Abstract
Primary ovarian insufficiency (POI) is a heterogeneous disease resulting from non-functional ovaries in women before the age of 40. It is characterized by primary amenorrhea or secondary amenorrhea. As regards its etiology, although many POI cases are idiopathic, menopausal age is a heritable trait and genetic factors play an important role in all POI cases with known causes, accounting for approximately 20% to 25% of cases. This paper reviews the selected genetic causes implicated in POI and examines their pathogenic mechanisms to show the crucial role of genetic effects on POI. The genetic factors that can be found in POI cases include chromosomal abnormalities (e.g., X chromosomal aneuploidies, structural X chromosomal abnormalities, X-autosome translocations, and autosomal variations), single gene mutations (e.g., newborn ovary homeobox gene (NOBOX), folliculogenesis specific bHLH transcription factor (FIGLA), follicle-stimulating hormone receptor (FSHR), forkhead box L2 (FOXL2), bone morphogenetic protein 15 (BMP15), etc., as well as defects in mitochondrial functions and non-coding RNAs (small ncRNAs and long ncRNAs). These findings are beneficial for doctors to diagnose idiopathic POI cases and predict the risk of POI in women.
1. Introduction
Female infertility refers to the inability to conceive after 6 months (for women over the age of 35) to 1 year of regular and unprotected sex. According to the Office on Women’s Health (OWH) of America, premature ovarian insufficiency (POI) is one of the most common causes of female infertility. POI, which is also known as primary ovarian insufficiency or premature ovarian failure, refers to female amenorrhea before the age of 40 due to non-functional ovaries caused by follicle atresia and the rapid loss of germ cells [1]. POI is manifested as primary or secondary amenorrhea with hormonal changes, such as increased gonadotropin levels (FSH > 25 IU/L) and decreased estradiol and anti-Müllerian hormone levels [1,2]. Moreover, there are many clinical presentations in POI women. Hot flashes, night sweats, and insomnia are all classic symptoms of POI, which coincide with reduced estrogen condition [3]. The increase in the POI cases, among which there is a vast number of POI women with unclear genetic diagnoses, justifies investigating the etiology of POI, which may be critical in the early diagnosis, treatment, and prevention [3].
Although POI is heterogenous and the causes of many cases remain unclear, various types of etiologies, such as genetic, autoimmune, iatrogenic, infectious, environmental, chemotherapeutic, and radiotherapeutic causes, have been determined [4,5,6]. Previous research established a relationship between the genetic effects and POI by studying POI in families (the prevalence of familial POI ranges from 4% to 31%) [7,8,9]. Moreover, there is an increasing prevalence of POI in adolescents. In a recent study, the research group gathered information on women under 21 years of age diagnosed with POI in 2000–2016 from all pediatric endocrinology units in Israel [10]. Among the 130 women with POI, the most common cause was Turner syndrome/mosaicism, accounting for 43% of cases. For non-Turner POI cases in this group, a significant increase in the incidence of POI was observed. This is due to new and more effective gene technology and the frequent occurrence of autoimmune diseases. The incidence rate of new POI diagnoses per 100,000 person-years increased year-by-year, especially in 2009–2016, indicating the remarkable incidence rate of POI in adolescents. Furthermore, the overall prevalence of genetic-associated POI is approximately 20–25% [11]. Therefore, taking genetic factors into account often leads to a more straightforward POI diagnosis. Previous reviews on the genetic factors of POI often focus on one or more aspects, such as the POI-related genes involved in meiosis or DNA repair, or they reveal a certain type of chromosomal mutation associated with POI, such as X-autosome translocations. In addition, the broader reviews only pay attention to the single gene mutations of POI and so on. In this review, we reasonably classify the genetic factors and non-syndromic POI-related genes (according to the biological process in which genes participate), based on previous research, so that the genetic factors and corresponding mechanisms of POI are more comprehensively and carefully summarized. We divided genetic causes of POI into four categories after reviewing and analyzing the content of other previous literatures: chromosomal abnormalities, single gene variants in non-syndromic and syndromic POI, mitochondrial dysfunction, and abnormal levels of non-coding RNAs (Figure 1). Thus, the present review provides essential information to help us better understand POI caused by genetic factors. We hope that this will act to improve the efficacy of diagnosis and treatment for POI patients.
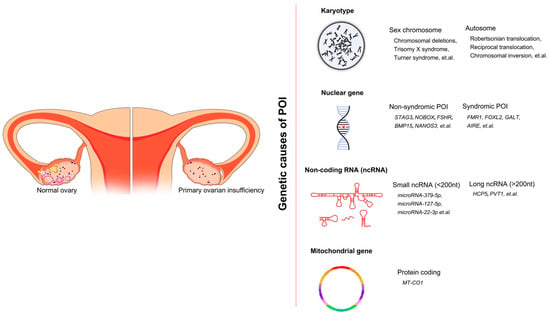
Figure 1.
The overall genetic causes of POI, showing the conditions of ovaries in healthy women and POI women. Four categories of genetic factors and corresponding examples are included.
2. Methods
We entered the following keywords “POI and genetic factors” (219), “POI and gene mutation” (217), “POI and variants” (209), “POI and a certain gene name”, “POI and mitochondria” (18), and “POI and non-coding RNA” (54) in Pubmed to search the articles and reviews on POI-related genetic factors in the past ten years. Overall, we searched for more than 717 articles. For the studies to be included in this review, the selected publications had to focus on the following: identifying the POI genetic factors in different POI populations via chromosomal analysis, candidate gene screening, genome wide association study, and various genome sequencing strategies. In addition, the role of POI candidate genes in animal models and studies on mitochondrial genes and non-coding RNAs associated with POI were also included. However, the literature identified was restricted to English language.
3. Chromosomal Abnormalities
Chromosomal abnormality is defined as a variation resulting from aneuploidy or structural defects in chromosomes. This can lead to many harmful and even lethal human genetic diseases, such as trisomies 21, 18, and 13 and sex chromosomes rearrangements. On this basis, researchers attempted to detect chromosomal variations using prenatal testing, which can be used to effectively avoid human genetic diseases caused by chromosomal abnormalities [12,13]. In addition, recent studies have shown that chromosomal abnormalities are responsible for POI [1,6,14]. The prevalence of POI caused by chromosomal abnormalities varies in different populations, with the values ranging from approximately 10% to 13% [4,15,16]. Chromosomal disorders cause POI via the depletion of primordial oocytes during early female development [1]. However, the mechanism involved in the loss of oocytes is not clearly understood. Moreover, defects in both the X chromosome and autosome can contribute to POI. Specifically, all X monosomies and trisomies, X chromosome deletions, X-autosome translocations, and autosomal translocations represent chromosomal abnormalities that can lead to POI.
3.1. X Chromosome Aneuploidies
Turner syndrome is a critical sex chromosomal disease in females with a 1 in 2500 incidence. It is caused by the complete or partial deletion of one sex chromosome [17,18]. TS has many hallmarks, including ovarian failure, and it is the most common genetic cause of POI, accounting for 4–5% of all POI cases [7,11,19]. Primordial follicle atresia and a reduction in the ovarian reserve are the circumstances under which TS causes POI, but its mechanism is unknown. A recent review suggested that TS-related POI was associated with the function and length of telomeres and epigenetic modifications [20]. Moreover, many scientists have shown that ovary-related genes were also responsible for the ovarian phenotype in TS patients [21]. The severity of TS patients’ symptoms depends on whether their genotype is 45X or mosaicism, such as 45X/46XX. The TS patients with a mosaic genotype are characterized by secondary amenorrhea, which means that this group of patients is fertile (producing a lower level of follicles) at first. However, fertility reduces over time, and eventually, POI develops [22,23]. In contrast, TS patients who lose a complete X chromosome are characterized by primary amenorrhea with a small chance of menarche [11].
Trisomy X syndrome (TXS) with a karyotype 47XXX is another sex chromosome aneuploidy that contributes to POI [24]. A relatively small body of literature is concerned with the connection between X trisomy and POI. In 2020, Shanlee et al. performed a case-control study and demonstrated that the level of anti-mullerian hormone (AMH) in TXS patients was lower than in healthy females, indicating that TXS females had a higher risk of suffering from POI [25]. In addition, in females with TXS, increasing follicle-stimulating hormone (FSH) and luteinizing hormone (LH) can cause menstrual cycle disturbance, which is associated with POI [26]. A previous survey also reported a patient with both blepharophimosis-ptosis-epicanthus syndrome (BPES) and TXS presenting with POI. However, this patient had a normal level of gonadotropins, which is rare in POI cases [27].
3.2. Structural X Chromosomal Abnormalities and X-Autosome Translocations
A number of studies have established a relationship between X chromosomal structural disorders (mainly X chromosomal deletions), X-autosomal translocations, and POI [11,26,28]. Furthermore, a recent study posited the existence of POI critical regions 1 and 2 on the X chromosome, which define the positions of the breakpoints for X chromosomal deletions and X-autosomal translocations related to ovarian functions, respectively [22]. The POI1 region consists of a part of the long arm of the X chromosome ranging from Xq24 to Xq27, but does not conclude the fragile X mental retardation 1 (FMR1) gene, while the Xq13.1 to Xq21.33 region belongs to the POI2 region. Although a considerable body of research has demonstrated the pathogenic role of structural X chromosomal abnormalities in POI, much less attention has been paid to this area over the past decade. By comparing and analyzing the statistics from four studies, the reasonable prevalence of X chromosome structural anomalies and X-autosome translocations related to POI was calculated to range from 4.2% to 12.0% [29,30,31,32]. Moreover, many POI candidate genes on the X chromosome can be found by analyzing the X-autosome translocations, some of which are introduced below.
3.3. Autosomal Abnormalities
Autosomal translocations, microdeletions, specific gene mutations, epistasis, and epigenetics associated with autosomal genes are all responsible for POI [33,34]. As has been previously reported, for chromosome variations, most POI cases are caused by X chromosome variations, while autosomal variations account for only a small number of POI cases [16,29,35]. The majority of previous and current studies focus on autosomal translocations and gene variants. According to a recent literature review, only 23 cases exhibited autosomal abnormalities (Robertsonian translocation, reciprocal translocation, and chromosomal inversion) associated with POI, which were identified in different POI populations with different ethnicities, including Chinese, Thai, and American [36]. Many autosomal genes associated with ovarian functions are discussed in the next section.
4. Single Gene Variants and Non-Syndromic POI
Aside from chromosomal abnormalities, single gene variations can also cause POI. The classical candidate gene approach is based on genes with known functions and experimental models in mice. In these, scientists establish a hypothesis regarding the connection between candidate genes and POI. Using this method, many genes, such as BMP15, NOBOX, and FMR1, have been discovered [37]. However, the range of candidate genes is restricted in this traditional approach. The appearance of many new strategies, such as genome-wide association studies (GWAS), can overcome the shortcomings of the traditional method. However, as a result of the low prevalence and high heterogeneity of POI, it is challenging to perform replicated experiments to prove the causation of these candidate genes using GWAS. Therefore, the majority of recent studies that investigate POI-related genes utilize another method, known as whole exome sequencing (WES). Moreover, next-generation sequencing (NGS) is also responsible for identifying variants. The associated genes identified in the last 10 years are classified according to the biological processes they participate in (Table 1), which are introduced below.

Table 1.
List of candidate genes implicated in non-syndromic POI.
4.1. Meiosis and DNA Replication and Repair
A normal oocyte reserve is essential for females of reproductive age to give birth to a healthy baby. However, if any error occurs during meiosis, DNA replication, or DNA repair, the genetic information is negatively affected, leading to germ cell apoptosis and infertility. Therefore, collecting and investigating the genes involved in the critical processes of meiosis, DNA replication, and DNA repair is beneficial for obtaining a better understanding of POI.
4.1.1. Helicase for Meiosis 1 (HFM1)
HFM1 exists on chromosome 1p22 and encodes DNA helicase, which is only expressed in the ovary and testis. There are numerous studies that demonstrate the connection between HFM1 and POI. A recent study conducted WES in a Chinese POI cohort containing two POI patients (the proband and her mother) and found a novel missense mutation of HFM1 (c.3470G> A), which can affect mRNA transcription [88]. However, its role in protein levels needs further investigation. Moreover, one out of twenty-four POI patients in a cohort harbored two disease-causing variants in HFM1 (c.3100G > A and c.1006 + 1G > T) [41]. Similar to c.3470G> A, c.1006 + 1G > T also disrupts RNA splicing. In addition, another mutation, c.3100G > A, alters the amino acid of the protein (p.G1034S). The way in which mutant HFM1 causes POI is its ability to damage meiosis in the oocyte. However, the definite role of the HFM1 gene in meiosis remains uncertain. The majority of associated studies have shown that the HFM1 gene is involved in homologous recombination (HR) and synapsis [38,39,40]. Moreover, in 2020, Wang et al. was first to demonstrate that HFM1 gathers at the spindle pole and is responsible for normal spindle formation and function during meiosis in female mice oocytes via maintaining the usual activities of GM130 and p-Mapk proteins [89].
4.1.2. DNA Primase Subunit 1 (PRIM1)
PRIM1 is a protein-coding gene and is located at 12q13.3. It is one of the subunits of DNA primase, which is essential for DNA replication by synthesizing RNA oligonucleotide primers, so as to promote the production of new lagging and leading strands through DNA polymerase [42]. In addition, PRIM1 is also related to DNA repair [90]. As previously reported in a meta-analysis, many loci in the corresponding genes were identified using GWAS that are associated with natural menopause age in European women. In this, PRIM1 (SNP rs2277339) was the second strongest candidate gene [43]. Perry et al. proved that non-synonymous SNP rs2277339 in PRIM1 was responsible for POI [91]. Another meta-analysis also recognized the relationship between PRIM1 (SNP rs2277339) and natural menopause age in African American women [92]. However, the further consequences of mutations in SNP rs2277339 remain unclear. However, in 2016, Wang et al. published a paper concluding that perturbations in the coding region of PRIM1 were not common among Chinese POI patients [93].
4.1.3. Stromal Antigen 3 (STAG3)
STAG3 (7q22.1) is also an important candidate gene in POI. Much of the current literature on gene mutations that cause POI pays particular attention to the effects of STAG3 variants. Over the last 5 years, scientists have conducted WES in consanguineous families [94,95,96] and showed various novel homozygous pathogenic STAG3 variants that cause POI, which included a missense variant (NM_012447.3:c.962G > A), two novel in-frame variants (c.877_885del, p.293_295del; c.891_893dupTGA, p.297_298insAsp), and a donor splice site variant (NM_012447.2: c.1573 + 5G > A). In addition, a series of reports focusing on different ethnicities (Senegalese, white British, and Brazilian POI patients) also revealed many POI-related pathogenic mutations in STAG3 (c.3381_3384delAGAA, p.Glu1128Metfs*42; c.1336G > T, p.Glu446Ter; c.291dupC, p.Asn98Glnfs*2; and c.1950C > A, p.Tyr650*) [45,97,98]. As such an important causative gene, understanding the mechanism of action of STAG3 is crucial. Synapsis mainly occurs during prophase I of meiosis, and it refers to the pairing of homologous chromosomes for DNA exchange of non-sister chromatids, also known as crossover. The synaptonemal complex (SC) acts as a zipper and is responsible for normal synapsis. Cohesin is another essential protein that contributes to establishing SC, ensuring the correct separation of pairing chromatids, DNA repair, and transcriptional regulation [59,99]. Moreover, STAG3 encodes the corresponding protein, which functions as a subunit of the cohesin complex [44]. Therefore, defects in STAG3 will lead to abnormal folliculogenesis and, eventually, POI.
4.1.4. Minichromosome Maintenance 8 (MCM8) and Minichromosome Maintenance 9 (MCM9)
MCM8 and MCM9 are homologous to the MCM 2-7 complex, and all belong to the MCM family. Similar to other members of the MCM family, MCM8 and MCM9 contain the highly conserved helicase domain that can open up DNA strands [100]. MCM8 dimerizes with MCM9, giving rise to a hexameric helicase that is responsible for homologous recombination (HR) initiated by DNA double strand breaks (DSB) and facilitating DNA synthesis in a RAD51-dependent manner [46]. The presence of DSB can lead to a loss of DNA integrity, eventually causing follicular apoptosis and degeneration. Therefore, mutations in MCM8 and MCM9 are associated with infertility. In female mice, loss of MCM8 and MCM9 contributes to sterility [101]. By performing the NGS approach (mainly WES) in POI consanguineous families from various ethnicities (Han Chinese, Arab, Turkish, and Tunisian), new deleterious homozygous mutations in MCM8 or MCM9 were identified (c.351_354delAAAG, p.Lys118Glufs*5; c.1483G > T, p.E495*, and c. 482A > C, p.His161Pro) [48,102,103]. These variants cause chromosomal instability and reduce the DNA repairing capacity. In addition, the members of these consanguineous families with heterozygous variants of MCM8 and MCM9 are healthy. However, a considerable body of work investigating POI causal genes in several cohorts from different ethnic groups also provides evidence for heterozygous mutations in MCM8 and MCM9, such as c.2488G > A, p.A830T; c.482A.G, p.His161Arg; c.548A.G, p.Asn183Ser; c.686T.G, p.Val229Gly, etc. [41,104,105,106,107,108]. However, not all of the aforementioned mutations have been shown to be harmful. The nature of certain variations has not been determined, and some of the variants are benign. Moreover, The T allele and C allele in two MCM8 single-nucleotide polymorphisms (SNPs) (rs16991615 and rs451417) were found to be associated with susceptibility to POI in a cross-sectional study [109]. Moreover, Wang et al. reported the first family presenting with a disease-causing MCM8 mutation (c.724T > C, p.C242R, and c.1334C > A, p.S445*) in adolescence and childhood [47].
4.1.5. DNA Meiotic Recombinase 1 (DMC1)
DMC1 is an autosomal POI candidate gene that is located at 22q13.1. Its protein product works with RAD51 and RPA to repair DSBs during mammalian meiosis. In this repairing process, DMC1 plays an important role in strand exchange [49]. In addition, DMC1 is associated with fertility capacity. Failure of folliculogenesis and spermatogenesis can be observed in DMC1 knockout murine models [110]. Moreover, previous studies emphasized its disease-causing role in POI. He et al. investigated DMC1 mutation in a consanguineous family with POI and non-obstructive azoospermia (NOA) members through WES and found a new missense mutation in DMC1, which was the causal mutation in both POI and NOA (c.106G > A, p.Asp36Asn) [50]. Another study performed NGS in 269 POI patients from Caucasian, sub-Saharan African, North African, and Asian origin to screen mutant genes. It was reported that 7% of POI patients in this cohort harbored DMC1 variants (c.449G > A, p.Gly150Asp; c.598A > G, p.Met200Val) [111]. According to the results of the mutation taster algorithm, c.449G > A is a disease-causing alteration and c.598A > G is a polymorphism. These scientists also used sorting intolerant from the tolerant (SIFT) and PolyPhen-2 algorithms, with all the results indicating that the two DMC variants were damaging, except for the PolyPhen-2 result for c.598A > G (benign). In contrast to these results, many articles report no connection between DMC1 and POI or female mice fertility. Thus, the level of influence DMC1 has in female sterility remains unclear [110,112,113].
4.1.6. Myeloid Cell Leukemia-1 (MCL-1)
MCL-1 exists at chromosome 1 and can encode an antiapoptotic protein, a BCL-2 family member, necessary for regulating the cell cycle. In oocytes, MCL-1 mutations affect the transition from mitosis to meiosis, resulting in reduced original follicles [51]. Previous research demonstrated the association between female fertility and MCL-1. In a study investigating the therapeutic effect of optimized platelet-rich plasma in POI mice, the MCL-1 expression level was lower in the POI group than in the controls, while MCL-1 had a higher level of expression after treatment [114]. In contrast to the protective factor, a deleterious factor for female fertility, known as cadmium (Cd), could cause decreased MCL-1 expression [115]. Cadmium is a heavy metal and is a known toxin with effects on the reproductive system. It has many effects on various cellular processes. In addition, it was demonstrated in other studies that MCL-1 is expressed in primordial and preantral follicles, contributing to the normal development of follicles [116]. Moreover, MCL-1-depleted mice exhibited similar presentations to POI patients [52]. However, the authors in one work of literature failed to reveal the causal connection between the MCL-1 gene and Chinese idiopathic POI patients [51].
4.1.7. MutS Homolog 4 (MSH4) and Muts Homolog 5 (MSH5)
Recently, MSH4 (1p31) and MSH5 (6p21.31) became POI candidate genes, due to their roles in chromosomal synapsis and meiotic recombination through forming a dimer [38]. Both are members of the MutS family, which is associated with DNA mismatch repair. By performing WES in Iranian, Chinese, and Colombian families, two homozygous missense variants from MSH4 and MSH5 (NM_002440.4: c.2261C > T, p.Ser754Leu; ENST00000375755: c.1459G > T, p.D487Y) and a homozygous donor splice-site MSH4 variant (p.Ile743_Lys785del) were identified [53,117,118]. These two mutations in MSH4 are harmful to the ATP binding site, affecting the gene’s normal function. In a MSH5 variant-finding study, the scientists also identified infertile female mice with a mutation of the gene homologous to MSH5 p.D487Y. In addition, three other unconfirmed heterozygous MSH5 variants were also revealed by screening 200 patients with sporadic POI. Moreover, two novel mutations in MSH5 were recently identified in two POI cohorts (c.1264C > T, p.Arg422Cys and c.C1051G, p.R351G) [54,119]. Scientists in these studies explored the effects of these variants via yeast assay and C. elegans, respectively. However, only the c. C1051G; p.R351G variant, tested using C. elegans, exhibited obvious defects.
4.1.8. Meiosis Specific with OB Domain (MEIOB)
MEIOB (16p13.3) is another mandatory gene associated with RAD51 and DMC1 stabilization, meiotic homologous recombination, and DSB repairing. The MEIOB protein binds to single-strand DNA and dimerizes with spermatogenesis-associated 22 (SPATA22) [55]. Both are important for fertility in males and females. Two recent studies identified two homozygous MEIOB mutations that affect female fertility in Arab and Pakistani consanguineous families (c.1218G > A and c.683-1G > A) [120,121]. They also revealed infertile female mice with homozygous deletion of MEIOB. The mutation identified in the Arab family was responsible for the failure of MEIOB–SPATA22 binding and then POI, while the other variant was unrelated to the interaction between these two proteins. As regards the relationship between MEIOB and SPATA22, another study showed that SPATA22 contributed to the localization of MEIOB [122]. In addition, a research group found many new pathogenic homozygous MEIOB variants (c.258_259del, c.1072_1073del and c.814C > T) in three consanguineous Chinese families containing POI and NOA patients [56]. All the variants give rise to truncated proteins, the functions of which are affected.
4.1.9. PSMC3 Interaction Protein (PSMC3IP)
PSMC3IP (17q21.2) is also known as homologous-pairing protein 2 (HOP2), and its protein product forms a complex with Mnd1 to activate DMC1 and Rad51 recombinases using the same protein domains [57]. Therefore, it is also necessary in HR and fertility. In PSMC3IP knockout female mice, smaller ovaries and a depletion of follicles can be observed [40]. In female members with POI and primary amenorrhea from a Yemeni consanguineous family, a deleterious homozygous stop gain mutation of PSMC3IP (c.489 C.G, p.Tyr163Ter) was identified, which caused the partial deletion of the C-terminal portion in the PSMC3IP protein [58]. This deletion led to failed interaction with RAD51 and DMC1, contributing to impaired HR and DNA repair. Aside from the POI patients with primary amenorrhea, two compound heterozygous PSMC3IP mutations (c.206_208delAGA and c.189 G > T) in a secondary amenorrhea POI patient was discovered in 2022 [123]. Another two compound heterozygous mutations (c.597 + 1G > T and c.268G > C p.D90H) in a Chinese POI woman have also been reported [124]. Conversely, no PSMC3IP mutations were observed in a POI cohort from Sweden, which indicates that more studies are required to establish causal PSMC3IP variants in POI cohorts from different populations and containing different ethnicities [125].
4.1.10. Fanconi Anemia Complementation (FANC) Group Genes
Fanconi anemia (FA) is a heterogeneous recessive human genetic disease caused by mutations of one of the genes in the FANC group. These mutations usually lead to impaired meiosis and folliculogenesis [59]. Among genes in the FANC group, many are associated with female fertility. It was reported that FANCI, FANCB, FANCA, and FANCE mutations could affect fertility in female mice [126,127,128,129]. In addition, the FANCL, FANCA, and FANCM variants were identified in non-syndromic POI patients (c.1048_1051delGTCT, p.Gln350Valfs*18; c.739dupA, p.Met247Asnfs*4; c.1772G > A, p.R591Q; c.3887A > G, p.E1296G; and c.5101C > T, p.Gln1701*) [60,130,131,132].
4.1.11. Other Genes
There exist other rare and novel POI candidate meiotic genes (SYCP2L, HSF2BP, and ZSWIM7) [61,62,63,64,133]. These studies expand the range of POI-causing genes.
4.2. Transcription Factor
Although transcription factors cannot directly participate in many vital biological processes, they all play crucial parts in regulating physical activities by controlling various target genes. As regards female fertility, they control the expression time, the site, and the expression level of reproductive genes to ensure the smooth functioning of every reproductive process.
4.2.1. Nuclear Receptor Subfamily 5, Group A, Member 1 (NR5A1) or Steroidogenic Factor-1 (SF-1)
NR5A1 is located at 9q33.3 and encodes an orphan nuclear receptor. It is expressed in the gonads and adrenals, and the NR5A1 protein acts as a transcription factor, regulating the expression of genes involved in steroidogenesis, reproduction, and gonadal and adrenal development in human, such as Eps15 homology domain-containing protein 3 (EHD3), anti-Mullerian hormone (AMH), and Wilms’ tumor 1 (WT1), etc. In addition, a previous research study emphasized the importance of screening NR5A1 variants for POI women with a family member suffering from disorders related to gonadal development [134]. In women, NR5A1 is mainly expressed in the granulosa and theca cells to control ovarian folliculogenesis and steroidogenesis [135], but the POI pathogenic role in NR5A1 variants is controversial. Certain NR5A1 variants (p.Ser54Arg, p.Pro198Leu, p.Pro129Leu, and p.Gly123Ala) found in POI patients are not pathogenic, and the pathogenicity of some mutations, such as c.437G > C, IVS4-20C > T, has not been confirmed via functional tests [136,137]. There also exist other NR5A1 variants that have been shown to be deleterious. For example, a heterozygous missense NR5A1 variant (c.74A > G, p.Y25C), a rare missense NR5A1 variant (c.1063G > A, p.(Val355Met)), and the p.Val15Met NR5A1 variant were demonstrated to be deleterious in POI patients [65,138,139].
4.2.2. Newborn Ovary Homeobox Gene (NOBOX)
NOBOX (7q35) is expressed in oocytes and granulosa cells and is responsible for early folliculogenesis. In one study, which established a NOBOX-deficient female mice model, a reduced number of primordial follicles (due to abnormal germ cell cysts) and adherens junctions between unseparated oocytes was observed, which indicated the important role of NOBOX in female mice fertility [66]. In addition, it also indicated the significance of oocyte–somatic cells signaling for mice POI. KIT-L is one of the target genes of NOBOX and is present in granulosa cells. Kit-L can transmit the signals of granulosa cells to oocytes via the phosphatidylinositol 3-kinase/AKT pathway [140]. Moreover, other ovarian-specific genes, including growth and differentiation factor 9 (GDF9), bone morphogenetic protein 15 (BMP15), and POU class 5 Homeobox1 (POU5F1) are under the control of NOBOX [141,142,143]. Aside from regulating target genes, NOBOX can also interact with FOXL2; however, mutant NOBOX affects this interaction, leading to POI [144]. A previous study conducted in China demonstrated that a homozygous NOBOX mutation (c.567delG, p.T190Hfs*13) failed to arrest G2/M, causing disrupted cell cycles in meiotic oocytes [67]. According to recent articles, NOBOX is a strong candidate gene for causing POI in humans, due to its high prevalence in different POI populations (5.6–6.5%) [140,142,145]. In addition, these researchers confirmed various NOBOX variants (p.Gly91Thr, p.Gly111Arg, p.Arg117Trp, p.Lys371Thr, p.Pro619Leu, p.Gly91Trp, p.R44L, p.G91W, p.G111R, p.G152R, p.K273*, p.R449*, and p.D452N) to be deleterious.
4.2.3. Spermatogenesis-and Oogenesis-Specific Basic Helix-Loop-Helix 1 (SOHLH1) and 2 (SOHLH2)
SOHLH1 (9q34.3) and SOHLH2 (13q13.3) are only expressed in germ cells, primordial follicles, and primary follicles, and their protein products can form a heterodimer, which functions as master–master regulators of other master transcription factors to ensure the normal development of gonadal glands and primordial follicles [68,70]. In a murine model, deficiency of at least one of these two genes caused reduced ovarian sizes and a decreased number of primordial follicles and primary follicles [146]. SOHLH1 and SOHLH2 are POI candidate genes, but few studies have investigated their POI-causative roles in the last 10 years. POI-related SOHLH1 variants (p.Ser317Phe, p.Glu376Lys, and c.*118C > T) were first observed in 2015 [69]. These three deleterious variants were identified in Chinese POI cases, while the variants in Serbian POI cases were synonymous. Two causative variants, p.Ser317Phe and p.Glu376Lys, altered the transactivation capacity of the SOHLH1 gene, resulting in lower expression levels of its target genes, including LIM homeobox 8 (LHX8) and zona pellucida glycoprotein 1 (ZP1) and 3 (ZP3). Additionally, scientists identified five heterozygous nonsynonymous SOHLH2 mutations in a POI cohort of the same ethnicity as the cohort discussed before [70]. They were c.235G > A, p.Glu79Lys; c.314A > G, p.Glu105Gly; c.961A > C, p.Thr321Pro; c.360A > T, p.Leu120Phe and c.610C > T, p.Leu204Phe. Among these variants, only c.235G > A and C.314A > G proved to be deleterious. Moreover, this study firstly demonstrated the association between SOHLH2 and idiopathic POI. Furthermore, two more recent studies performed next-generation sequencing in a POI cohort containing 100 patients and a cohort containing 36 Turkish families with POI members, and both showed deleterious SOHLH1 mutations [72,147].
4.2.4. LIM Homeobox 8 (LHX8)
LHX8 (1p31.1) is expressed in germ cells and is essential for early oogenesis. In mice ovaries without the LHX8 gene, remarkably decreased expression levels of other genes associated with oogenesis are observed, which indicates that LHX8 functions as a transcription factor [71]. This study also demonstrates that LHX8, SOHLH1, and FIGLA can interact with each other, forming a nuclear complex and regulating the expression of various oogenesis-related genes. Aside from mice, LHX8 contributes to oogenesis in rainbow trout [148]. In 2016, Bouily reported a missense mutation of LHX8 (c.974C > T, p.A325V) in a Caucasian POI women cohort and verified its damaging effect on POI [72]. In addition, a recent study also revealed a disease-causing LHX8 variant (c.974C > T p.Ala325Val) in POI patients [111]. However, it was reported that there were no causative LHX8 mutations in 95 US Caucasian women with POI [149]. Therefore, LHX8 may be a relatively rare candidate POI-causing gene in the US Caucasian population, and more research is needed to this end.
4.2.5. Folliculogenesis Specific BHLH Transcription Factor (FIGLA)
FIGLA is located at 2p13.3. It is mainly expressed in female germ cells and plays an important role in oogenesis. In female mice without FIGLA, the genes involved in different processes (meiosis, growth, and differentiation) of oogenesis are downregulated, such as Rad51, SYCP3, NOBOX, LHX8, SOHLH1, and SOHLH2, which indicates an association between FIGLA and female fertility [71]. In addition, zona pellucida glycoprotein genes, which are important in folliculogenesis, also fall under the control of FIGLA [14,28]. Aside from oogenesis disruption, impaired secondary follicles maturation may also be a pathogenesis of POI caused by FIGLA variations. A recent article showed that FIGLA had the ability to support the development of secondary follicles in mature female mice [150]. Moreover, many researchers have attempted to evaluate the impact of FIGLA mutations on POI, with various deleterious FIGLA variants (c.364del p.Glu122Lysis*45 and c.2 T > C, p.Met1Thr) in consanguineous and non-consanguineous families being reported [73,139,151]. All these variants were homozygous with the autosomal recessive inheritance mode.
4.3. Signaling Molecules and Receptors
Signaling molecules, such as hormones, have the ability to transmit information between cells to regulate cellular activities through binding to receptors outside or inside cells. A variety of hormones and receptors are involved in many important processes related to female reproduction and fertility capacity. Their expression levels in human bodies can be considered indicators of female reproductive status and have strong diagnostic significance.
4.3.1. Follicle-Stimulating Hormone Receptor
Follicle-stimulating hormone (FSH) is a glycoprotein essential for the development of antral follicles. It also promotes hormone secretion, such as estradiol, and indicates the ovarian reserve level by binding to FSHR. Therefore, mutations in the FSHR gene may negatively affect FSH-FSHR signaling, arresting folliculogenesis and causing POI. Data from several studies identified detrimental FSHR variants associated with POI. A novel missense FSHR mutation (I423T), a homozygous FSHR variant (c.1253T > G, p.Ile418Ser), and two compound heterozygous missense FSHR variants (c.646 G > A, p.Gly216Arg and c.1313C > T, p.Thr438Ile) were observed in different POI women from various ethnicities [74,75,152]. All these mutations can affect the expression of FSHR, forming different partial FSHRs at granulosa cell membranes with impaired function. However, various studies failed to establish a causative relationship between FSHR mutations and some POI cases [153,154,155]. Woad et al. found no harmful FSHR variants in exons 7 and 10 from a population of POI women from New Zealand. Another two studies revealed both positive and negative results. In 192 Han Chinese participants with POI, the p.L5971 variant was deleterious, while mutation p.M265V was harmless. In addition, according to a meta-analysis, a FSHR polymorphism (rs6166) was regarded as a genetic biomarker exclusively in POI cases from Asia. In summary, FSHR is a rare POI candidate gene in certain ethnic populations, and researchers should focus on screening FSHR mutations in more POI patients from different countries to provide further evidence of the role of FSHR in POI.
4.3.2. Inhibin Family
Inhibin genes can be divided into two types: inhibin alpha (INHA) (2p35) and inhibin beta (INHB), which includes inhibin beta A (INHBA) (7p15-p13) and inhibin beta B (INHBB) (2cen-q13). They encode the corresponding subunits to give rise to two forms of dimeric glycoproteins (inhibin A and inhibin B) belonging to the transforming growth factor-beta (TGF-beta) family. As regards the mechanism of function, inhibins cause downregulated FSH through the inhibin/beta glycan complex, and they compete with activins (a FSH promoting factor) for binding to ACVR2 [33]. Inhibins and activins act opposingly, but regulate the FSH level together, associating with folliculogenesis. However, there is only evidence of a causative link between inhibin genes and POI. In a 136 Korean POI population, the effect of the T–G haplotype and the CT + TT/GG genotype of two INHA polymorphisms (c.-16C > T and c.-124A > G) were related to the susceptibility to POI [156]. In a recent study, both homozygous and heterozygous c.769G > A variants of INHA were shown to be correlated with POI patients from a Kashmiri cohort [76]. This study also showed increased FSH levels in POI patients, which caused accelerated follicle recruitment and premature loss of the follicular pool. This phenomenon is consistent with the characteristics observed in female mice, with an inactivated INHA mutation. An increased FSH expression level, a high ovulation rate, and impaired ovarian function appeared in the affected mice [157]. However, INHA is heterogeneous in different populations. For example, an INHA variant (G769A) is uncommon in Brazilian and Argentina POI patients, while there is a higher prevalence of G796A in Indian and Italian POI women [158]. Moreover, Ma et al. suggested that INHBB mutation (c.1095C > A) may be the cause of POI cases in Chinese Hui women [159]. Further research is warranted to elucidate the causative role of INHBB.
4.3.3. Anti-Mullerian Hormone (AMH) and Anti-Mullerian Hormone Receptor 2 (AMHR2)
The AMH gene is located at chromosome 7, and its protein product can only bind to AMHR2. This ensures the successful control of the AMH signaling pathway, which plays a crucial role in the development of follicles. It has been suggested that AMH can work synergistically with INHA to prevent the formation of FSH-initiated progesterone and estradiol and eventually contribute to mice reproduction [77]. Various recent studies investigated the connection between AMHR2 and POI. In 2014, two novel missense AMHR2 variants (p.I209N and p.L354F) were identified in a Chinese POI cohort [78]. However, the functional assays were not performed to establish their role in AMH signaling. Their effects were confirmed by silico analysis in 2016 [160]. The scientists chose another Chinese Han POI patient population and found a novel missense AMHR2 variant, p.Ala17Glu (A17E). Moreover, they demonstrated that p.I209N, p.L354F, and p.Ala17Glu (A17E) were all detrimental, but only the 1209N mutation harmed the AMH signaling pathway. In addition, it has been shown that other deleterious rare missense AMH mutations are associated with POI [161].
4.3.4. Bone Morphogenetic Protein 15 (BMP15) and Growth Differentiation Factor 9 (GDF9)
BMP15 (Xq11.22) and GDF9 (5q31.1) belong to the TGF-beta superfamily and are expressed exclusively in oocytes. They are essential for many processes associated with female fertility via forming heterodimers or homodimers to induce downstream signaling pathways [162]. BMP15 is involved in promoting follicle development during primordial gonadotropin-independent phases, controlling the sensitivity of GCs to FSH and ovulation, and avoiding depletion of GCs. GDF19 is responsible for the maturation of follicles from the primary to the secondary stage, stimulating GC proliferation, regulating various GC enzymes related to cumulus expansion, etc. [1,80]. To date, many researchers have attempted to investigate the impact of GDF9 mutations on POI. Two homozygous variants from GDF9 (c.783delC and c.604C > T, p.Gln202*) were discovered in one Brazilian POI patient and two Caucasian siblings with POI [81,163]. These two mutations caused truncated GDF9 proteins, which negatively affected the biological function of the GDF9 protein. Furthermore, the first likely POI-causing mutation influencing the regulatory region of GDF9 was identified in 2014 via high-resolution array comparative genomic hybridization (CGH) analysis [164]. As regards BMP15, its heterozygous variants are the second-most frequent causative mutant gene causes of POI. Numerous studies have assessed the pathogenesis of BMP15 heterozygous variants. Similar to GDF9, BMP15 mutations were found in two POI siblings (c.791G > A, p.R264Q, and c.1076C > T, p.P359L) [79]. These variants conformationally affect protein-surrounding water molecules and the thermal stability of BMP15. Scientists also conducted in vitro cell line experiments, which showed impaired BMP15 function. Another two studies also identified deleterious heterozygous BMP15 variants (p.N103K, p.M184T, and c.406G > C (V136L) in POI patients [165,166]. Moreover, homozygous BMP15 mutations are also implicated in POI. Zhang et al. firstly predicted the pathogenic role of its homozygous variants (c.G1070A, p.C357Y) in a Chinses POI girl from a consanguineous family [167]. More recently, another biallelic missense variant (c.1076C >T, p.Pro359Leu) was identified in a case report [168]. Although the prevalence of GDF9 and BMP15 variants in POI patients is high, there still exist idiopathic POI cases without causative mutations in these two genes, indicating the presence of heterogeneity [169].
4.3.5. Other Novel Candidate Genes
Many studies have focused on finding new POI-causing genes, which provide idiopathic POI patients with better diagnoses and treatments and broaden the spectrum of the genetic factors implicated in POI. For example, linkers for the activation of T cells (LAT), vascular endothelial growth factor A (VEGFA), and bone morphogenic protein receptors 1A (BMPR1A) and 1B (BMPR1B) were first linked to POI in the 5 five years [82,83,84].
4.4. RNA Metabolism and Translation
RNAs connect DNAs and proteins. Therefore, impaired RNA activities, including metabolism and translation, have an adverse impact on protein production. These proteins may play important roles in various biological processes, such as folliculogenesis, steroidogenesis, cell proliferation, apoptosis, etc.
4.4.1. Nanos C2HC-Type Zinc Finger 3 (NANOS3)
It is known that the migration, development, and maintenance of primordial germ cells (PGCs) cannot be finished successfully without the involvement of NANOS3 (19p13.13). NANOS3 encodes an RNA-binding protein with anti-apoptotic and translation repressive abilities. In addition, mutations in NANOS3 are associated with POI, based on the function of this gene. According to a recent article, a homozygous mutation of NANOS3 (c.358G > A, p.Glu120Lys) was found in two sisters from a Brazilian POI cohort using a mutational analysis [85]. This mutation is located at the zinc finger domain and affects the interaction of the NANOS3 protein and its target mRNA. Moreover, in vitro experiments were also conducted to evaluate and confirm the accelerated apoptosis of PGCs caused by a p.Glu120Lys mutation. However, no causative mutation of NANOS3 was found in another population of Brazilian women with POI, which indicates the heterogenicity of NANOS3, depending on different populations [170]. Aside from the Brazilian population, a novel pathogenic heterozygous NANOS3 variant (c.457C4T; p.Arg153Trp) was identified in a Chinses POI cohort [171]. The researchers also established heterozygous and homozygous p.Arg153Trp knockout mice models and identified the relationship between the dosage of functional NANOS3 and the normal development of PGCs.
4.4.2. Eukaryotic Translation Initiation Factor 4E Nuclear Import Factor 1 (EIF4ENIF1)
EIF4ENIF1 (22q11.2) encodes a nucleocytoplasmic shuttle protein, which can transport a translation-induced protein called eIF4E to repress translation via interrupting eIF4E–eIF4G binding [86]. The disrupted translation activities caused by EIF4NIF1 mutations lead to increased mRNA expression and stability, which may result in disrupted ovarian follicles and enhanced oocyte apoptosis [87]. It has been demonstrated that EIF4ENIF1 mutations are implicated in POI. The first heterozygous EIF4NIF1 mutation (c. 1286C > G, p.Ser429X) was found in all POI patients from the same family in 2013 [87]. An additional pathogenic heterozygous variant (c.2525A > C, p.Q842P) was identified in both diminished ovarian reserve (DOR) and POI cases through whole-exome and Sanger sequencing [8]. Moreover, the secondary structure of the EIF4ENIF1 protein with a p.Q842P mutation revealed the abnormal structure and length of the alpha-helix, which can influence EIF4ENIF1’s ability to regulate the translation of mRNA. In addition, two more rare EIF4ENIF1 variants (c.9_11delGAG, p.R4del and c.2861G > C p.G954A) from two Han Chinese POI women were reported in 2022 [172]. However, multiple bioinformatic tools showed that only p.G954A was detrimental.
5. Single Gene Mutations in Syndromic and Pleiotropic POI
Unlike non-syndromic POI, POI can also be present in syndromic or pleiotropic Mendelian diseases. Therefore, the gene mutations that cause POI-relevant Mendelian inheritable disorders can be considered to be the genetic factors contributing to POI.
5.1. Fragile X Mental Retardation 1 (FMR1)
The fragile X mental retardation protein (FMRP) encoded by FMR1 (Xq27.3) acts as a type of RNA-binding protein and plays an important role in regulating translation. Any deleterious mutations in FMR1 can lead to abnormally expressed FMRP. However, the expansion of trinucleotide (CGG) repeats at 5′UTR in FMR1 is the most common cause [173]. This expansion can be regarded as a pathogenic variant that contributes to different and unrelated pathologies, depending on the expanded length of CGG repeats (pleiotropy). Normally, there is an AGG triplet after every nine to ten CGG repeats, which is called AGG interruption. This is due to the existence of a sufficient number of AGG triplets for the number of CGG repeats to be maintained within a stable range, i.e., the normal alleles contain 4–55 CGG repeats [174]. The most frequent CGG length varies among different populations [175,176]. Women with 45–54 CGG repeats and 55–200 CGG repeats have gray zone (GM)/intermediate alleles and premutation (PM) alleles, respectively. Both are related to fragile X-associated primary ovarian insufficiency (FXPOI) in women from various countries, such as Turkey, India, Argentina, Brazil, and China [175,176,177,178,179,180]. According to these studies, the prevalence of POI patients with GM and/or PM alleles ranges from 1% to 9.6%. This mutation is rare in Chinese and south Indian POI patients. Nevertheless, the premutation range of the FMR1 gene is the most common aetiology among POI cases caused by single gene mutations [7,14]. Approximately 16% of women with PM alleles suffer from POI [181]. In addition, a higher percentage (34.2%) was reported in a large Turkish cohort [177]. As compared to PM alleles, the GM/intermediate repeat size seems less implicated in POI. One recent meta-analysis failed to establish a connection between the GM CGG repeat length of FMR1 and susceptibility to, or severity of, POI [182]. Moreover, several studies also indicated that there was no correlation between GM alleles and POI [183,184]. In addition, the fully mutated alleles of FMR1 (>200 CGG repeats) initiated via maternal transmission from premutation or an intermediate size are significantly correlated with fragile X syndrome [173].
To date, numerous researchers have investigated how FMR1 variants cause FXPOI, and they have attempted to explain the potential mechanisms involved. Recently, a case report reported an FXPOI woman who became spontaneously pregnant with two healthy babies [185]. In this study, a FMR1 premutation mice model was also established. These experimental mice carried a normal number of primordial follicles, a reduced number of antral follicles and corpora lutea, and an increased number of atretic large antral follicles. Therefore, it was the disrupted follicular function, not the exhausted primordial follicles, that led to FXPOI. Furthermore, another murine model in which premutation FMR1 alleles were introduced was established by scientists in 2012 [186]. It showed the impaired development of immature follicles, except for primordial follicles, which is consistent with the results of the aforementioned article. In addition, impaired luteinizing hormone (LH) and Akt/mTOR-mediated biological pathways were also associated with inducing FXPOI. Finally, abnormal FMR1 alleles were shown to produce a higher risk of reduced functional ovarian reserve (FOR) in 30–38-year-old women because they were associated with skewed X-chromosome inactivation, which contributed to a low level of AMH [187].
5.2. Forkhead Box L2 (FOXL2)
FOXL2 is located at 3q23, and it can be found in the ovary. It encodes the forkhead transcription factor, which is essential for maintaining ovarian somatic cells’ (GCs and theca cells) identities by preventing the transdifferention to their testicular counterpart and regulating the expression of genes involved in estrogen production, folliculogenesis, and steroidogenesis [188,189]. Mice without FOXL2 manifest impaired maturation of follicles and altered gonadotrophic production, thus affecting their fertility [190,191]. In addition, the high expression level of FOXL2 in female mice can also affect their reproductive ability by destroying the differentiation of granulosa and theca cells, influencing steroidogenesis, etc. [192]. FOXL2 mutations are associated with a type of rare and autosomal dominant syndrome, known as blepharophimosis-ptosis-epicanthus-inversus syndrome (BPES). Two types of BRES are classified according to the consequences of different mutations. POI can only be identified in BPES type I [188]. Therefore, FOXL2 is responsible for syndromic POI, and it represents the first autosomal gene related to syndromic POI [191]. A great deal of previous research into FOXL2-implicated syndromic POI is focused on Chinese populations. In Chinese families, scientists discovered various disease-causing FOXL2 variants (c.307C > T p.Arg103Cys, c.462_468del, and c.988_989insG) from BPES type I members with POI [193,194]. Moreover, researches showed two new FOXL2 mutations in Chinese families with BPES type II, but failed to link this to POI [195]. However, a case report conducted in 2019 revealed one of the two aforementioned FOXL2 mutations, showing that the FOXL2 variant (c.223C > T p.Leu75Phe) was associated with women with typical POI hormonal alteration from a BPES type I Polish family [196]. Aside from syndromic POI, FOXL2 mutations are also responsible for non-syndromic POI [72,197].
5.3. Galactose 1-Phosphate Uridyl Transferase (GALT)
The GALT gene exists at 9q13 and encodes one of the enzymes necessary in the main galactose metabolism pathway (the Leloir pathway). The other two enzymes are galactokinase (GALK) and UDP galactose 4-epimerase (GALE). The functional-affecting disorders of any of these three enzymes can lead to impaired galactose metabolism and eventually galactosemia. Recently, pathogenic mutations in the galactose mutarotase (GALM) gene were identified in patients with unexplained galactosemia, giving rise to a new type of galactosemic [198]. Classical galactosemia (GC) and Duarte galactosemia (DG) are caused by mutations in GALT. As a symptom of hypergonadotropic hypogonadism, POI is one of the long-term complications of GC, while it has not been observed in DG female patients [199,200,201]. Moreover, the prevalence of POI in GC patients is high. Over 90% of GC patients exhibited signs of POI [202]. Thus far, the exact mechanism of POI in galactosemia has not been discovered. However, studies over the past 10 years have highlighted many potential mechanisms, including (1) ovarian damage caused by the toxic effects from accumulated galactoses and/or their metabolisms; (2) alteration of the function of FSH resulting from impaired glycosylation; (3) defects in the development of follicles, germ cells functions, and steroidogenesis, due to low levels of UDP-glucose (UDP-Glc) pyrophosphorylase and UDP galactose (UDP-Gal); (4) aberration of cell signaling pathways, such as the PI3K/AKT/mTOR signaling pathway; and (5) epigenetic mechanisms [203,204]. As regards specific GALT variants in galactosemia patients with POI, p.Q188R and p.K285N are the most common mutations, accounting for 70% of cases [7]. Although a low pregnancy rate (5–10%) is generally reported among POI patients, women with POI and galactosemia can still become pregnant spontaneously [205,206].
5.4. Autoimmune Regulator (AIRE)
Polyendocrinopathy candidiasis ectodermal dystrophy (APECED)/autoimmune polyendocrinopathy syndrome type I (APS1) is a rare monogenic autoimmune disease. It is characterized by two out of three of the following clinical manifestations: chronic mucocutaneous candidiasis (CMC), hypoparathyroidism, and primary adrenal insufficiency (Addison’s disease). In addition, deleterious mutations in the autoimmune regulator gene (AIRE) can confirm the diagnosis of APECED [207]. Aside from these three major clinical manifestations, other features, such as POI, are also observed in APECED patients. The literature review revealed that, in a large cohort of APECED patients from various countries, the prevalence of gonadal failure ranged from 0 to 70% [208]. It was also reported that the female-to-male ratio of hypergonadotropic hypogonadism was 7:1 in North America. Moreover, there is a considerable body of literature on finding AIRE variants in APECED patients. Various homozygous and heterozygous pathogenic AIRE variants (c.967_979delCTGTCCCCTCCGC, p.(L323SfsX51); c.995 + (3_5)delGAGinsTAT, NM_000383.2: c.623G > T, NP_000374.1: p.Gly208Val; c.967_979del13bp; c.396G > C (p.Arg132Ser; p.R132S) and (c.47C > T, p.Thr16Met)) were observed in APS1 women with POI as a symptom [209,210,211,212,213]. As regards the toxic effects of AIRE mutations on the ovaries, loss-of-function AIRE can cause ovarian autoimmune disease by repressing the antigen expression levels specific to the ovaries. In female mice without AIRE, follicular depletion and an exhausted ovarian reserve were observed, indicating a potential mechanism in female infertility [214].
5.5. Other Syndromic Disorders
Aside from the pleiotropic and syndromic disorders that were discussed in detail previously, there are also many studies from the last 10 years that revealed gene mutations that cause various syndromes (ataxia telangiectasia, Nijmegen breakage syndrome, Alagille syndrome, Mulibrey nanism disorder, and congenital disorders of glycosylation) that are characterized with POI [108,215,216,217,218].
6. Mitochondrial Dysfunction in POI
The mitochondrion is a common organelle in most eukaryotic cells. It plays an essential role in producing energy via oxidative phosphorylation. Therefore, abnormalities in mitochondrial functions are associated with a wide range of human diseases, including POI. In fact, any perturbations in mitochondria can severely affect the ovaries because the ovaries contain the maximum number of mitochondria. As regards the genetic factors related to mitochondrial dysfunction, mutations in nuclear and mitochondrial genes are responsible for POI. According to recent studies, required for meiotic nuclear division 1 homolog (RMND1), mitochondrial cytochrome c oxidase 1 (MT-CO1), nuclear-encoded gene mitochondrial ribosomal protein S22, (MRPS22), caseinolytic peptidase B (CLPB), and mitochondrial transcription factor A (TFAM), variants were reported in POI, and their pathogenic roles were confirmed [219,220,221,222,223] (Table 2). In addition, mutations in the genes that regulate mitochondrial activities or functions can also lead to POI. In 2021, Feng et al. demonstrated that a leucyl-tRNA synthetase 2 (LARS2) variation increased the amount of mitochondrial reactive oxygen species (ROS), decreased the number of mitochondrial DNA (mtDNA) copies and ATP, and reduced the expression level of a mitochondrial fusion-related gene known as mitofusin-2 (Mfn-2), which is one of the pathogenic mechanisms of POI (impaired mitochondrial function in granulosa cells) [224]. Furthermore, by establishing a POI mice model, scientists observed conformational changes (swollen mitochondria, decreased matrix density, and marginally shifted cristae) in mitochondria from granulosa cells of POI mice [225]. In addition, a low level of mitochondrial oxidative phosphorylation complexes (OXPHOS) also existed in the POI group. This resulted from a Sirtuin 1 (SIRT1) mutation. Additionally, a new variation in alanyl-tRNA synthetase 2 (AARS2) was identified in a Chinese consanguineous family, resulting in impaired translation activity in mitochondria implicated in POI [226].
Table 2.
List of genes associated with POI (initiated via mitochondrial dysfunction) and their MOA.
7. Non-Coding RNA in POI
Establishing the role of non-coding RNAs (ncRNAs) in POI is an emerging research area. It will help us in understanding the genetic effects of POI and provide important information on POI pathogenesis. NcRNAs can be divided into small ncRNAs (sncRNAs) and long ncRNAs (lncRNA), and they contribute to regulating various biological processes, such as cell proliferation and apoptosis, rather than giving rise to proteins [227]. They usually have an influence on ovarian function by interacting with their corresponding target genes. MicroRNAs (miRNAs), as one essential sncRNA type, are closely related to POI. Increasing levels of both microRNA-379-5p and microRNA-127-5p were observed in the GCs of biological POI (bPOI) patients. They had an adverse impact on GCs proliferation and the ability to repair DNA damage through targeting poly ADP-ribose polymerase 1 (PARP1) (microRNA-379-5p), X-ray repair cross complementing 6 (XRCC6) (microRNA-379-5p), and high mobility group box 2 (HMGB2) (microRNA-127-5p) [228,229]. In addition, the suppression of the B-cell lymphoma 9 (BCL9) level initiated by upregulated microRNA-122-5p was shown to promote GCs apoptosis in POI mice [230]. Distinct from the miRNA mentioned previously, microRNA-22-3p acts as a protective factor for POI, which was demonstrated in a receiver operating characteristic (ROC) curve, logistic binary regression, and bioinformatics analysis [231]. Aside from miRNAs, lncRNAs are also critical in POI development. According to a recent study, accelerated GCs apoptosis was observed due to downregulated lncRNA HCP5 in a Chinese bPOI cohort, which led to a lower nuclear level of YB1 and eventually induced the obstructed transcription of a critical POI candidate gene, known as MSH5 [232]. Moreover, the lower expression of another lncRNA, known as PVT1, was also shown to disrupt ovarian function in POI patients. These deleterious effects were demonstrated in a POI mice model [233].
8. Discussion and Conclusions
Many strategies are used to establish the genetic factors related to POI, and these factors will guide future POI prediction, diagnosis, and treatment protocols. Chromosome analysis (karyotyping) is the main method for discovering abnormalities in chromosomes. Approximately 10–13% of POI cases are associated with chromosomal abnormalities, such as mosaicism 45, X/46, XX. Therefore, chromosome analysis is not only helpful in identifying POI-related chromosomal variations, but also plays an important role in the clinical evaluation and diagnosis of POI. Moreover, for small duplications and deletions that cannot be detected under a microscope, array comparative genomic hybridization (CGH) is the better choice. In 2022, CGH was performed in a Tunisian family [234]. The study suggested that EIF1AX duplication might lead to POI after a familial tandem duplication in Xp22.12 was identified. In addition to chromosome variation, GWAS, WES, and NGS are used to more effectively and efficiently detect single gene mutations than classical methods. In addition, the strong POI component and the similar genetic background among family members make family analyses important in investigating the genetic causes of POI. As a result of GWAS, many loci that are potentially responsible for POI have been disclosed in Chinese, Korean, and Dutch women [14]. Moreover, via NGS, various POI-related genes involved in meiosis and DNA repair have been identified, enriching the genetic etiology of POI [235]. The contributions of GWAS and NGS are indisputable, and they will certainly be applied more widely in the future. As regards WES, it has revealed many POI-related genes involved in DNA damage repair and HR, and its application will also be of great importance in the future. Recently, several in vitro cellular models have been successfully used to demonstrate that certain rare genomic variants can cause mutations and dysfunction of the corresponding proteins, thus confirming the association between these variants and POI [236,237]. Once new deleterious variations are found, they can be used to predict the age of menopause [236]. Thus, this review may be useful in large genetic screening research for POI and in regulating fertility in women. Taken together, the genetic factors of POI confirmed in different tests can be used as the basis for POI diagnosis and risk prediction protocols. Reproductive and genetic counseling is essential for women with POI or those at risk of POI. It can help women select when to attempt pregnancy and increase the possibility of pregnancy through other methods, such as assisted reproductive technologies, oocyte or embryo cryopreservation, etc. [11].
In order to fully understand the genetic causes of POI, many challenges remain. Future research will be designed to this end. First, the mechanisms involved in POI that are caused by certain genetic factors are unclear. For example, although TS is ubiquitous in POI patients, the exact pathogenesis of TS-causing POI remains unknown. Second, less attention has been paid by scientists in recent years to certain areas, such as TXS and structural chromosomal abnormalities. Third, many genes, such as MCL-1, have been shown to be associated with POI, but their causative role has not been confirmed. Even for genes whose mutations have been demonstrated to cause POI, many of their variants have not been connected with POI. Fourth, because of the heterogeneous nature of POI and the multiple modes of mutational spread, the prevalence and type of gene mutations are distinct among POI women of different ethnicities and different POI populations. Therefore, in the future, population stratification will be vital when analyzing genetic alterations. Fifth, the experimental results obtained from mouse models are not all applicable to humans because there are genetic and physiological differences between mice and humans. Moreover, the mouse models cannot deal with the extremely complex interactions among molecules, cells, organs, organisms, and the environment. Finally, according to many recent studies, digenic and oligogenic effects on POI have been observed, demonstrating that POI may not be a completely monogenic disease [40,72]. These are the questions that need to be addressed in the future.
In conclusion, the present review summarizes the genetic effects of POI in different fields (chromosomal variations, single gene mutations, impaired mitochondrial functions, and abnormal levels of non-coding RNAs). The findings clearly indicate that examining various genetic factors is crucial in determining the underlying etiologies of idiopathic POI cases. Therefore, this work summarizes and enriches our knowledge of POI etiology by providing the latest information concerning the selected genetic causes of POI obtained from patients and experimental animal models. However, this article is limited, as it only focuses on genetic causes.
Author Contributions
Conceptualization and supervision, C.Z., M.C. and H.J.; writing—original draft preparation, M.C.; writing—review and editing, M.C. and H.J. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China, grant number Nos. 81960272 and 32160176.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yatsenko, S.A.; Rajkovic, A. Genetics of human female infertility. Biol. Reprod. 2019, 101, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Webber, L.; Davies, M.; Anderson, R.; Bartlett, J.; Braat, D.; Cartwright, B.; Cifkova, R.; de Muinck Keizer-Schrama, S.; Hogervorst, E.; Janse, F.; et al. ESHRE Guideline: Management of women with premature ovarian insufficiency. Hum. Reprod. 2016, 31, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Chon, S.J.; Umair, Z.; Yoon, M.S. Premature Ovarian Insufficiency: Past, Present, and Future. Front. Cell Dev. Biol. 2021, 9, 672890. [Google Scholar] [CrossRef]
- Ishizuka, B. Current Understanding of the Etiology, Symptomatology, and Treatment Options in Premature Ovarian Insufficiency (POI). Front. Endocrinol. 2021, 12, 626924. [Google Scholar] [CrossRef]
- Laven, J.S. Primary Ovarian Insufficiency. Semin. Reprod. Med. 2016, 34, 230–234. [Google Scholar] [CrossRef]
- Takahashi, A.; Yousif, A.; Hong, L.; Chefetz, I. Premature ovarian insufficiency: Pathogenesis and therapeutic potential of mesenchymal stem cell. J. Mol. Med. 2021, 99, 637–650. [Google Scholar] [CrossRef]
- Rossetti, R.; Ferrari, I.; Bonomi, M.; Persani, L. Genetics of primary ovarian insufficiency. Clin. Genet. 2017, 91, 183–198. [Google Scholar] [CrossRef]
- Zhao, M.; Feng, F.; Chu, C.; Yue, W.; Li, L. A novel EIF4ENIF1 mutation associated with a diminished ovarian reserve and premature ovarian insufficiency identified by whole-exome sequencing. J. Ovarian Res. 2019, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, M.; Moore, B.; Mosbruger, T.; Neklason, D.W.; Yandell, M.; Jorde, L.B.; Welt, C.K. POLR2C Mutations Are Associated with Primary Ovarian Insufficiency in Women. J. Endocr. Soc. 2017, 1, 162–173. [Google Scholar] [CrossRef]
- Gruber, N.; Kugler, S.; de Vries, L.; Brener, A.; Zung, A.; Eyal, O.; Rachmiel, M.; Koren, I.; Tenenbaum-Rakover, Y.; Hershkovitz, E.; et al. Primary Ovarian Insufficiency Nationwide Incidence Rate and Etiology Among Israeli Adolescents. J. Adolesc. Health 2020, 66, 603–609. [Google Scholar] [CrossRef]
- Barros, F.; Carvalho, F.; Barros, A.; Dória, S. Premature ovarian insufficiency: Clinical orientations for genetic testing and genetic counseling. Porto Biomed. J. 2020, 5, e62. [Google Scholar] [CrossRef] [PubMed]
- Viotti, M. Preimplantation Genetic Testing for Chromosomal Abnormalities: Aneuploidy, Mosaicism, and Structural Rearrangements. Genes 2020, 11, 602. [Google Scholar] [CrossRef]
- Oneda, B.; Rauch, A. Microarrays in prenatal diagnosis. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 42, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiao, X.; Simpson, J.L.; Chen, Z.J. Genetics of primary ovarian insufficiency: New developments and opportunities. Hum. Reprod. Update 2015, 21, 787–808. [Google Scholar] [CrossRef] [PubMed]
- Cordts, E.B.; Christofolini, D.M.; Dos Santos, A.A.; Bianco, B.; Barbosa, C.P. Genetic aspects of premature ovarian failure: A literature review. Arch. Gynecol. Obstet. 2011, 283, 635–643. [Google Scholar] [CrossRef]
- Kalantari, H.; Madani, T.; Zari Moradi, S.; Mansouri, Z.; Almadani, N.; Gourabi, H.; Mohseni Meybodi, A. Cytogenetic analysis of 179 Iranian women with premature ovarian failure. Gynecol. Endocrinol. 2013, 29, 588–591. [Google Scholar] [CrossRef]
- Oktay, K.; Bedoschi, G.; Berkowitz, K.; Bronson, R.; Kashani, B.; McGovern, P.; Pal, L.; Quinn, G.; Rubin, K. Fertility Preservation in Women with Turner Syndrome: A Comprehensive Review and Practical Guidelines. J. Pediatr. Adolesc. Gynecol. 2016, 29, 409–416. [Google Scholar] [CrossRef]
- Hook, E.B.; Warburton, D. Turner syndrome revisited: Review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014, 133, 417–424. [Google Scholar] [CrossRef]
- Mastellari, E.; Marca, L.A.A. Genetic conditions impairing female fertility. Panminerva Med. 2020, 62, 260–267. [Google Scholar] [CrossRef]
- Jackson-Cook, C. A hypothesis: Could telomere length and/or epigenetic alterations contribute to infertility in females with Turner syndrome? Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 108–116. [Google Scholar] [CrossRef]
- Castronovo, C.; Rossetti, R.; Rusconi, D.; Recalcati, M.P.; Cacciatore, C.; Beccaria, E.; Calcaterra, V.; Invernizzi, P.; Larizza, D.; Finelli, P.; et al. Gene dosage as a relevant mechanism contributing to the determination of ovarian function in Turner syndrome. Hum. Reprod. 2014, 29, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Di-Battista, A.; Moysés-Oliveira, M.; Melaragno, M.I. Genetics of premature ovarian insufficiency and the association with X-autosome translocations. Reproduction 2020, 160, R55–R64. [Google Scholar] [CrossRef]
- La Marca, A.; Mastellari, E. Fertility preservation for genetic diseases leading to premature ovarian insufficiency (POI). J. Assist. Reprod. Genet. 2021, 38, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Franić-Ivanišević, M.; Franić, D.; Ivović, M.; Tančić-Gajić, M.; Marina, L.; Barac, M.; Vujović, S. Genetic Etiology of Primary Premature Ovarian Insufficiency. Acta Clin. Croat. 2016, 55, 629–635. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Davis, S.M.; Soares, K.; Howell, S.; Cree-Green, M.; Buyers, E.; Johnson, J.; Tartaglia, N.R. Diminished Ovarian Reserve in Girls and Adolescents with Trisomy X Syndrome. Reprod. Sci. 2020, 27, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Rafique, M.; AlObaid, S.; Al-Jaroudi, D. 47, XXX syndrome with infertility, premature ovarian insufficiency, and streak ovaries. Clin. Case Rep. 2019, 7, 1238–1241. [Google Scholar] [CrossRef]
- Schlade-Bartusiak, K.; Brown, L.; Lomax, B.; Bruyère, H.; Gillan, T.; Hamilton, S.; McGillivray, B.; Eydoux, P. BPES with atypical premature ovarian insufficiency, and evidence of mitotic recombination, in a woman with trisomy X and a translocation t(3;11)(q22.3;q14.1). Am. J. Med. Genet. A 2012, 158a, 2322–2327. [Google Scholar] [CrossRef]
- Bilgin, E.M.; Kovanci, E. Genetics of premature ovarian failure. Curr. Opin. Obstet. Gynecol. 2015, 27, 167–174. [Google Scholar] [CrossRef]
- Lakhal, B.; Braham, R.; Berguigua, R.; Bouali, N.; Zaouali, M.; Chaieb, M.; Veitia, R.A.; Saad, A.; Elghezal, H. Cytogenetic analyses of premature ovarian failure using karyotyping and interphase fluorescence in situ hybridization (FISH) in a group of 1000 patients. Clin. Genet. 2010, 78, 181–185. [Google Scholar] [CrossRef]
- Ceylaner, G.; Altinkaya, S.O.; Mollamahmutoglu, L.; Ceylaner, S. Genetic abnormalities in Turkish women with premature ovarian failure. Int. J. Gynaecol. Obstet. 2010, 110, 122–124. [Google Scholar] [CrossRef]
- Geckinli, B.B.; Toksoy, G.; Sayar, C.; Soylemez, M.A.; Yesil, G.; Aydın, H.; Karaman, A.; Devranoglu, B. Prevalence of X-aneuploidies, X-structural abnormalities and 46,XY sex reversal in Turkish women with primary amenorrhea or premature ovarian insufficiency. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 182, 211–215. [Google Scholar] [PubMed]
- Jiao, X.; Qin, C.; Li, J.; Qin, Y.; Gao, X.; Zhang, B.; Zhen, X.; Feng, Y.; Simpson, J.L.; Chen, Z.J. Cytogenetic analysis of 531 Chinese women with premature ovarian failure. Hum. Reprod. 2012, 27, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Vichinsartvichai, P. Primary ovarian insufficiency associated with autosomal abnormalities: From chromosome to genome-wide and beyond. Menopause 2016, 23, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; He, M.; Fang, Y.; Zhu, J.; Liang, L.; Wang, C. Autosomal chromosome microdeletions in three adolescent girls with premature ovarian insufficiency: A case report. Turk. J. Pediatr. 2022, 64, 729–735. [Google Scholar] [CrossRef]
- Baronchelli, S.; Conconi, D.; Panzeri, E.; Bentivegna, A.; Redaelli, S.; Lissoni, S.; Saccheri, F.; Villa, N.; Crosti, F.; Sala, E.; et al. Cytogenetics of premature ovarian failure: An investigation on 269 affected women. J. Biomed. Biotechnol. 2011, 2011, 370195. [Google Scholar] [CrossRef]
- Vichinsartvichai, P.; Manolertthewan, C.; Promrungrueng, P. Premature ovarian failure with 46,XX,t(1;4)(p34.1;q34): First case report and literature review. Climacteric 2015, 18, 656–658. [Google Scholar] [CrossRef]
- Jiao, X.; Ke, H.; Qin, Y.; Chen, Z.J. Molecular Genetics of Premature Ovarian Insufficiency. Trends Endocrinol. Metab. 2018, 29, 795–807. [Google Scholar] [CrossRef]
- Biswas, L.; Tyc, K.; El Yakoubi, W.; Morgan, K.; Xing, J.; Schindler, K. Meiosis interrupted: The genetics of female infertility via meiotic failure. Reproduction 2021, 161, R13–R35. [Google Scholar] [CrossRef]
- Tanaka, K.; Miyamoto, N.; Shouguchi-Miyata, J.; Ikeda, J.E. HFM1, the human homologue of yeast Mer3, encodes a putative DNA helicase expressed specifically in germ-line cells. DNA Seq. 2006, 17, 242–246. [Google Scholar] [CrossRef]
- Veitia, R.A. Primary ovarian insufficiency, meiosis and DNA repair. Biomed. J. 2020, 43, 115–123. [Google Scholar] [CrossRef]
- Liu, H.; Wei, X.; Sha, Y.; Liu, W.; Gao, H.; Lin, J.; Li, Y.; Tang, Y.; Wang, Y.; Wang, Y.; et al. Whole-exome sequencing in patients with premature ovarian insufficiency: Early detection and early intervention. J. Ovarian Res. 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, M.L.; De Piccoli, G.; Perera, R.L.; Labib, K.; Pellegrini, L. A conserved motif in the C-terminal tail of DNA polymerase α tethers primase to the eukaryotic replisome. J. Biol. Chem. 2012, 287, 23740–23747. [Google Scholar] [CrossRef] [PubMed]
- Stolk, L.; Perry, J.R.; Chasman, D.I.; He, C.; Mangino, M.; Sulem, P.; Barbalic, M.; Broer, L.; Byrne, E.M.; Ernst, F.; et al. Meta-analyses identify 13 loci associated with age at menopause and highlight DNA repair and immune pathways. Nat. Genet. 2012, 44, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Le Quesne Stabej, P.; Williams, H.J.; James, C.; Tekman, M.; Stanescu, H.C.; Kleta, R.; Ocaka, L.; Lescai, F.; Storr, H.L.; Bitner-Glindzicz, M.; et al. STAG3 truncating variant as the cause of primary ovarian insufficiency. Eur. J. Hum. Genet. 2016, 24, 135–138. [Google Scholar] [CrossRef]
- França, M.M.; Nishi, M.Y.; Funari, M.F.A.; Lerario, A.M.; Baracat, E.C.; Hayashida, S.A.Y.; Maciel, G.A.R.; Jorge, A.A.L.; Mendonca, B.B. Two rare loss-of-function variants in the STAG3 gene leading to primary ovarian insufficiency. Eur. J. Med. Genet. 2019, 62, 186–189. [Google Scholar] [CrossRef]
- Hustedt, N.; Saito, Y.; Zimmermann, M.; Álvarez-Quilón, A.; Setiaputra, D.; Adam, S.; McEwan, A.; Yuan, J.Y.; Olivieri, M.; Zhao, Y.; et al. Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes Dev. 2019, 33, 1397–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, S.; Li, P. Two novel mutations in the MCM8 gene shared by two Chinese siblings with primary ovarian insufficiency and short stature. Mol. Genet. Genom. Med. 2020, 8, e1396. [Google Scholar] [CrossRef]
- Fauchereau, F.; Shalev, S.; Chervinsky, E.; Beck-Fruchter, R.; Legois, B.; Fellous, M.; Caburet, S.; Veitia, R.A. A non-sense MCM9 mutation in a familial case of primary ovarian insufficiency. Clin. Genet. 2016, 89, 603–607. [Google Scholar] [CrossRef]
- Hinch, A.G.; Becker, P.W.; Li, T.; Moralli, D.; Zhang, G.; Bycroft, C.; Green, C.; Keeney, S.; Shi, Q.; Davies, B.; et al. The Configuration of RPA, RAD51, and DMC1 Binding in Meiosis Reveals the Nature of Critical Recombination Intermediates. Mol. Cell 2020, 79, 689–701.e10. [Google Scholar] [CrossRef]
- He, W.B.; Tu, C.F.; Liu, Q.; Meng, L.L.; Yuan, S.M.; Luo, A.X.; He, F.S.; Shen, J.; Li, W.; Du, J.; et al. DMC1 mutation that causes human non-obstructive azoospermia and premature ovarian insufficiency identified by whole-exome sequencing. J. Med. Genet. 2018, 55, 198–204. [Google Scholar] [CrossRef]
- Cao, J.; He, Y.; Cai, W.; Zhou, W.; Cong, J.; Tan, R.; Ge, H.; Pu, D.; Wu, J. Analysis of the MCL-1 gene in Chinese women with idiopathic premature ovarian insufficiency. Climacteric 2021, 24, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Omari, S.; Waters, M.; Naranian, T.; Kim, K.; Perumalsamy, A.L.; Chi, M.; Greenblatt, E.; Moley, K.H.; Opferman, J.T.; Jurisicova, A. Mcl-1 is a key regulator of the ovarian reserve. Cell Death Dis. 2015, 6, e1755. [Google Scholar] [CrossRef]
- Akbari, A.; Padidar, K.; Salehi, N.; Mashayekhi, M.; Almadani, N.; Sadighi Gilani, M.A.; Bashambou, A.; McElreavey, K.; Totonchi, M. Rare missense variant in MSH4 associated with primary gonadal failure in both 46, XX and 46, XY individuals. Hum. Reprod. 2021, 36, 1134–1145. [Google Scholar] [CrossRef]
- Macaisne, N.; Touzon, M.S.; Rajkovic, A.; Yanowitz, J.L. Modeling primary ovarian insufficiency-associated loci in C. elegans identifies novel pathogenic allele of MSH5. J. Assist. Reprod. Genet. 2022, 39, 1255–1260. [Google Scholar] [CrossRef]
- Souquet, B.; Abby, E.; Hervé, R.; Finsterbusch, F.; Tourpin, S.; Le Bouffant, R.; Duquenne, C.; Messiaen, S.; Martini, E.; Bernardino-Sgherri, J.; et al. MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 2013, 9, e1003784. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.; Tan, C.; Meng, G.; Meng, L.; Nie, H.; Du, J.; Lu, G.X.; Lin, G.; He, W.B.; et al. Novel MEIOB variants cause primary ovarian insufficiency and non-obstructive azoospermia. Front. Genet. 2022, 13, 936264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Sung, P. Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res. 2015, 43, 4055–4066. [Google Scholar] [CrossRef] [PubMed]
- Al-Agha, A.E.; Ahmed, I.A.; Nuebel, E.; Moriwaki, M.; Moore, B.; Peacock, K.A.; Mosbruger, T.; Neklason, D.W.; Jorde, L.B.; Yandell, M.; et al. Primary Ovarian Insufficiency and Azoospermia in Carriers of a Homozygous PSMC3IP Stop Gain Mutation. J. Clin. Endocrinol. Metab. 2018, 103, 555–563. [Google Scholar] [CrossRef]
- Yang, Q.; Mumusoglu, S.; Qin, Y.; Sun, Y.; Hsueh, A.J. A kaleidoscopic view of ovarian genes associated with premature ovarian insufficiency and senescence. FASEB J. 2021, 35, e21753. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, T.; Liu, R.; Ke, H.; Xu, W.; Zhao, S.; Qin, Y. FANCL gene mutations in premature ovarian insufficiency. Hum. Mutat. 2020, 41, 1033–1041. [Google Scholar] [CrossRef]
- He, W.B.; Tan, C.; Zhang, Y.X.; Meng, L.L.; Gong, F.; Lu, G.X.; Lin, G.; Du, J.; Tan, Y.Q. Homozygous variants in SYCP2L cause premature ovarian insufficiency. J. Med. Genet. 2021, 58, 168–172. [Google Scholar] [CrossRef]
- Felipe-Medina, N.; Caburet, S.; Sánchez-Sáez, F.; Condezo, Y.B.; de Rooij, D.G.; Gómez, H.L.; Garcia-Valiente, R.; Todeschini, A.L.; Duque, P.; Sánchez-Martin, M.A.; et al. A missense in HSF2BP causing primary ovarian insufficiency affects meiotic recombination by its novel interactor C19ORF57/BRME1. eLife 2020, 9, e56996. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, W.; Xu, B.; Gao, S.; Zhang, Q.; Qin, Y.; Guo, T. Pathogenic Variations of Homologous Recombination Gene HSF2BP Identified in Sporadic Patients with Premature Ovarian Insufficiency. Front. Cell Dev. Biol. 2021, 9, 768123. [Google Scholar] [CrossRef] [PubMed]
- McGlacken-Byrne, S.M.; Le Quesne Stabej, P.; Del Valle, I.; Ocaka, L.; Gagunashvili, A.; Crespo, B.; Moreno, N.; James, C.; Bacchelli, C.; Dattani, M.T.; et al. ZSWIM7 Is Associated With Human Female Meiosis and Familial Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2022, 107, e254–e263. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Todeschini, A.L.; Caburet, S.; Perets, L.P.; Mila, M.; Younis, J.S.; Shalev, S.; Veitia, R.A. An exome-wide exploration of cases of primary ovarian insufficiency uncovers novel sequence variants and candidate genes. Clin. Genet. 2020, 98, 293–298. [Google Scholar] [CrossRef]
- Lechowska, A.; Bilinski, S.; Choi, Y.; Shin, Y.; Kloc, M.; Rajkovic, A. Premature ovarian failure in nobox-deficient mice is caused by defects in somatic cell invasion and germ cell cyst breakdown. J. Assist. Reprod. Genet. 2011, 28, 583–589. [Google Scholar] [CrossRef]
- Li, L.; Wang, B.; Zhang, W.; Chen, B.; Luo, M.; Wang, J.; Wang, X.; Cao, Y.; Kee, K. A homozygous NOBOX truncating variant causes defective transcriptional activation and leads to primary ovarian insufficiency. Hum. Reprod. 2017, 32, 248–255. [Google Scholar]
- Lim, E.J.; Choi, Y. Transcription factors in the maintenance and survival of primordial follicles. Clin. Exp. Reprod. Med. 2012, 39, 127–131. [Google Scholar] [CrossRef]
- Zhao, S.; Li, G.; Dalgleish, R.; Vujovic, S.; Jiao, X.; Li, J.; Simpson, J.L.; Qin, Y.; Ivanisevic, M.; Ivovic, M.; et al. Transcription factor SOHLH1 potentially associated with primary ovarian insufficiency. Fertil. Steril. 2015, 103, 548–553.e5. [Google Scholar] [CrossRef]
- Qin, Y.; Jiao, X.; Dalgleish, R.; Vujovic, S.; Li, J.; Simpson, J.L.; Al-Azzawi, F.; Chen, Z.J. Novel variants in the SOHLH2 gene are implicated in human premature ovarian failure. Fertil. Steril. 2014, 101, 1104–1109.e6. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, C.Y.; Zhao, Y.; Dean, J. FIGLA, LHX8 and SOHLH1 transcription factor networks regulate mouse oocyte growth and differentiation. Nucleic Acids Res. 2020, 48, 3525–3541. [Google Scholar] [CrossRef]
- Bouilly, J.; Beau, I.; Barraud, S.; Bernard, V.; Azibi, K.; Fagart, J.; Fèvre, A.; Todeschini, A.L.; Veitia, R.A.; Beldjord, C.; et al. Identification of Multiple Gene Mutations Accounts for a new Genetic Architecture of Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2016, 101, 4541–4550. [Google Scholar] [CrossRef]
- Yuan, P.; He, Z.; Sun, S.; Li, Y.; Wang, W.; Liang, X.; Xie, X.; Jiang, Y.; Yang, D. Bi-allelic recessive loss-of-function mutations in FIGLA cause premature ovarian insufficiency with short stature. Clin. Genet. 2019, 95, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Zariñán, T.; Mayorga, J.; Jardón-Valadez, E.; Gutiérrez-Sagal, R.; Maravillas-Montero, J.L.; Mejía-Domínguez, N.R.; Martínez-Luis, I.; Yacini-Torres, O.G.; Cravioto, M.D.; Reiter, E.; et al. A Novel Mutation in the FSH Receptor (I423T) Affecting Receptor Activation and Leading to Primary Ovarian Failure. J. Clin. Endocrinol. Metab. 2021, 106, e534–e550. [Google Scholar] [CrossRef] [PubMed]
- Katari, S.; Wood-Trageser, M.A.; Jiang, H.; Kalynchuk, E.; Muzumdar, R.; Yatsenko, S.A.; Rajkovic, A. Novel Inactivating Mutation of the FSH Receptor in Two Siblings of Indian Origin with Premature Ovarian Failure. J. Clin. Endocrinol. Metab. 2015, 100, 2154–2157. [Google Scholar] [CrossRef]
- Zargar, M.H.; Shafia, S.; Masoodi, S.R.; Mahajan, Q.; Khan, N.; Ahmad, R. Variations in the inhibin gene in Kashmiri women with primary ovarian insufficiency. Hum. Fertil. 2020, 23, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Qiao, T.; Hua, L.; Liu, S.; Zhao, X.; Lv, C.; Zhao, X.; Wang, J.; Han, L.; Yang, L.; et al. Synergistic Regulatory Effect of Inhibin and Anti-Müllerian Hormone on Fertility of Mice. Front. Vet. Sci. 2021, 8, 747619. [Google Scholar] [CrossRef]
- Qin, C.; Yuan, Z.; Yao, J.; Zhu, W.; Wu, W.; Xie, J. AMH and AMHR2 genetic variants in Chinese women with primary ovarian insufficiency and normal age at natural menopause. Reprod. Biomed. Online 2014, 29, 311–318. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, Q.; Shen, Q.; Jiang, C.; Zou, F.; Shen, Y.; Wang, Y. Identification of novel biallelic variants in BMP15 in two siblings with premature ovarian insufficiency. J. Assist. Reprod. Genet. 2022, 39, 2125–2134. [Google Scholar] [CrossRef]
- Liu, M.N.; Zhang, K.; Xu, T.M. The role of BMP15 and GDF9 in the pathogenesis of primary ovarian insufficiency. Hum. Fertil. 2021, 24, 325–332. [Google Scholar] [CrossRef]
- Verma, K.P.; Thompson, B.; Wolfe, J.; Price, S.; Djukiadmodjo, F.; Trainer, A. A homozygous truncating variant in GDF9 in siblings with primary ovarian insufficiency. J. Assist. Reprod. Genet. 2021, 38, 1539–1543. [Google Scholar] [CrossRef]
- Chu, K.; He, Y.; Li, Z.; Jiang, Z.; Wang, L.; Ji, Y.; Wang, X.; Pang, W.; Sun, N.; Yang, F.; et al. Novel LAT Pathogenic Variants in a POI Family and Its Role in the Ovary. Front. Genet. 2021, 12, 764160. [Google Scholar] [CrossRef]
- Li, Y.; Liu, F.; Li, S.; Tan, S. Association of Variants in Vascular Endothelial Growth Factor A Gene and VEGFA Serum Levels with the Risk of Primary Ovarian Insufficiency: A Case-Control Study. Gynecol. Obstet. Investig. 2021, 86, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Renault, L.; Patiño, L.C.; Magnin, F.; Delemer, B.; Young, J.; Laissue, P.; Binart, N.; Beau, I. BMPR1A and BMPR1B Missense Mutations Cause Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2020, 105, e1449–e1457. [Google Scholar] [CrossRef]
- Santos, M.G.; Machado, A.Z.; Martins, C.N.; Domenice, S.; Costa, E.M.; Nishi, M.Y.; Ferraz-de-Souza, B.; Jorge, S.A.; Pereira, C.A.; Soardi, F.C.; et al. Homozygous inactivating mutation in NANOS3 in two sisters with primary ovarian insufficiency. BioMed Res. Int. 2014, 2014, 787465. [Google Scholar] [CrossRef]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Kasippillai, T.; MacArthur, D.G.; Kirby, A.; Thomas, B.; Lambalk, C.B.; Daly, M.J.; Welt, C.K. Mutations in eIF4ENIF1 are associated with primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 2013, 98, E1534–E1539. [Google Scholar] [CrossRef]
- Zhe, J.; Chen, S.; Chen, X.; Liu, Y.; Li, Y.; Zhou, X.; Zhang, J. A novel heterozygous splice-altering mutation in HFM1 may be a cause of premature ovarian insufficiency. J. Ovarian Res. 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhong, C.; Yang, R.; Yin, Y.; Tan, R.; Gao, L.; Gao, C.; Cui, Y.; Pu, D.; Wu, J. Hfm1 participates in Golgi-associated spindle assembly and division in mouse oocyte meiosis. Cell Death Dis. 2020, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Zhao, S.; Bian, Y.; Ning, Y.; Qin, Y. Mutational analysis of theFAM175A gene in patients with premature ovarian insufficiency. Reprod. Biomed. Online 2019, 38, 943–950. [Google Scholar] [CrossRef]
- Perry, J.R.; Corre, T.; Esko, T.; Chasman, D.I.; Fischer, K.; Franceschini, N.; He, C.; Kutalik, Z.; Mangino, M.; Rose, L.M.; et al. A genome-wide association study of early menopause and the combined impact of identified variants. Hum. Mol. Genet. 2013, 22, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Liu, C.T.; Chen, G.K.; Andrews, J.S.; Arnold, A.M.; Dreyfus, J.; Franceschini, N.; Garcia, M.E.; Kerr, K.F.; Li, G.; et al. Meta-analysis of loci associated with age at natural menopause in African-American women. Hum. Mol. Genet. 2014, 23, 3327–3342. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, L.; Zhang, J.; Qin, Y.; Zhao, S.; Chen, Z.J. Variation analysis of PRIM1 gene in Chinese patients with primary ovarian insufficiency. Reprod. Biomed. Online 2016, 33, 587–591. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jaillard, S.; McElreavy, K.; Robevska, G.; Akloul, L.; Ghieh, F.; Sreenivasan, R.; Beaumont, M.; Bashamboo, A.; Bignon-Topalovic, J.; Neyroud, A.S.; et al. STAG3 homozygous missense variant causes primary ovarian insufficiency and male non-obstructive azoospermia. Mol. Hum. Reprod. 2020, 26, 665–677. [Google Scholar] [CrossRef]
- Xiao, W.J.; He, W.B.; Zhang, Y.X.; Meng, L.L.; Lu, G.X.; Lin, G.; Tan, Y.Q.; Du, J. In-Frame Variants in STAG3 Gene Cause Premature Ovarian Insufficiency. Front. Genet. 2019, 10, 1016. [Google Scholar] [CrossRef]
- He, W.B.; Banerjee, S.; Meng, L.L.; Du, J.; Gong, F.; Huang, H.; Zhang, X.X.; Wang, Y.Y.; Lu, G.X.; Lin, G.; et al. Whole-exome sequencing identifies a homozygous donor splice-site mutation in STAG3 that causes primary ovarian insufficiency. Clin. Genet. 2018, 93, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Mellone, S.; Zavattaro, M.; Vurchio, D.; Ronzani, S.; Caputo, M.; Leone, I.; Prodam, F.; Giordano, M. A Long Contiguous Stretch of Homozygosity Disclosed a Novel STAG3 Biallelic Pathogenic Variant Causing Primary Ovarian Insufficiency: A Case Report and Review of the Literature. Genes 2021, 12, 1709. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.M.; Boetje, E.; Edgerley, J.J.; Miles, E.; Fitzgerald, C.T.; Busby, G.; Beaman, G.M.; O’Sullivan, J.; O’Keefe, R.T.; Newman, W.G. Biallelic loss of function variants in STAG3 result in primary ovarian insufficiency. Reprod. Biomed. Online 2021, 43, 899–902. [Google Scholar] [CrossRef]
- Chavda, A.P.; Ang, K.; Ivanov, D. The torments of the cohesin ring. Nucleus 2017, 8, 261–267. [Google Scholar] [CrossRef][Green Version]
- Griffin, W.C.; Trakselis, M.A. The MCM8/9 complex: A recent recruit to the roster of helicases involved in genome maintenance. DNA Repair 2019, 76, 1–10. [Google Scholar] [CrossRef]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; He, W.B.; Xiao, W.J.; Meng, L.L.; Tan, C.; Du, J.; Lu, G.X.; Lin, G.; Tan, Y.Q. Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol. Genet. Genom. Med. 2020, 8, e1165. [Google Scholar] [CrossRef] [PubMed]
- Bouali, N.; Francou, B.; Bouligand, J.; Imanci, D.; Dimassi, S.; Tosca, L.; Zaouali, M.; Mougou, S.; Young, J.; Saad, A.; et al. New MCM8 mutation associated with premature ovarian insufficiency and chromosomal instability in a highly consanguineous Tunisian family. Fertil. Steril. 2017, 108, 694–702. [Google Scholar] [CrossRef]
- Desai, S.; Wood-Trageser, M.; Matic, J.; Chipkin, J.; Jiang, H.; Bachelot, A.; Dulon, J.; Sala, C.; Barbieri, C.; Cocca, M.; et al. MCM8 and MCM9 Nucleotide Variants in Women with Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2017, 102, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zheng, Y.; Li, G.; Zhao, S.; Ma, J.; Qin, Y. Novel pathogenic mutations in minichromosome maintenance complex component 9 (MCM9) responsible for premature ovarian insufficiency. Fertil. Steril. 2020, 113, 845–852. [Google Scholar] [CrossRef]
- Shen, J.; Qu, D.; Gao, Y.; Sun, F.; Xie, J.; Sun, X.; Wang, D.; Ma, X.; Cui, Y.; Liu, J.; et al. Genetic etiologic analysis in 74 Chinese Han women with idiopathic premature ovarian insufficiency by combined molecular genetic testing. J. Assist. Reprod. Genet. 2021, 38, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Touraine, P.; Desai, S.; Humphreys, G.; Jiang, H.; Yatsenko, A.; Rajkovic, A. Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J. Assist. Reprod. Genet. 2019, 36, 39–45. [Google Scholar] [CrossRef]
- França, M.M.; Funari, M.F.A.; Lerario, A.M.; Santos, M.G.; Nishi, M.Y.; Domenice, S.; Moraes, D.R.; Costalonga, E.F.; Maciel, G.A.R.; Maciel-Guerra, A.T.; et al. Screening of targeted panel genes in Brazilian patients with primary ovarian insufficiency. PLoS ONE 2020, 15, e0240795. [Google Scholar] [CrossRef]
- Mirinezhad, M.R.; Khosroabadi, N.; Rahpeyma, M.; Khayami, R.; Hashemi, S.R.; Ghazizadeh, H.; Ferns, G.A.; Pasdar, A.; Ghayour-Mobarhan, M.; Hamzehloei, T. Genetic Determinants of Premature Menopause in A Mashhad Population Cohort. Int. J. Fertil. Steril. 2021, 15, 26–33. [Google Scholar] [PubMed]
- Cao, D.; Shi, F.; Guo, C.; Liu, Y.; Lin, Z.; Zhang, J.; Li, R.H.W.; Yao, Y.; Liu, K.; Ng, E.H.Y.; et al. A pathogenic DMC1 frameshift mutation causes nonobstructive azoospermia but not primary ovarian insufficiency in humans. Mol. Hum. Reprod. 2021, 27, gaab058. [Google Scholar] [CrossRef]
- Eskenazi, S.; Bachelot, A.; Hugon-Rodin, J.; Plu-Bureau, G.; Gompel, A.; Catteau-Jonard, S.; Molina-Gomes, D.; Dewailly, D.; Dodé, C.; Christin-Maitre, S.; et al. Next Generation Sequencing Should Be Proposed to Every Woman With “Idiopathic” Primary Ovarian Insufficiency. J. Endocr. Soc. 2021, 5, bvab032. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, M.; Qin, Y.; Xia, T.; Ma, J.; Chen, Z.J. Mutations in DMC1 are not responsible for premature ovarian failure in Chinese women. Reprod. Biomed. Online 2013, 26, 175–178. [Google Scholar] [CrossRef]
- Tran, T.N.; Schimenti, J.C. A putative human infertility allele of the meiotic recombinase DMC1 does not affect fertility in mice. Hum. Mol. Genet. 2018, 27, 3911–3918. [Google Scholar] [CrossRef] [PubMed]
- El Bakly, W.; Medhat, M.; Shafei, M.; Tash, R.; Elrefai, M.; Shoukry, Y.; Omar, N.N. Optimized platelet rich plasma releasate (O-rPRP) repairs galactosemia-induced ovarian follicular loss in rats by activating mTOR signaling and inhibiting apoptosis. Heliyon 2020, 6, e05006. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Liu, S.; Huang, M.; Jiang, X.; Yang, M. Cadmium induces apoptosis of human granulosa cell line KGN via mitochondrial dysfunction-mediated pathways. Ecotoxicol. Environ. Saf. 2021, 220, 112341. [Google Scholar] [CrossRef]
- Hartley, P.S.; Bayne, R.A.; Robinson, L.L.; Fulton, N.; Anderson, R.A. Developmental changes in expression of myeloid cell leukemia-1 in human germ cells during oogenesis and early folliculogenesis. J. Clin. Endocrinol. Metab. 2002, 87, 3417–3427. [Google Scholar] [CrossRef][Green Version]
- Guo, T.; Zhao, S.; Zhao, S.; Chen, M.; Li, G.; Jiao, X.; Wang, Z.; Zhao, Y.; Qin, Y.; Gao, F.; et al. Mutations in MSH5 in primary ovarian insufficiency. Hum. Mol. Genet. 2017, 26, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Carlosama, C.; Elzaiat, M.; Patiño, L.C.; Mateus, H.E.; Veitia, R.A.; Laissue, P. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum. Mol. Genet. 2017, 26, 3161–3166. [Google Scholar] [CrossRef]
- Luo, W.; Guo, T.; Li, G.; Liu, R.; Zhao, S.; Song, M.; Zhang, L.; Wang, S.; Chen, Z.J.; Qin, Y. Variants in Homologous Recombination Genes EXO1 and RAD51 Related with Premature Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2020, 105, e3566–e3574. [Google Scholar] [CrossRef]
- Caburet, S.; Todeschini, A.L.; Petrillo, C.; Martini, E.; Farran, N.D.; Legois, B.; Livera, G.; Younis, J.S.; Shalev, S.; Veitia, R.A. A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. EBioMedicine 2019, 42, 524–531. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Murtaza, G.; Zhou, J.; Jiao, Y.; Gong, C.; Hu, C.; Han, Q.; Zhang, H.; Zhang, Y.; et al. Whole-exome sequencing of consanguineous families with infertile men and women identifies homologous mutations in SPATA22 and MEIOB. Hum. Reprod. 2021, 36, 2793–2804. [Google Scholar] [CrossRef]
- Hays, E.; Majchrzak, N.; Daniel, V.; Ferguson, Z.; Brown, S.; Hathorne, K.; La Salle, S. Spermatogenesis associated 22 is required for DNA repair and synapsis of homologous chromosomes in mouse germ cells. Andrology 2017, 5, 299–312. [Google Scholar] [CrossRef]
- Sirchia, F.; Giorgio, E.; Cucinella, L.; Valente, E.M.; Nappi, R.E. Biallelic mutations in PSMC3IP are associated with secondary amenorrhea: Expanding the spectrum of premature ovarian insufficiency. J. Assist. Reprod. Genet. 2022, 39, 1177–1181. [Google Scholar] [CrossRef]
- Mei, L.; Huang, L.; Huang, Y.; Wu, X.; He, H.; He, X.; Su, Z.; Li, P. Two novel biallelic mutations in PSMC3IP in a patient affected by premature ovarian insufficiency. Mol. Med. Rep. 2022, 25, 45. [Google Scholar] [CrossRef]
- Norling, A.; Hirschberg, A.L.; Karlsson, L.; Rodriguez-Wallberg, K.A.; Iwarsson, E.; Wedell, A.; Barbaro, M. No mutations in the PSMC3IP gene identified in a Swedish cohort of women with primary ovarian insufficiency. Sex. Dev. 2014, 8, 146–150. [Google Scholar] [CrossRef]
- Dubois, E.L.; Guitton-Sert, L.; Béliveau, M.; Parmar, K.; Chagraoui, J.; Vignard, J.; Pauty, J.; Caron, M.C.; Coulombe, Y.; Buisson, R.; et al. A Fanci knockout mouse model reveals common and distinct functions for FANCI and FANCD2. Nucleic Acids Res. 2019, 47, 7532–7547. [Google Scholar] [CrossRef] [PubMed]
- Cen, C.; Chen, J.; Lin, L.; Chen, M.; Dong, F.; Shen, Z.; Cui, X.; Hou, X.; Gao, F. Fancb deficiency causes premature ovarian insufficiency in mice†. Biol. Reprod. 2022, 107, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yang, X.; Zhang, F.; Chen, S.; Zhou, Z.; Yin, H.; Ma, H.; Shang, L.; Yang, J.; Li, G.; et al. A heterozygous hypomorphic mutation of Fanca causes impaired follicle development and subfertility in female mice. Mol. Genet. Genom. 2021, 296, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Begum, K.; Overbeek, P.A. Primary Ovarian Insufficiency Induced by Fanconi Anemia E Mutation in a Mouse Model. PLoS ONE 2016, 11, e0144285. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, X.; Jiao, J.; Zhang, F.; Pan, Y.; Wang, Q.; Chen, Q.; Cai, B.; Tang, S.; Zhou, Z.; et al. Rare variants in FANCA induce premature ovarian insufficiency. Hum. Genet. 2019, 138, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Mendonca, B.B. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101594. [Google Scholar] [CrossRef]
- Fouquet, B.; Pawlikowska, P.; Caburet, S.; Guigon, C.; Mäkinen, M.; Tanner, L.; Hietala, M.; Urbanska, K.; Bellutti, L.; Legois, B.; et al. A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. eLife 2017, 6, e30490. [Google Scholar] [CrossRef]
- Yatsenko, S.A.; Gurbuz, F.; Topaloglu, A.K.; Berman, A.J.; Martin, P.M.; Rodrigue-Escribà, M.; Qin, Y.; Rajkovic, A. Pathogenic Variants in ZSWIM7 Cause Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2022, 107, e2359–e2364. [Google Scholar] [CrossRef] [PubMed]
- Bertrand-Delepine, J.; Manouvrier-Hanu, S.; Cartigny, M.; Paris, F.; Mallet, D.; Philibert, P.; Morel, Y.; Lefevre, C.; Dewailly, D.; Catteau-Jonard, S. In cases of familial primary ovarian insufficiency and disorders of gonadal development, consider NR5A1/SF-1 sequence variants. Reprod. Biomed. Online 2020, 40, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Buaas, F.W.; Gardiner, J.R.; Clayton, S.; Val, P.; Swain, A. In vivo evidence for the crucial role of SF1 in steroid-producing cells of the testis, ovary and adrenal gland. Development 2012, 139, 4561–4570. [Google Scholar] [CrossRef]
- Voican, A.; Bachelot, A.; Bouligand, J.; Francou, B.; Dulon, J.; Lombès, M.; Touraine, P.; Guiochon-Mantel, A. NR5A1 (SF-1) mutations are not a major cause of primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 2013, 98, E1017–E1021. [Google Scholar] [CrossRef] [PubMed]
- Ramos, L. WT1, NR0B1, NR5A1, LHX9, ZFP92, ZNF275, INSL3, and NRIP1 Genetic Variants in Patients with Premature Ovarian Insufficiency in a Mexican Cohort. Genes 2022, 13, 611. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, S.; Sreenivasan, R.; Beaumont, M.; Robevska, G.; Dubourg, C.; Knarston, I.M.; Akloul, L.; van den Bergen, J.; Odent, S.; Croft, B.; et al. Analysis of NR5A1 in 142 patients with premature ovarian insufficiency, diminished ovarian reserve, or unexplained infertility. Maturitas 2020, 131, 78–86. [Google Scholar] [CrossRef]
- Cattoni, A.; Spano, A.; Tulone, A.; Boneschi, A.; Masera, N.; Maitz, S.; Di Blasio, A.M.; Persani, L.; Guizzardi, F.; Rossetti, R. The Potential Synergic Effect of a Complex Pattern of Multiple Inherited Genetic Variants as a Pathogenic Factor for Ovarian Dysgenesis: A Case Report. Front. Endocrinol. 2020, 11, 540683. [Google Scholar] [CrossRef] [PubMed]
- Bouilly, J.; Roucher-Boulez, F.; Gompel, A.; Bry-Gauillard, H.; Azibi, K.; Beldjord, C.; Dodé, C.; Bouligand, J.; Mantel, A.G.; Hécart, A.C.; et al. New NOBOX mutations identified in a large cohort of women with primary ovarian insufficiency decrease KIT-L expression. J. Clin. Endocrinol. Metab. 2015, 100, 994–1001. [Google Scholar] [CrossRef]
- Pan, L.; Gong, W.; Zhou, Y.; Li, X.; Yu, J.; Hu, S. A comprehensive transcriptomic analysis of infant and adult mouse ovary. Genom. Proteom. Bioinform. 2014, 12, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Bouali, N.; Francou, B.; Bouligand, J.; Lakhal, B.; Malek, I.; Kammoun, M.; Warszawski, J.; Mougou, S.; Saad, A.; Guiochon-Mantel, A. NOBOX is a strong autosomal candidate gene in Tunisian patients with primary ovarian insufficiency. Clin. Genet. 2016, 89, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Bayne, R.A.; Kinnell, H.L.; Coutts, S.M.; He, J.; Childs, A.J.; Anderson, R.A. GDF9 is transiently expressed in oocytes before follicle formation in the human fetal ovary and is regulated by a novel NOBOX transcript. PLoS ONE 2015, 10, e0119819. [Google Scholar] [CrossRef] [PubMed]
- Bouilly, J.; Veitia, R.A.; Binart, N. NOBOX is a key FOXL2 partner involved in ovarian folliculogenesis. J. Mol. Cell Biol. 2014, 6, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, I.; Bouilly, J.; Beau, I.; Guizzardi, F.; Ferlin, A.; Pollazzon, M.; Salerno, M.; Binart, N.; Persani, L.; Rossetti, R. Impaired protein stability and nuclear localization of NOBOX variants associated with premature ovarian insufficiency. Hum. Mol. Genet. 2016, 25, 5223–5233. [Google Scholar]
- Shin, Y.H.; Ren, Y.; Suzuki, H.; Golnoski, K.J.; Ahn, H.W.; Mico, V.; Rajkovic, A. Transcription factors SOHLH1 and SOHLH2 coordinate oocyte differentiation without affecting meiosis I. J. Clin. Investig. 2017, 127, 2106–2117. [Google Scholar] [CrossRef]
- Jolly, A.; Bayram, Y.; Turan, S.; Aycan, Z.; Tos, T.; Abali, Z.Y.; Hacihamdioglu, B.; Coban Akdemir, Z.H.; Hijazi, H.; Bas, S.; et al. Exome Sequencing of a Primary Ovarian Insufficiency Cohort Reveals Common Molecular Etiologies for a Spectrum of Disease. J. Clin. Endocrinol. Metab. 2019, 104, 3049–3067. [Google Scholar] [CrossRef]
- Fu, L.; Koganti, P.P.; Wang, J.; Wang, L.; Wang, C.L.; Yao, J. Lhx8 interacts with a novel germ cell-specific nuclear factor containing an Nbl1 domain in rainbow trout (Oncorhynchus mykiss). PLoS ONE 2017, 12, e0170760. [Google Scholar] [CrossRef]
- Qin, Y.; Zhao, H.; Kovanci, E.; Simpson, J.L.; Chen, Z.J.; Rajkovic, A. Analysis of LHX8 mutation in premature ovarian failure. Fertil. Steril. 2008, 89, 1012–1014. [Google Scholar] [CrossRef][Green Version]
- Okunomiya, A.; Horie, A.; Tani, H.; Sato, Y.; Takamatsu, S.; Brown, J.B.; Sugimoto, M.; Hamanishi, J.; Kondoh, E.; Matsumura, N.; et al. Figla promotes secondary follicle growth in mature mice. Sci. Rep. 2021, 11, 9842. [Google Scholar] [CrossRef]
- Chen, B.; Li, L.; Wang, J.; Li, T.; Pan, H.; Liu, B.; Zhou, Y.; Cao, Y.; Wang, B. Consanguineous familial study revealed biallelic FIGLA mutation associated with premature ovarian insufficiency. J. Ovarian Res. 2018, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Sassi, A.; Désir, J.; Janssens, V.; Marangoni, M.; Daneels, D.; Gheldof, A.; Bonduelle, M.; Van Dooren, S.; Costagliola, S.; Delbaere, A. Novel inactivating follicle-stimulating hormone receptor mutations in a patient with premature ovarian insufficiency identified by next-generation sequencing gene panel analysis. F S Rep. 2020, 1, 193–201. [Google Scholar] [CrossRef]
- Woad, K.J.; Prendergast, D.; Winship, I.M.; Shelling, A.N. FSH receptor gene variants are rarely associated with premature ovarian failure. Reprod. Biomed. Online 2013, 26, 396–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, W.; Cao, Y.; Shi, L. Effects of FSHR polymorphisms on premature ovarian insufficiency in human beings: A meta-analysis. Reprod. Biol. Endocrinol. 2019, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, T.; Gong, Z.; Yu, Y.; Zhang, Y.; Zhao, S.; Qin, Y. Novel FSHR mutations in Han Chinese women with sporadic premature ovarian insufficiency. Mol. Cell Endocrinol. 2019, 492, 110446. [Google Scholar] [CrossRef]
- Rah, H.; Jeon, Y.J.; Ko, J.J.; Kim, J.H.; Kim, Y.R.; Cha, S.H.; Choi, Y.; Lee, W.S.; Kim, N.K. Association of inhibin α gene promoter polymorphisms with risk of idiopathic primary ovarian insufficiency in Korean women. Maturitas 2014, 77, 163–167. [Google Scholar] [CrossRef]
- Walton, K.L.; Goney, M.P.; Peppas, Z.; Stringer, J.M.; Winship, A.; Hutt, K.; Goodchild, G.; Maskey, S.; Chan, K.L.; Brûlé, E.; et al. Inhibin Inactivation in Female Mice Leads to Elevated FSH Levels, Ovarian Overstimulation, and Pregnancy Loss. Endocrinology 2022, 163, bqac025. [Google Scholar] [CrossRef]
- Christofolini, D.M.; Cordts, E.B.; Santos-Pinheiro, F.; Kayaki, E.A.; Dornas, M.C.F.; Santos, M.C.; Bianco, B.; Barbosa, C.P. How polymorphic markers contribute to genetic diseases in different populations? The study of inhibin A for premature ovarian insufficiency. Einstein 2017, 15, 269–272. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Y.; Mei, S.; Liu, C.; Ma, X.; Li, Y.; Jiang, Y.; Ha, L.; Xu, X. Single nucleotide polymorphisms in premature ovarian failure-associated genes in a Chinese Hui population. Mol. Med. Rep. 2015, 12, 2529–2538. [Google Scholar] [CrossRef]
- Li, L.; Zhou, X.; Wang, X.; Wang, J.; Zhang, W.; Wang, B.; Cao, Y.; Kee, K. A dominant negative mutation at the ATP binding domain of AMHR2 is associated with a defective anti-Müllerian hormone signaling pathway. Mol. Hum. Reprod. 2016, 22, 669–678. [Google Scholar] [CrossRef]
- Alvaro Mercadal, B.; Imbert, R.; Demeestere, I.; Gervy, C.; De Leener, A.; Englert, Y.; Costagliola, S.; Delbaere, A. AMH mutations with reduced in vitro bioactivity are related to premature ovarian insufficiency. Hum. Reprod. 2015, 30, 1196–1202. [Google Scholar] [CrossRef]
- Peng, J.; Li, Q.; Wigglesworth, K.; Rangarajan, A.; Kattamuri, C.; Peterson, R.T.; Eppig, J.J.; Thompson, T.B.; Matzuk, M.M. Growth differentiation factor 9:bone morphogenetic protein 15 heterodimers are potent regulators of ovarian functions. Proc. Natl. Acad. Sci. USA 2013, 110, E776–E785. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Funari, M.F.A.; Nishi, M.Y.; Narcizo, A.M.; Domenice, S.; Costa, E.M.F.; Lerario, A.M.; Mendonca, B.B. Identification of the first homozygous 1-bp deletion in GDF9 gene leading to primary ovarian insufficiency by using targeted massively parallel sequencing. Clin. Genet. 2018, 93, 408–411. [Google Scholar] [CrossRef]
- Norling, A.; Hirschberg, A.L.; Rodriguez-Wallberg, K.A.; Iwarsson, E.; Wedell, A.; Barbaro, M. Identification of a duplication within the GDF9 gene and novel candidate genes for primary ovarian insufficiency (POI) by a customized high-resolution array comparative genomic hybridization platform. Hum. Reprod. 2014, 29, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Afkhami, F.; Shahbazi, S.; Farzadi, L.; Danaei, S. Novel bone morphogenetic protein 15 (BMP15) gene variants implicated in premature ovarian insufficiency. Reprod. Biol. Endocrinol. 2022, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, E.; De Marco, G.; Orsolini, F.; Gianetti, E.; Benelli, E.; Fruzzetti, F.; Simoncini, T.; Agretti, P.; Tonacchera, M. Characterization of a novel mutation V136L in bone morphogenetic protein 15 identified in a woman affected by POI. J. Ovarian Res. 2021, 14, 85. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, J.; Wang, X.; Li, L.; Pan, H.; Chen, B.; Zhu, Y.; Li, T.; Cao, Y.; Wang, B. A novel homozygous mutation of bone morphogenetic protein 15 identified in a consanguineous marriage family with primary ovarian insufficiency. Reprod. Biomed. Online 2018, 36, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Demain, L.A.M.; Metcalfe, K.; Boetje, E.; Clayton, P.; Martindale, E.A.; Busby, G.; O’Keefe, R.T.; Newman, W.G. Homozygous missense variants in BMPR15 can result in primary ovarian insufficiency. Reprod. Biomed. Online 2022, 45, 727–729. [Google Scholar] [CrossRef]
- Juárez-Rendón, K.J.; García-Ortiz, J.E. Evaluation of four genes associated with primary ovarian insufficiency in a cohort of Mexican women. J. Assist. Reprod. Genet. 2018, 35, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Sousa, B.L.; Nishi, M.Y.; Santos, M.G.; Brito, V.N.; Domenice, S.; Mendonca, B.B. Mutation analysis of NANOS3 in Brazilian women with primary ovarian failure. Clinics 2016, 71, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, B.; Dong, Z.; Zhou, S.; Liu, Z.; Shi, G.; Cao, Y.; Xu, Y. A NANOS3 mutation linked to protein degradation causes premature ovarian insufficiency. Cell Death Dis. 2013, 4, e825. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Ren, S.; Yang, X.; Zhang, F.; Jin, L.; Zhang, X.; Wu, Y. EIF4ENIF1 variants in two patients with non-syndromic premature ovarian insufficiency. Eur. J. Med. Genet. 2022, 65, 104597. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, K.G.; Lyon, E.; Spector, E.B. ACMG Standards and Guidelines for fragile X testing: A revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet. Med. 2013, 15, 575–586. [Google Scholar] [CrossRef]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V. Laboratory testing for fragile X, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 799–812. [Google Scholar] [CrossRef]
- Ramos, C.; Ocampos, M.; Barbato, I.T.; Niehues, V.M.S.; Bicalho, M.D.G.; Nisihara, R. Association between mutations in the FMR1 gene and ovarian dysfunction in Brazilian patients. JBRA Assist. Reprod. 2022, 26, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Yu, Q. The significance of FMR1 CGG repeats in Chinese women with premature ovarian insufficiency and diminished ovarian reserve. Reprod. Biol. Endocrinol. 2020, 18, 82. [Google Scholar] [CrossRef]
- Utine, G.E.; Şimşek-Kiper, P.; Akgün-Doğan, Ö.; Ürel-Demir, G.; Alanay, Y.; Aktaş, D.; Boduroğlu, K.; Tunçbilek, E.; Alikaşifoğlu, M. Fragile x-associated premature ovarian failure in a large Turkish cohort: Findings of Hacettepe Fragile X Registry. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 221, 76–80. [Google Scholar] [CrossRef]
- Dean, D.D.; Agarwal, S.; Kapoor, D.; Singh, K.; Vati, C. Molecular Characterization of FMR1 Gene by TP-PCR in Women of Reproductive Age and Women with Premature Ovarian Insufficiency. Mol. Diagn. Ther. 2018, 22, 91–100. [Google Scholar] [CrossRef]
- Espeche, L.D.; Chiauzzi, V.; Ferder, I.; Arrar, M.; Solari, A.P.; Bruque, C.D.; Delea, M.; Belli, S.; Fernández, C.S.; Buzzalino, N.D.; et al. Distribution of FMR1 and FMR2 Repeats in Argentinean Patients with Primary Ovarian Insufficiency. Genes 2017, 8, 194. [Google Scholar] [CrossRef]
- Lu, C.L.; Li, R.; Chen, X.N.; Xu, Y.Y.; Yan, L.Y.; Yan, J.; Zhang, Y.Y.; Jin, H.Y.; Zhang, W.X.; Qiao, J.; et al. The ‘normal’ range of FMR1 triple CGG repeats may be associated with primary ovarian insufficiency in China. Reprod. Biomed. Online 2017, 34, 175–180. [Google Scholar] [CrossRef]
- Barasoain, M.; Barrenetxea, G.; Huerta, I.; Télez, M.; Criado, B.; Arrieta, I. Study of the Genetic Etiology of Primary Ovarian Insufficiency: FMR1 Gene. Genes 2016, 7, 123. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, W.; Liu, Y.; Liu, Y.; Wang, J.; Jiang, H. Association between the FMR1 CGG repeat lengths and the severity of idiopathic primary ovarian insufficiency: A meta analysis. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3116–3122. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Schoemaker, M.J.; Bennett, C.E.; Ennis, S.; Macpherson, J.N.; Jones, M.; Morris, D.H.; Orr, N.; Ashworth, A.; Jacobs, P.A.; et al. Population-based estimates of the prevalence of FMR1 expansion mutations in women with early menopause and primary ovarian insufficiency. Genet. Med. 2014, 16, 19–24. [Google Scholar] [CrossRef]
- Voorhuis, M.; Onland-Moret, N.C.; Janse, F.; Ploos van Amstel, H.K.; Goverde, A.J.; Lambalk, C.B.; Laven, J.S.; van der Schouw, Y.T.; Broekmans, F.J.; Fauser, B.C. The significance of fragile X mental retardation gene 1 CGG repeat sizes in the normal and intermediate range in women with primary ovarian insufficiency. Hum. Reprod. 2014, 29, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.A.; Nelson, L.M.; Pyeritz, R.; Johnson, J.; Sherman, S.L.; Cohen, Y.; Elizur, S.E. Fragile X Associated Primary Ovarian Insufficiency (FXPOI): Case Report and Literature Review. Front. Genet. 2018, 9, 529. [Google Scholar] [CrossRef]
- Lu, C.; Lin, L.; Tan, H.; Wu, H.; Sherman, S.L.; Gao, F.; Jin, P.; Chen, D. Fragile X premutation RNA is sufficient to cause primary ovarian insufficiency in mice. Hum. Mol. Genet. 2012, 21, 5039–5047. [Google Scholar] [CrossRef] [PubMed]
- Barad, D.H.; Darmon, S.; Weghofer, A.; Latham, G.J.; Filipovic, S.; Wang, Q.; Kushnir, V.A.; Albertini, D.F.; Gleicher, N. Association of skewed X-chromosome inactivation with FMR1 CGG repeat length and anti-Mullerian hormone levels: A cohort study. Reprod. Biol. Endocrinol. 2017, 15, 34. [Google Scholar] [CrossRef][Green Version]
- Georges, A.; Auguste, A.; Bessière, L.; Vanet, A.; Todeschini, A.L.; Veitia, R.A. FOXL2: A central transcription factor of the ovary. J. Mol. Endocrinol. 2014, 52, R17–R33. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Treier, M. Forkhead transcription factors in ovarian function. Reproduction 2011, 142, 489–495. [Google Scholar] [CrossRef]
- Li, Y.; Schang, G.; Wang, Y.; Zhou, X.; Levasseur, A.; Boyer, A.; Deng, C.X.; Treier, M.; Boehm, U.; Boerboom, D.; et al. Conditional Deletion of FOXL2 and SMAD4 in Gonadotropes of Adult Mice Causes Isolated FSH Deficiency. Endocrinology 2018, 159, 2641–2655. [Google Scholar] [CrossRef]
- Méjécase, C.; Nigam, C.; Moosajee, M.; Bladen, J.C. The Genetic and Clinical Features of FOXL2-Related Blepharophimosis, Ptosis and Epicanthus Inversus Syndrome. Genes 2021, 12, 364. [Google Scholar] [CrossRef]
- Nicol, B.; Rodriguez, K.; Yao, H.H. Aberrant and constitutive expression of FOXL2 impairs ovarian development and functions in mice. Biol. Reprod. 2020, 103, 966–977. [Google Scholar] [CrossRef]
- Li, F.; Chen, H.; Wang, Y.; Yang, J.; Zhou, Y.; Song, X.; Fan, J. Functional Studies of Novel FOXL2 Variants in Chinese Families with Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome. Front. Genet. 2021, 12, 616112. [Google Scholar] [CrossRef]
- Yang, X.W.; He, W.B.; Gong, F.; Li, W.; Li, X.R.; Zhong, C.G.; Lu, G.X.; Lin, G.; Du, J.; Tan, Y.Q. Novel FOXL2 mutations cause blepharophimosis-ptosis-epicanthus inversus syndrome with premature ovarian insufficiency. Mol. Genet. Genom. Med. 2018, 6, 261–267. [Google Scholar] [CrossRef]
- Xue, M.; Zheng, J.; Zhou, Q.; Hejtmancik, J.F.; Wang, Y.; Li, S. Novel FOXL2 mutations in two Chinese families with blepharophimosis-ptosis-epicanthus inversus syndrome. BMC Med. Genet. 2015, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Grzechocińska, B.; Warzecha, D.; Wypchło, M.; Ploski, R.; Wielgoś, M. Premature ovarian insufficiency as a variable feature of blepharophimosis, ptosis, and epicanthus inversus syndrome associated with c.223C > T p.(Leu75Phe) FOXL2 mutation: A case report. BMC Med. Genet. 2019, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Thanatsis, N.; Kaponis, A.; Koika, V.; Georgopoulos, N.A.; Decavalas, G.O. Reduced Foxo3a, FoxL2, and p27 mRNA expression in human ovarian tissue in premature ovarian insufficiency. Hormones 2019, 18, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. 2019, 21, 1286–1294. [Google Scholar] [CrossRef]
- Badik, J.R.; Castañeda, U.; Gleason, T.J.; Spencer, J.B.; Epstein, M.P.; Ficicioglu, C.; Fitzgerald, K.; Fridovich-Keil, J.L. Ovarian function in Duarte galactosemia. Fertil. Steril. 2011, 96, 469–473.e1. [Google Scholar] [CrossRef]
- Anderson, S. GALT Deficiency Galactosemia. MCN Am. J. Matern Child. Nurs. 2018, 43, 44–51. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Nicholas, C.; Siddiqi, A.; Chen, W.; Bales, E.; Feng, M.; Johnson, J.; Lai, K. Reversal of aberrant PI3K/Akt signaling by Salubrinal in a GalT-deficient mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3286–3293. [Google Scholar] [CrossRef]
- Colhoun, H.O.; Rubio Gozalbo, E.M.; Bosch, A.M.; Knerr, I.; Dawson, C.; Brady, J.; Galligan, M.; Stepien, K.; O’Flaherty, R.; Catherine Moss, C.; et al. Fertility in classical galactosaemia, a study of N-glycan, hormonal and inflammatory gene interactions. Orphanet J. Rare Dis. 2018, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Feldman, G.; Puscheck, E.E. Primary ovarian insufficiency in classic galactosemia: Current understanding and future research opportunities. J. Assist. Reprod. Genet. 2018, 35, 3–16. [Google Scholar] [CrossRef]
- Abidin, Z.; Treacy, E.P. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. Int. J. Mol. Sci. 2019, 20, 5236. [Google Scholar] [CrossRef]
- Kruszewska, J.; Laudy-Wiaderny, H.; Krzywdzinska, S.; Grymowicz, M.; Smolarczyk, R.; Meczekalski, B. Two consecutive pregnancies in a patient with premature ovarian insufficiency in the course of classic galactosemia and a review of the literature. Gynecol. Endocrinol. 2022, 38, 186–189. [Google Scholar] [CrossRef]
- van Erven, B.; Berry, G.T.; Cassiman, D.; Connolly, G.; Forga, M.; Gautschi, M.; Gubbels, C.S.; Hollak, C.E.M.; Janssen, M.C.; Knerr, I.; et al. Fertility in adult women with classic galactosemia and primary ovarian insufficiency. Fertil. Steril. 2017, 108, 168–174. [Google Scholar] [CrossRef]
- Ferré, E.M.N.; Schmitt, M.M.; Lionakis, M.S. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Front. Pediatr. 2021, 9, 723532. [Google Scholar] [CrossRef]
- Guo, C.J.; Leung, P.S.C.; Zhang, W.; Ma, X.; Gershwin, M.E. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (APS-1). Autoimmun. Rev. 2018, 17, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Streeten, E.A.; Chan, A.; Lwin, W.; Tian, L.; Pellegrino da Silva, R.; Kim, C.E.; Anderson, M.S.; Hakonarson, H.; Levine, M.A. Exome Sequencing Reveals Mutations in AIRE as a Cause of Isolated Hypoparathyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 1726–1733. [Google Scholar] [CrossRef]
- Zheng, W.B.; Li, L.J.; Zhao, D.C.; Wang, O.; Jiang, Y.; Xia, W.B.; Li, M. A novel variant in AIRE causing a rare, non-classical autoimmune polyendocrine syndrome type 1. Mol. Med. Rep. 2020, 22, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.K.; Zaidi, G.; Srivastava, R.; Sarangi, A.N.; Bharti, N.; Eriksson, D.; Bensing, S.; Kämpe, O.; Aggarwal, A.; Aggarwal, R.; et al. Identification of autoimmune polyendocrine syndrome type 1 in patients with isolated hypoparathyroidism. Clin. Endocrinol. 2016, 85, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Bellacchio, E.; Dhamo, R.; Frasca, F.; Betterle, C.; Fierabracci, A. A Novel Homozygous Mutation of the AIRE Gene in an APECED Patient from Pakistan: Case Report and Review of the Literature. Front. Immunol. 2018, 9, 1835. [Google Scholar] [CrossRef] [PubMed]
- Halabi, I.; Barohom, M.N.; Peleg, S.; Trougouboff, P.; Elias-Assad, G.; Agbaria, R.; Tenenbaum-Rakover, Y. Case Report: Severe Hypocalcemic Episodes Due to Autoimmune Enteropathy. Front. Endocrinol. 2021, 12, 645279. [Google Scholar] [CrossRef]
- Jasti, S.; Warren, B.D.; McGinnis, L.K.; Kinsey, W.H.; Petroff, B.K.; Petroff, M.G. The autoimmune regulator prevents premature reproductive senescence in female mice. Biol. Reprod. 2012, 86, 110. [Google Scholar] [CrossRef] [PubMed]
- Szeliga, A.; Zysnarska, A.; Szklarska, Z.; Truszkowska, E.; Podfigurna, A.; Czyzyk, A.; Genazzani, A.R.; Chrzanowska, K.; Meczekalski, B. A case of premature ovarian insufficiency in Nijmegen breakage syndrome patient and review of literature. From gene mutation to clinical management. Gynecol. Endocrinol. 2019, 35, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Patiño, L.C.; Beau, I.; Morel, A.; Delemer, B.; Young, J.; Binart, N.; Laissue, P. Functional evidence implicating NOTCH2 missense mutations in primary ovarian insufficiency etiology. Hum. Mutat. 2019, 40, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, S.; Tiitinen, A.; Alfthan, H.; Lipsanen-Nyman, M. Premature ovarian insufficiency and early depletion of the ovarian reserve in the monogenic Mulibrey nanism disorder. Hum. Reprod. 2018, 33, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Masunaga, Y.; Mochizuki, M.; Kadoya, M.; Wada, Y.; Okamoto, N.; Fukami, M.; Kato, F.; Saitsu, H.; Ogata, T. Primary ovarian insufficiency in a female with phosphomannomutase-2 gene (PMM2) mutations for congenital disorder of glycosylation. Endocr. J. 2021, 68, 605–611. [Google Scholar] [CrossRef]
- Boros, E.; Elilié Mawa Ongoth, F.; Heinrichs, C.; Mansbach, A.L.; Seneca, S.; Aeby, A.; Ismaïli, K.; Brachet, C. Primary ovarian insufficiency in RMND1 mitochondrial disease. Mitochondrion 2022, 66, 51–53. [Google Scholar] [CrossRef]
- Zhen, X.; Wu, B.; Wang, J.; Lu, C.; Gao, H.; Qiao, J. Increased Incidence of Mitochondrial Cytochrome C Oxidase 1 Gene Mutations in Patients with Primary Ovarian Insufficiency. PLoS ONE 2015, 10, e0132610. [Google Scholar] [CrossRef]
- Chen, A.; Tiosano, D.; Guran, T.; Baris, H.N.; Bayram, Y.; Mory, A.; Shapiro-Kulnane, L.; Hodges, C.A.; Akdemir, Z.C.; Turan, S.; et al. Mutations in the mitochondrial ribosomal protein MRPS22 lead to primary ovarian insufficiency. Hum. Mol. Genet. 2018, 27, 1913–1926. [Google Scholar] [PubMed]
- Tucker, E.J.; Baker, M.J.; Hock, D.H.; Warren, J.T.; Jaillard, S.; Bell, K.M.; Sreenivasan, R.; Bakhshalizadeh, S.; Hanna, C.A.; Caruana, N.J.; et al. Premature ovarian insufficiency in CLPB deficiency: Transcriptomic, proteomic and phenotypic insights. J. Clin. Endocrinol. Metab. 2022, 107, 3328–3340. [Google Scholar] [CrossRef]
- Ullah, F.; Rauf, W.; Khan, K.; Khan, S.; Bell, K.M.; de Oliveira, V.C.; Tariq, M.; Bakhshalizadeh, S.; Touraine, P.; Katsanis, N.; et al. A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Hum. Genet. 2021, 140, 1733–1751. [Google Scholar] [CrossRef]
- Feng, S.; Wan, S.; Liu, S.; Wang, W.; Tang, M.; Bai, L.; Zhu, Y. LARS2 Regulates Apoptosis via ROS-Mediated Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Ovarian Granulosa Cells. Oxid. Med. Cell. Longev. 2022, 2022, 5501346. [Google Scholar] [CrossRef]
- Guo, L.; Liu, X.; Chen, H.; Wang, W.; Gu, C.; Li, B. Decrease in ovarian reserve through the inhibition of SIRT1-mediated oxidative phosphorylation. Aging 2022, 14, 2335–2347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, B.; Li, L.; Pan, H.; Liu, B.; Li, T.; Wang, R.; Ma, X.; Wang, B.; Cao, Y. Novel alanyl-tRNA synthetase 2 (AARS2) homozygous mutation in a consanguineous Chinese family with premature ovarian insufficiency. Fertil. Steril. 2019, 112, 569–576.e2. [Google Scholar] [CrossRef]
- Pankiewicz, K.; Laudański, P.; Issat, T. The Role of Noncoding RNA in the Pathophysiology and Treatment of Premature Ovarian Insufficiency. Int. J. Mol. Sci. 2021, 22, 9336. [Google Scholar] [CrossRef]
- Dang, Y.; Wang, X.; Hao, Y.; Zhang, X.; Zhao, S.; Ma, J.; Qin, Y.; Chen, Z.J. MicroRNA-379-5p is associate with biochemical premature ovarian insufficiency through PARP1 and XRCC6. Cell Death Dis. 2018, 9, 106. [Google Scholar] [CrossRef]
- Zhang, X.; Dang, Y.; Liu, R.; Zhao, S.; Ma, J.; Qin, Y. MicroRNA-127-5p impairs function of granulosa cells via HMGB2 gene in premature ovarian insufficiency. J. Cell. Physiol. 2020, 235, 8826–8838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, R.; Hao, J.; Huang, X.; Liu, M.; Lv, M.; Su, C.; Mu, Y.L. miRNA-122-5p in POI ovarian-derived exosomes promotes granulosa cell apoptosis by regulating BCL9. Cancer Med. 2022, 11, 2414–2426. [Google Scholar] [CrossRef]
- Dang, Y.; Zhao, S.; Qin, Y.; Han, T.; Li, W.; Chen, Z.J. MicroRNA-22-3p is down-regulated in the plasma of Han Chinese patients with premature ovarian failure. Fertil. Steril. 2015, 103, 802–807.e1. [Google Scholar] [PubMed]
- Wang, X.; Zhang, X.; Dang, Y.; Li, D.; Lu, G.; Chan, W.Y.; Leung, P.C.K.; Zhao, S.; Qin, Y.; Chen, Z.J. Long noncoding RNA HCP5 participates in premature ovarian insufficiency by transcriptionally regulating MSH5 and DNA damage repair via YB1. Nucleic Acids Res. 2020, 48, 4480–4491. [Google Scholar] [CrossRef]
- Wang, F.; Chen, X.; Sun, B.; Ma, Y.; Niu, W.; Zhai, J.; Sun, Y. Hypermethylation-mediated downregulation of lncRNA PVT1 promotes granulosa cell apoptosis in premature ovarian insufficiency via interacting with Foxo3a. J. Cell Physiol. 2021, 236, 5162–5175. [Google Scholar] [CrossRef]
- Sakka, R.; Abdelhedi, F.; Sellami, H.; Pichon, B.; Lajmi, Y.; Mnif, M.; Kebaili, S.; Derbel, R.; Kamoun, H.; Gdoura, R.; et al. An unusual familial Xp22.12 microduplication including EIF1AX: A novel candidate dosage-sensitive gene for premature ovarian insufficiency. Eur. J. Med. Genet. 2022, 65, 104613. [Google Scholar] [CrossRef] [PubMed]
- França, M.M.; Mendonca, B.B. Genetics of Primary Ovarian Insufficiency in the Next-Generation Sequencing Era. J. Endocr. Soc. 2020, 4, bvz037. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, G.; Zhang, J.; Gao, F.; Li, W.; Qin, Y.; Chen, Z.J. Novel WT1 Missense Mutations in Han Chinese. Women with Premature Ovarian Failure. Sci. Rep. 2015, 5, 13983. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Han, T.; Yan, L.; Cheng, L.; Qin, Y.; Liu, W.; Zhao, S.; Chen, Z.J. A novel homozygous mutation in the FSHR gene is causative for primary ovarian insufficiency. Fertil. Steril. 2017, 108, 1050–1055.e2. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).