models of Alzheimer’s disease: A systematic review of pre-clinical studies. BMC Complement. Altern. Med. 2019, 19, 109. [Google Scholar] [CrossRef]
- Ji, H.-F.; Shen, L. Berberine: A Potential Multipotent Natural Product to Combat Alzheimer’s Disease. Molecules 2011, 16, 6732–6740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.A.; Singh, K.S.; Nandi, K.M.; Mishra, G.; Maurya, A.; Rai, A.; Rai, K.G.; Awasthi, R.; Sharma, B.; Kulkarni, T.G. Berberine: A Plant-derived Alkaloid with Therapeutic Potential to Combat Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2019, 19, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Durairajan, S.S.; Liu, L.F.; Lu, J.H.; Chen, L.L.; Yuan, Q.; Chung, S.K.; Huang, L.; Li, X.S.; Huang, J.D.; Li, M. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 2012, 33, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Qian, C. Berberine chloride can ameliorate the spatial memory impairment and increase the expression of interleukin-1beta and inducible nitric oxide synthase in the rat model of Alzheimer’s disease. BMC Neurosci. 2006, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Kalalian-Moghaddam, H.; Baluchnejadmojarad, T.; Roghani, M.; Goshadrou, F.; Ronaghi, A. Hippocampal synaptic plasticity restoration and anti-apoptotic effect underlie berberine improvement of learning and memory in streptozotocin-diabetic rats. Eur. J. Pharmacol. 2013, 698, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, D.A.; Khalil, S.; Hafez, H.S.; Bajorath, J.; Ahmed, H.E.A.; Sarhan, E.; Elwakeel, E.; El-Demellawy, M.A. Berberine Reduces Neurotoxicity Related to Nonalcoholic Steatohepatitis in Rats. Evid.-Based Complement. Altern. Med. 2015, 2015, 361847. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Sur, B.; Shim, I.; Lee, H.; Hahm, D.-H. Phellodendron amurense and Its Major Alkaloid Compound, Berberine Ameliorates Scopolamine-Induced Neuronal Impairment and Memory Dysfunction in Rats. Korean J. Physiol. Pharmacol. 2012, 16, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Bhutada, P.; Mundhada, Y.; Bansod, K.; Tawari, S.; Patil, S.; Dixit, P.; Umathe, S.; Mundhada, D. Protection of cholinergic and antioxidant system contributes to the effect of berberine ameliorating memory dysfunction in rat model of streptozotocin-induced diabetes. Behav. Brain Res. 2011, 220, 30–41. [Google Scholar] [CrossRef]
- Huang, M.; Jiang, X.; Liang, Y.; Liu, Q.; Chen, S.; Guo, Y. Berberine improves cognitive impairment by promoting autophagic clearance and inhibiting production of β-amyloid in APP/tau/PS1 mouse model of Alzheimer’s disease. Exp. Gerontol. 2017, 91, 25–33. [Google Scholar] [CrossRef]
- Ye, C.; Liang, Y.; Chen, Y.; Xiong, Y.; She, Y.; Zhong, X.; Chen, H.; Huang, M. Berberine Improves Cognitive Impairment by Simultaneously Impacting Cerebral Blood Flow and β-Amyloid Accumulation in an APP/tau/PS1 Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 1161. [Google Scholar] [CrossRef]
- Shaker, F.H.; El-Derany, M.O.; Wahdan, S.A.; El-Demerdash, E.; El-Mesallamy, H.O. Berberine ameliorates doxorubicin-induced cognitive impairment (chemobrain) in rats. Life Sci. 2021, 269, 119078. [Google Scholar] [CrossRef]
- Panahi, N.; Mahmoudian, M.; Mortazavi, P.; Hashjin, G.S. Effects of berberine on β-secretase activity in a rabbit model of Alzheimer’s disease. Arch. Med. Sci. 2013, 9, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, C.; Cao, G.; Guo, L.; Zhang, S.; Liang, Y.; Qin, C.; Su, P.; Li, H.; Zhang, W. Berberine modulates amyloid-β peptide generation by activating AMP-activated protein kinase. Neuropharmacology 2017, 125, 408–417. [Google Scholar] [CrossRef]
- Zhu, F.; Wu, F.; Ma, Y.; Liu, G.; Li, Z.; Sun, Y.A.; Pei, Z. Decrease in the production of beta-amyloid by berberine inhibition of the expression of beta-secretase in HEK293 cells. BMC Neurosci. 2011, 12, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wang, C.; Chen, Y.; He, Y.; Cai, Z. Berberine attenuates cognitive impairment and ameliorates tau hyperphosphorylation by limiting the self-perpetuating pathogenic cycle between NF-κB signaling, oxidative stress and neuroinflammation. Pharmacol. Rep. 2017, 69, 1341–1348. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Liang, Y.; Chen, H.; Ji, X.; Huang, M. Berberine mitigates cognitive decline in an Alzheimer’s Disease Mouse Model by targeting both tau hyperphosphorylation and autophagic clearance. Biomed. Pharmacother. 2020, 121, 109670. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, Y.; Tian, Q.; Liu, R.; Wang, Q.; Wang, J.-Z.; Wang, X. Berberine Attenuates Calyculin A-Induced Cytotoxicity and Tau Hyperphosphorylation in HEK293 Cells. J. Alzheimer’s Dis. 2011, 24, 525–535. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Li, F.; An, L. Berberine alleviates postoperative cognitive dysfunction by suppressing neuroinflammation in aged mice. Int. Immunopharmacol. 2016, 38, 426–433. [Google Scholar] [CrossRef]
- Deng, H.; Jia, Y.; Pan, D.; Ma, Z. Berberine alleviates rotenone-induced cytotoxicity by antioxidation and activation of PI3K/Akt signaling pathway in SH-SY5Y cells. Neuroreport 2020, 31, 41–47. [Google Scholar] [CrossRef]
- Wang, X.; Wang, R.; Xing, D.; Su, H.; Ma, C.; Ding, Y.; Du, L. Kinetic difference of berberine between hippocampus and plasma in rat after intravenous administration of Coptidis rhizoma extract. Life Sci. 2005, 77, 3058–3067. [Google Scholar] [CrossRef]
- Meng, F.-C.; Wu, Z.-F.; Yin, Z.-Q.; Lin, L.-G.; Wang, R.; Zhang, Q.-W. Coptidis rhizoma and its main bioactive components: Recent advances in chemical investigation, quality evaluation and pharmacological activity. Chin. Med. 2018, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Lee, K.T.; Baek, N.-I.; Kim, S.-H.; Park, H.W.; Lim, J.P.; Shin, T.Y.; Eom, D.O.; Yang, J.H.; Eun, J.S. Acetylcholinesterase inhibitors from the aerial parts of Corydalis speciosa. Arch. Pharm. Res. 2004, 27, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.-T.; Peng, J.; Liang, Y.; Yang, J.; Bai, X.; Hao, X.-Y.; Yang, F.-M.; Sun, Q.-Y. Acetylcholinesterase inhibitors from Corydalis yanhusuo. Nat. Prod. Res. 2011, 25, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, D.; Kumar, V. Memory-Enhancing Activity of Palmatine in Mice Using Elevated Plus Maze and Morris Water Maze. Adv. Pharmacol. Sci. 2012, 2012, 357368. [Google Scholar] [CrossRef] [Green Version]
- Kiris, I.; Kukula-Koch, W.; Karayel-Basar, M.; Gurel, B.; Coskun, J.; Baykal, A.T. Proteomic alterations in the cerebellum and hippocampus in an Alzheimer’s disease mouse model: Alleviating effect of palmatine. Biomed. Pharmacother. 2023, 158, 114111. [Google Scholar] [CrossRef]
- Jia, W.; Su, Q.; Cheng, Q.; Peng, Q.; Qiao, A.; Luo, X.; Zhang, J.; Wang, Y. Neuroprotective Effects of Palmatine via the Enhancement of Antioxidant Defense and Small Heat Shock Protein Expression in Aβ-Transgenic Caenorhabditis elegans. Oxid. Med. Cell. Longev. 2021, 2021, 9966223. [Google Scholar] [CrossRef]
- Jeon, S.J.; Kwon, K.J.; Shin, S.; Lee, S.H.; Rhee, S.Y.; Han, S.-H.; Lee, J.; Kim, H.Y.; Cheong, J.H.; Ryu, J.H.; et al. Inhibitory effects of Coptis japonica alkaloids on the LPS-induced activation of BV2 microglial cells. Biomol. Ther. 2009, 17, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Song, J.; Zhong, L.; Liao, Y.; Liu, L.; Li, X. Palmatine: A review of its pharmacology, toxicity and pharmacokinetics. Biochimie 2019, 162, 176–184. [Google Scholar] [CrossRef]
- Wianowska, D.; Garbaczewska, S.; Cieniecka–Roslonkiewicz, A.; Typek, R.; Dawidowicz, A.L. Chemical composition and antifungal activity of Chelidonium majus extracts—Antagonistic action of chelerythrine and sanguinarine against Botrytis cinerea. Chem. Ecol. 2018, 34, 582–594. [Google Scholar] [CrossRef]
- Rouleau, J.; Iorga, B.I.; Guillou, C. New potent human acetylcholinesterase inhibitors in the tetracyclic triterpene series with inhibitory potency on amyloid β aggregation. Eur. J. Med. Chem. 2011, 46, 2193–2205. [Google Scholar] [CrossRef]
- Catto, M.; Aliano, R.; Carotti, A.; Cellamare, S.; Palluotto, F.; Purgatorio, R.; De Stradis, A.; Campagna, F. Design, synthesis and biological evaluation of indane-2-arylhydrazinylmethylene-1,3-diones and indol-2-aryldiazenylmethylene-3-ones as β-amyloid aggregation inhibitors. Eur. J. Med. Chem. 2010, 45, 1359–1366. [Google Scholar] [CrossRef]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Huyck, T.K.; Gradishar, W.; Manuguid, F.; Kirkpatrick, P. Eribulin mesylate. Nat. Rev. Drug Discov. 2011, 10, 173–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribble, G.W. Biological Activity of Recently Discovered Halogenated Marine Natural Products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, R.; Swanson, G.T. Recent progress in neuroactive marine natural products. Nat. Prod. Rep. 2014, 31, 273–309. [Google Scholar] [CrossRef] [Green Version]
- Hafez Ghoran, S.; Kijjoa, A. Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities. Mar. Drugs 2021, 19, 410. [Google Scholar] [CrossRef] [PubMed]
- Turk, T.; Maček, P.; Šuput, D. Inhibition of acetylcholinesterase by a pseudozoanthoxanthin-like compound isolated from the zoanthid Parazoanthus axinellae (O. Schmidt). Toxicon 1995, 33, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Stachel, S.J.; Coburn, C.A.; Rush, D.; Jones, K.L.G.; Zhu, H.; Rajapakse, H.; Graham, S.L.; Simon, A.; Katharine Holloway, M.; Allison, T.J.; et al. Discovery of aminoheterocycles as a novel beta-secretase inhibitor class: pH dependence on binding activity part 1. Bioorg. Med. Chem. Lett. 2009, 19, 2977–2980. [Google Scholar] [CrossRef]
- Vitale, R.M.; Rispoli, V.; Desiderio, D.; Sgammato, R.; Thellung, S.; Canale, C.; Vassalli, M.; Carbone, M.; Ciavatta, M.L.; Mollo, E.; et al. In Silico Identification and Experimental Validation of Novel Anti-Alzheimer’s Multitargeted Ligands from a Marine Source Featuring a “2-Aminoimidazole plus Aromatic Group” Scaffold. ACS Chem. Neurosci. 2018, 9, 1290–1303. [Google Scholar] [CrossRef]
- Galante, D.; Ruggeri, F.S.; Dietler, G.; Pellistri, F.; Gatta, E.; Corsaro, A.; Florio, T.; Perico, A.; D’Arrigo, C. A critical concentration of N-terminal pyroglutamylated amyloid beta drives the misfolding of Ab1-42 into more toxic aggregates. Int. J. Biochem. Cell Biol. 2016, 79, 261–270. [Google Scholar] [CrossRef]
- Galante, D.; Corsaro, A.; Florio, T.; Vella, S.; Pagano, A.; Sbrana, F.; Vassalli, M.; Perico, A.; D’Arrigo, C. Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1-42 and Aβpy3-42 beta-amyloids. Int. J. Biochem. Cell Biol. 2012, 44, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Naldi, M.; Fiori, J.; Valle, F.; Biscarini, F.; Nicolau, D.V.; Andrisano, V. Kinetic characterization of amyloid-beta 1-42 aggregation with a multimethodological approach. Anal. Biochem. 2011, 414, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Rispoli, V.; Rotiroti, D.; Carelli, V.; Liberatore, F.; Scipione, L.; Marra, R.; Tortorella, S.; Di Rienzo, B. Electroencephalographic effects induced by choline pivaloyl esters in scopolamine-treated or nucleus basalis magnocellularis lesioned rats. Pharmacol. Biochem. Behav. 2004, 78, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Rispoli, V.; Ragusa, S.; Nisticò, R.; Marra, R.; Russo, E.; Leo, A.; Felicitá, V.; Rotiroti, D. Huperzine A restores cortico-hippocampal functional connectivity after bilateral AMPA lesion of the nucleus basalis of Meynert. J. Alzheimer’s Dis. 2013, 35, 833–846. [Google Scholar] [CrossRef]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.-L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.Q.; Song, J.L.; Zhu, K.; Zhang, J.; Jiang, C.S.; Zhang, H. Total Synthesis of Pulmonarin B and Design of Brominated Phenylacetic Acid/Tacrine Hybrids: Marine Pharmacophore Inspired Discovery of New ChE and Aβ Aggregation Inhibitors. Mar. Drugs 2018, 16, 293. [Google Scholar] [CrossRef] [Green Version]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Alonso, D.; Castro, A.; Martinez, A. Marine compounds for the therapeutic treatment of neurological disorders. Expert Opin. Ther. Pat. 2005, 15, 1377–1386. [Google Scholar] [CrossRef]
- Gompel, M.; Leost, M.; De Kier Joffe, E.B.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Núñez-Pons, L.; Carbone, M.; Vázquez, J.; Rodríguez, J.; Nieto, R.M.; Varela, M.M.; Gavagnin, M.; Avila, C. Natural products from Antarctic colonial ascidians of the genera Aplidium and Synoicum: Variability and defensive role. Mar. Drugs 2012, 10, 1741–1764. [Google Scholar] [CrossRef] [Green Version]
- Llorach-Pares, L.; Rodriguez-Urgelles, E.; Nonell-Canals, A.; Alberch, J.; Avila, C.; Sanchez-Martinez, M.; Giralt, A. Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules 2020, 10, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Urgellés, E.; Sancho-Balsells, A.; Chen, W.; López-Molina, L.; Ballasch, I.; Del Castillo, I.; Avila, C.; Alberch, J.; Giralt, A. Meridianins Rescue Cognitive Deficits, Spine Density and Neuroinflammation in the 5xFAD Model of Alzheimer’s Disease. Front. Pharmacol. 2022, 13, 791666. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, M.; Svenson, J.; Sepčić, K.; Trembleau, L.; Engqvist, M.; Andersen, J.H.; Jaspars, M.; Stensvåg, K.; Haug, T. Isolation and Synthesis of Pulmonarins A and B, Acetylcholinesterase Inhibitors from the Colonial Ascidian Synoicum pulmonaria. J. Nat. Prod. 2014, 77, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbæk, L.; Breinholt, J. Circumdatins D, E, and F: Further Fungal Benzodiazepine Analogues from Aspergillus ochraceus. J. Nat. Prod. 1999, 62, 904–905. [Google Scholar] [CrossRef]
- Cui, C.-M.; Li, X.-M.; Li, C.-S.; Sun, H.-F.; Gao, S.-S.; Wang, B.-G. Benzodiazepine Alkaloids from Marine-Derived Endophytic Fungus Aspergillus ochraceus. Helv. Chim. Acta 2009, 92, 1366–1370. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, L.; Liu, D.; Huang, J.; Lin, W. Circumdatin D Exerts Neuroprotective Effects by Attenuating LPS-Induced Pro-Inflammatory Responses and Downregulating Acetylcholinesterase Activity In Vitro and In Vivo. Front. Pharmacol. 2020, 11, 760. [Google Scholar] [CrossRef]
- Xin, L.; Yamujala, R.; Wang, Y.; Wang, H.; Wu, W.H.; Lawton, M.A.; Long, C.; Di, R. Acetylcholinestarase-inhibiting alkaloids from Lycoris radiata delay paralysis of amyloid beta-expressing transgenic C. elegans CL4176. PLoS ONE 2013, 8, e63874. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AChE IC50 (µM) | BuChE IC50 (µM) | BACE-1 (% Inhibition at Given Conc.) | Aβ1–42 IC50 (µM) | MAO-A IC50 (µM) | CDK5 IC50 (µM) | GSK-3β IC50 (µM) | DYRK1A IC50 (µM) | PAMPA BBB Permeation Pe (×10−6 cm/s) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Harmine | 1.21 ± 0.04 b; 9.05 ± 1.08 b | 2.79 ± 0.27 d; 75.07 ± 1.29 d | 0 ± 13 at 10 µM | 2.5 ± 0.7 | 0.38 ± 0.21 | 20 | 31.1 ± 1.0 | 0.080 ± 0.007 | 44.6 (CNS+); 7.17 ± 1.29 (CNS+) | [70,71,72,73,74,75,76] |
| Harmaline | 1.95 ± 0.08 b; 10.58 ± 2.01 b | 5.38 ± 0.64 d; 101.39 ± 1.39 d | n.d. | n.d. | 0.10 ± 0.08 | Inhibition 7% at 50 µM | n.d. | 9 | n.d. | [70,72,76,77,78] |
| ZDWX-25 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 0.071 ± 0.009 | 0.103 ± 0.004 | 16.5 (CNS+) | [71] |
| Reserpine | 1.7 ± 2.08 b | 2.8 ± 1.84 d | 47 at 50 µM | 57% at 44 µM | Increase in activity | n.d. | n.d. | n.d. | 2.75 ± 0.25 (CNS+/−) | [68,75,79] |
| Ajmalicine | 3.5 ± 1.41 b | 5.44 ± 1.75 d | 69 at 50 µM | 69% at 44 µM | n.d. | n.d. | n.d. | n.d. | n.d. | [68] |
| Berberine | 0.520 ± 0.042 a; 0.61 ± 0.04 a; 0.7 ± 0.1 a; 0.44 ± 0.04 b; 2.74 ± 0.22 b | 30.7 ± 3.5 c; 6.40 ± 0.29 d; 3.44 ± 0.26 d | IC50 > 100 µM | 43.84 ± 6.09 | 126 | n.d. | n.d. | n.d. | 0.1 ± 0.1 (CNS−); 0.02 (CNS−) | [80,81,82,83,84,85,86] |
| Palmatine | 0.46 ± 0.013 a; 1.69 ± 0.11 a; 4.07 ± 0.09 b; 0.51 ± 0.00 b | >100 c; 6.84 ± 0.07 d | IC50 > 100 µM | 92.15 ± 3.42 | 63.86 ± 1.35 | n.d. | n.d. | n.d. | 0 (CNS−) | [80,81,82,83,85,87] |
| Avicine | 0.52 ± 0.05 a; 0.15 ± 0.01 b | 0.88 ± 0.08 d | n.d. | 5.56 ± 0.94 | 0.41 ± 0.02 | n.d. | n.d. | n.d. | n.d. | [86] |
| Nitidine | 1.25 ± 0.09 a; 0.65 ± 0.09 b | 5.73 ± 0.60 d | n.d. | 1.89 ± 0.40 | 1.89 ± 0.17 | n.d. | n.d. | n.d. | n.d. | [86] |
| Chelerythrine | 1.54 ± 0.07 a; 1.03 ± 0.11 b | 10.34 ± 0.24 c; 3.55 ± 0.18 d | n.d. | 4.20 ± 0.43 | 0.55 ± 0.042 | n.d. | 0% at 10 µM | n.d. | 3.25 (CNS+/−) | [82,85,86,88,89] |
| Compound | AChE IC50 (µM) | BuChE IC50 (µM) | BACE-1 IC50 (µM) | Aβ1–42 (% Inhibition at Given Conc.) | DYRK1A IC50 (µM) | Ref. |
|---|---|---|---|---|---|---|
| Pseudozoanthoxanthin | 12.2 ± 1.4 a | 14.6 ± 5.4 c | 0.9 ± 0.1 | 50% at 50 µM | n.d. | [143] |
| Stevensine | 7.8 ± 1.5 a | 141.6 ± 34.0 c | 1.4 ± 0.4 | n.d. | n.d. | [143] |
| Hymenialdisine | n.d. | n.d. | n.d. | n.d. | 0.0033 | [149] |
| Pulmonarin B | 37.02 ± 2.11 b | 30.70 ± 1.44 c | n.d. | 29.78 ± 1.45% at 10 µM | n.d. | [150] |
| Compound | CDK1 IC50 (µM) | CDK5 IC50 (µM) | GSK-3β IC50 (µM) | CK1 IC50 (µM) | Ref. |
|---|---|---|---|---|---|
| Hymenialdisine | 0.022 | 0.028 | 0.010 | 0.035 | [152] |
| Meridianin A | 2.50 | 3.00 | 1.30 | n.d. | [153] |
| Meridianin B | 1.50 | 1.00 | 0.50 | 1.00 | [153] |
| Meridianin C | 3.00 | 6.00 | 2.00 | 30.00 | [153] |
| Meridianin E | 0.18 | 0.15 | 2.50 | 100.00 | [153] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrabec, R.; Blunden, G.; Cahlíková, L. Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 4399. https://doi.org/10.3390/ijms24054399
Vrabec R, Blunden G, Cahlíková L. Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(5):4399. https://doi.org/10.3390/ijms24054399
Chicago/Turabian StyleVrabec, Rudolf, Gerald Blunden, and Lucie Cahlíková. 2023. "Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 5: 4399. https://doi.org/10.3390/ijms24054399
APA StyleVrabec, R., Blunden, G., & Cahlíková, L. (2023). Natural Alkaloids as Multi-Target Compounds towards Factors Implicated in Alzheimer’s Disease. International Journal of Molecular Sciences, 24(5), 4399. https://doi.org/10.3390/ijms24054399