
Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1
 ,
,
Abstract
:1. Introduction
2. Results
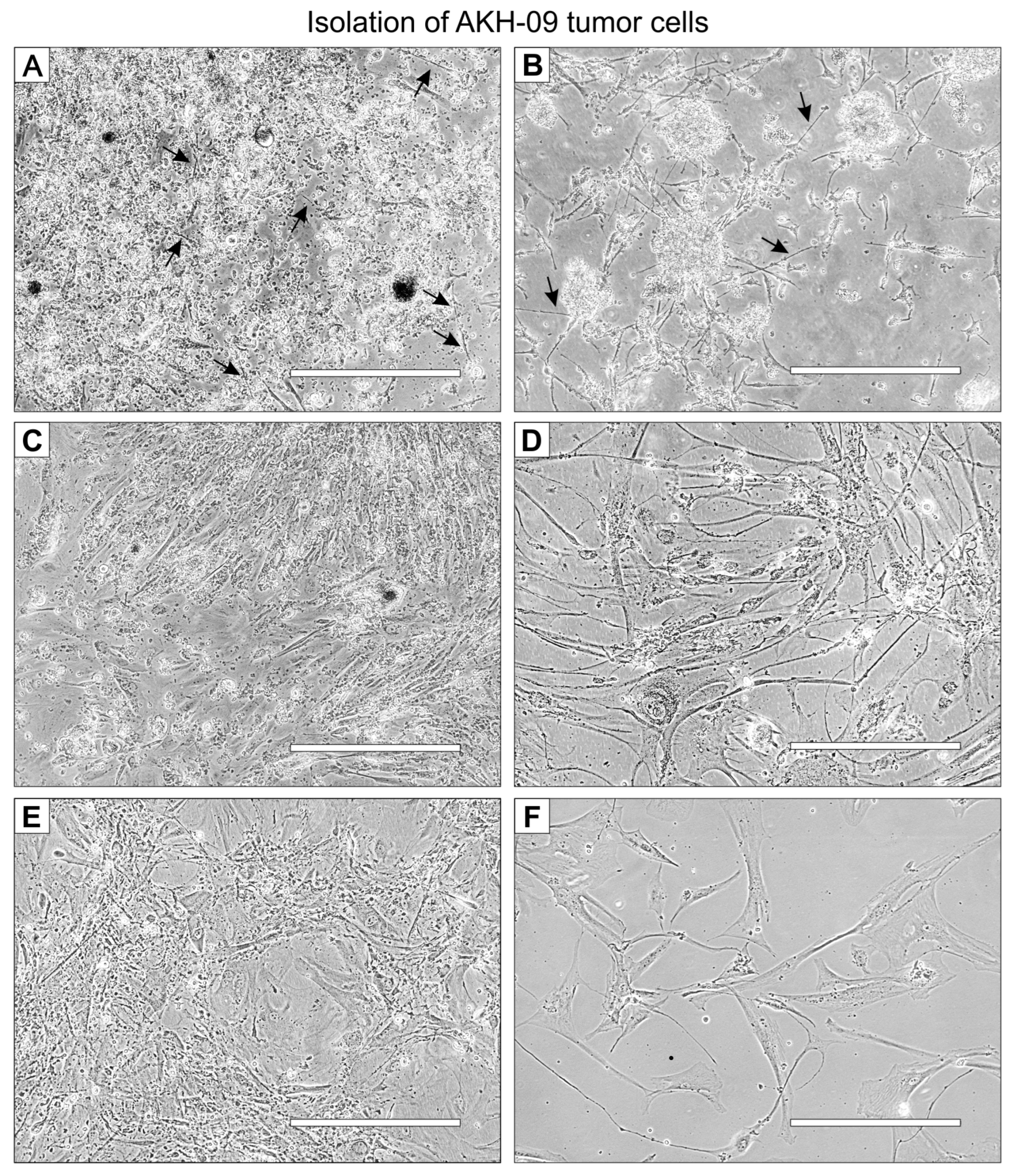
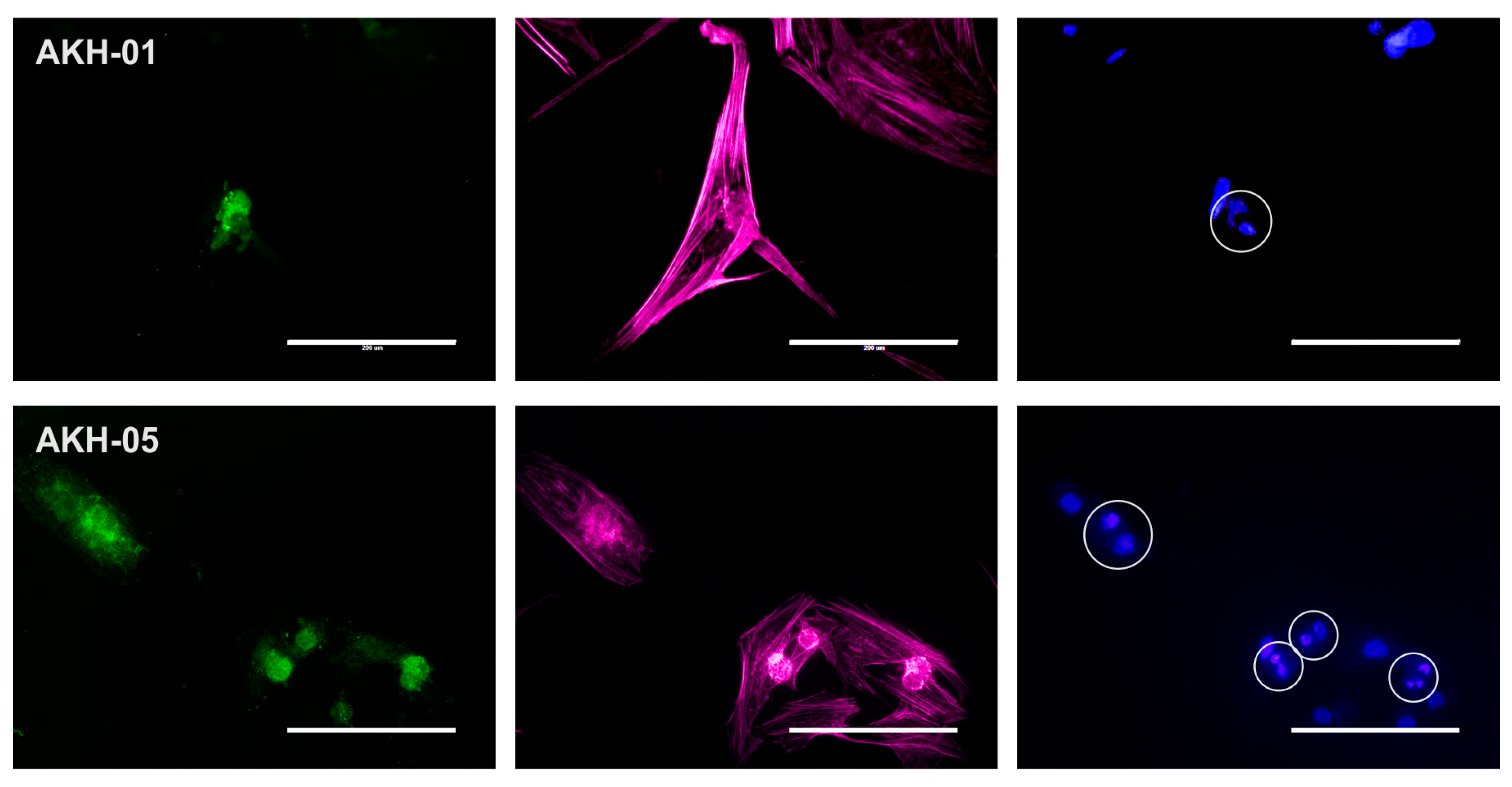
2.1. Isolation of GBM Cells from Tumor Samples
2.2. Detection of ZIKV Receptors Axl and Integrin αvβ5 on Cells Used for Pseudotype Infections
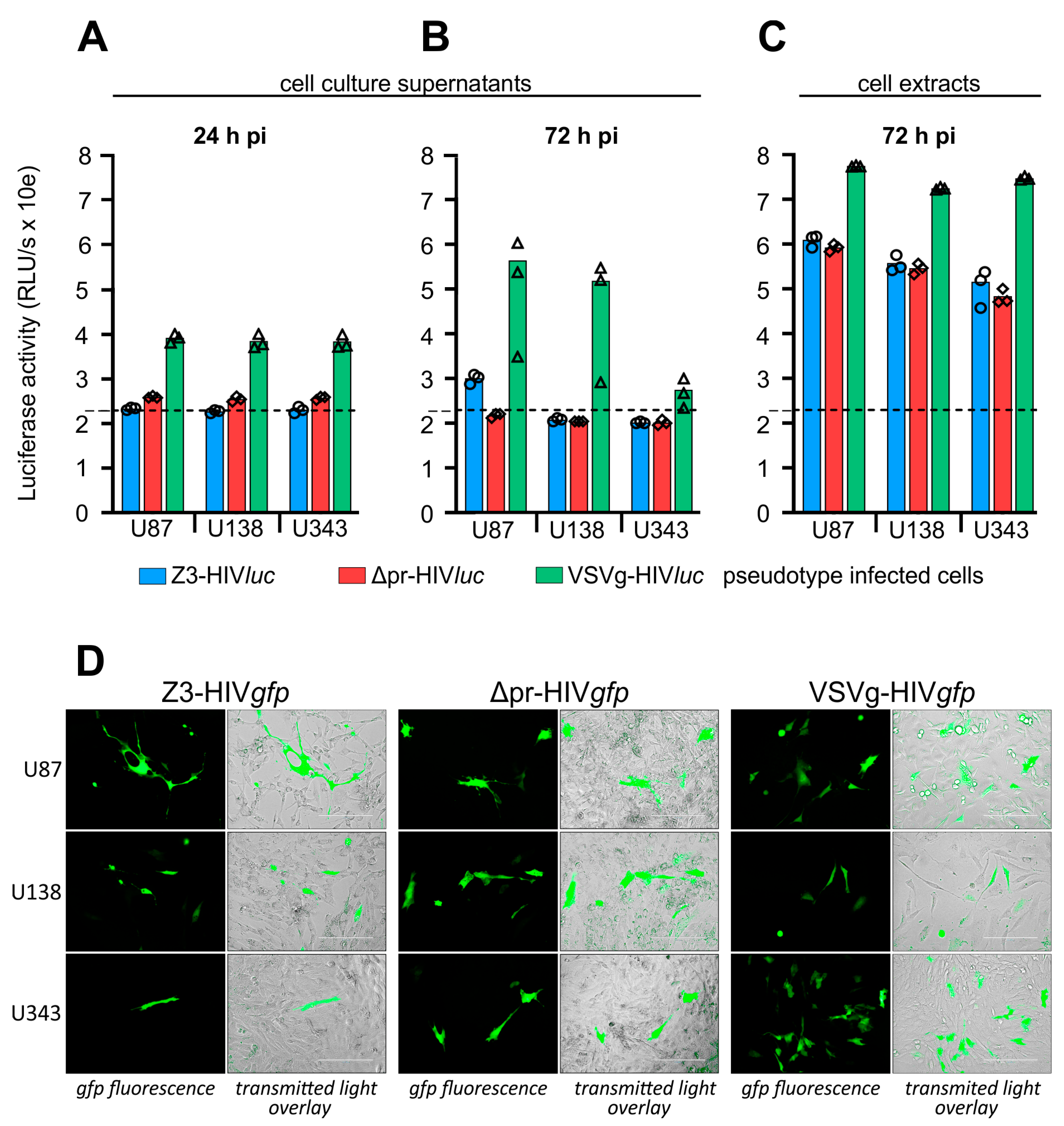
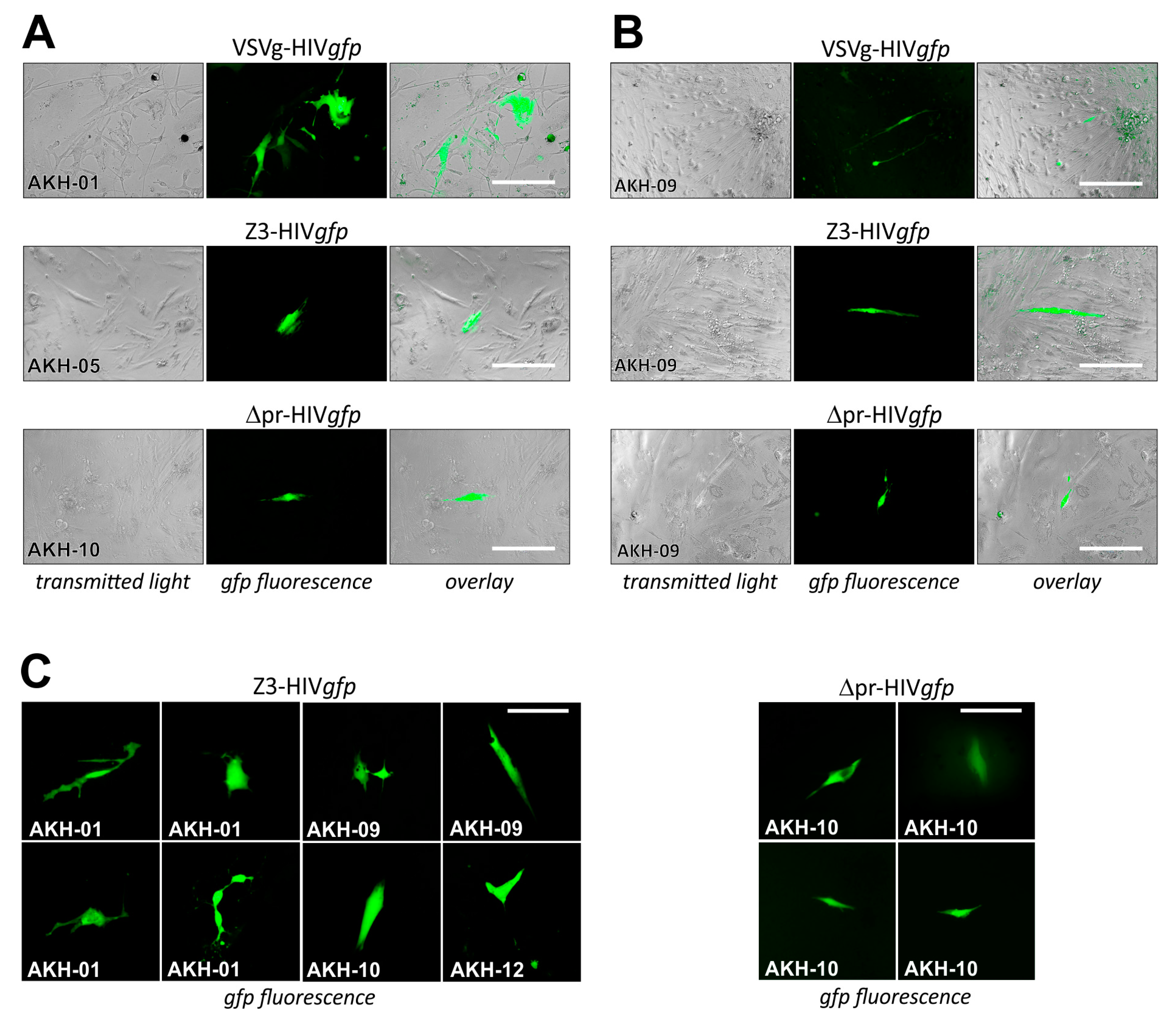
2.3. Infection of Glioma Cell Lines and AKH Tumor Cells with ZIKV-HIV Pseudotypes
3. Discussion
3.1. Isolation of Tumor Cells from Glioblastoma Multiforme Grade 4 Tissue Samples
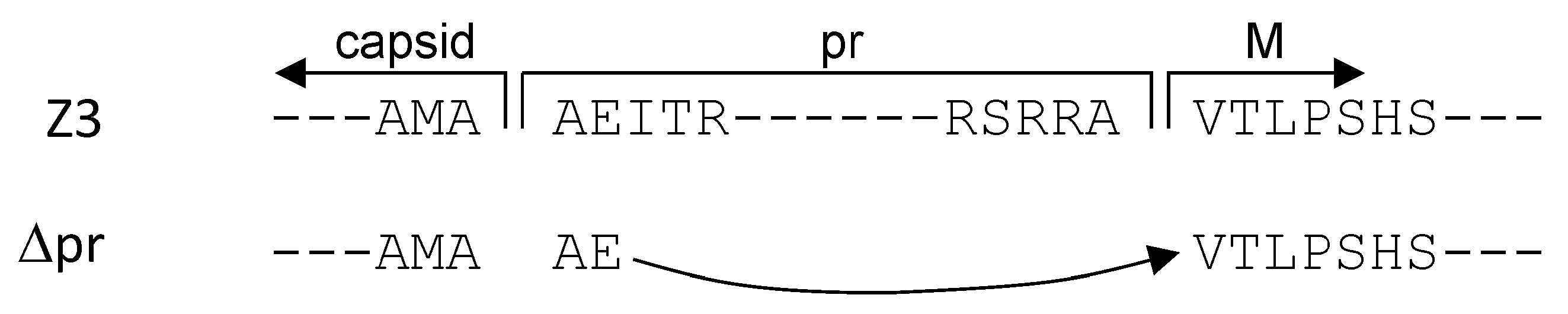
3.2. prME and ME Pseudotyped HIV-1 Particles for Tumor Cell Infections
4. Materials and Methods
4.1. Tumor Specimen
4.2. Preparation of Single-Cell Suspension from Tissue Samples
4.3. Cultivation of Tumor Cells
4.4. Immunostaining
4.5. Production of HIVluc and HIVgfp Pseudotype Particles
4.6. Infection of Tumor Cells by Pseudotype Particles
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From Molecular Pathology to Targeted Treatment. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma Stem-like Cells Give Rise to Tumour Endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Gilg, A.G.; Tye, S.L.; Tolliver, L.B.; Wheeler, W.G.; Visconti, R.P.; Duncan, J.D.; Kostova, F.V.; Bolds, L.N.; Toole, B.P.; Maria, B.L. Targeting Hyaluronan Interactions in Malignant Gliomas and Their Drug-Resistant Multipotent Progenitors. Clin. Cancer Res. 2008, 14, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Dey, M.; Ulasov, I.V.; Tyler, M.A.; Sonabend, A.M.; Lesniak, M.S. Cancer Stem Cells: The Final Frontier for Glioma Virotherapy. Stem Cell Rev. Rep. 2011, 7, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Park, D.M.; Rich, J.N. Biology of Glioma Cancer Stem Cells. Mol. Cells 2009, 28, 7–12. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2009, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2009, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Iannolo, G.; Conticello, C.; Memeo, L.; De Maria, R. Apoptosis in Normal and Cancer Stem Cells. Crit. Rev. Oncol. Hematol. 2008, 66, 42–51. [Google Scholar] [CrossRef]
- Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017, 37, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, J.P.; Chaichana, K.L.; Rincon-Torroella, J.; Quinones-Hinojosa, A. The Value of Extent of Resection of Glioblastomas: Clinical Evidence and Current Approach. Curr. Neurol. Neurosci. Rep. 2015, 15, 517. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Kirtane, A.R.; Miller, J.J.; Mazdiyasni, H.; Rogner, J.; Tai, T.; Williams, E.A.; Higuchi, F.; Juratli, T.A.; Tateishi, K.; et al. Genotype-Targeted Local Therapy of Glioma. Proc. Natl. Acad. Sci. USA 2018, 115, E8388–E8394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgöz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Döring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem. Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Lee, Y.; Shin, K.; Koo, H.; Kim, D.; Sa, J.K.; Cho, H.J.; mi Shin, H.; Lee, S.J.; Kim, H.; et al. Mutation-Specific Non-Canonical Pathway of PTEN as a Distinct Therapeutic Target for Glioblastoma. Cell Death Disease 2021, 12, 374. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular Stomatitis Virus G Glycoprotein Pseudotyped Retroviral Vectors: Concentration to Very High Titer and Efficient Gene Transfer into Mammalian and Nonmammalian Cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL Receptor Is Required for Zika Virus Strain MR-766 Infection in Human Glioblastoma Cell Lines. Mol. Oncolytics. 2021, 23, 447–457. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, R.M.R.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 Silencing in Glioblastoma Tumor-Initiating Cells Causes Stop of Proliferation and Loss of Tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, Q.; Tiwari, S.K.; Lichinchi, G.; Yau, E.H.; Hui, H.; Li, W.; Furnari, F.; Rana, T.M. Integrin Avβ5 Internalizes Zika Virus during Neural Stem Cells Infection and Provides a Promising Target for Antiviral Therapy. Cell Rep. 2020, 30, 969–983.e4. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, M.; Kadlubowska, P.; Hoffmann, D.; Schwalbe, B.; Auerswald, H.; Schreiber, M. Zikavirus Pr ME Envelope Pseudotyped Human Immunodeficiency Virus Type-1 as a Novel Tool for Glioblastoma-Directed Virotherapy. Cancers 2020, 12, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture Conditions Defining Glioblastoma Cells Behavior: What Is the Impact for Novel Discoveries? Oncotarget 2017, 8, 69185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, R.; Robert Snodgrass, S.; Johanson, C.E. A Balanced View of the Cerebrospinal Fluid Composition and Functions: Focus on Adult Humans. Exp. Neurol. 2015, 273, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñones-Hinojosa, A.; Sanai, N.; Gonzalez-Perez, O.; Garcia-Verdugo, J.M. The Human Brain Subventricular Zone: Stem Cells in This Niche and Its Organization. Neurosurg. Clin. N. Am. 2007, 18, 15–20. [Google Scholar] [CrossRef]
- Izsak, J.; Seth, H.; Theiss, S.; Hanse, E.; Illes, S. Stem Cell Reports Article Human Cerebrospinal Fluid Promotes Neuronal Circuit Maturation of Human Induced Pluripotent Stem Cell-Derived 3D Neural Aggregates. Stem Cell Rep. 2020, 14, 1044–1059. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, H.; Yuan, Y.; Yu, J.S.; Lou, Z.; Xue, Y.; Liu, Y. The Necessity for Standardization of Glioma Stem Cell Culture: A Systematic Review. Stem Cell Res. 2020, 11, 84. [Google Scholar] [CrossRef] [Green Version]
- Brewer, G.J.; Torricelli, J.R.; Evege, E.K.; Price, P.J. Optimized Survival of Hippocampal Neurons in B27-Supplemented Neurobasal, a New Serum-Free Medium Combination. J. Neurosci. Res. 1993, 35, 567–576. [Google Scholar] [CrossRef]
- Bardy, C.; van den Hurk, M.; Eames, T.; Marchand, C.; Hernandez, R.V.; Kellogg, M.; Gorris, M.; Galet, B.; Palomares, V.; Brown, J.; et al. Neuronal Medium That Supports Basic Synaptic Functions and Activity of Human Neurons in Vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E2725–E2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiber, H. Dynamics of Brain-Derived Proteins in Cerebrospinal Fluid. Clin. Chim. Acta 2001, 310, 173–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denysenko, T.; Gennero, L.; Roos, M.A.; Melcarne, A.; Juenemann, C.; Faccani, G.; Morra, I.; Cavallo, G.; Reguzzi, S.; Pescarmona, G.; et al. Glioblastoma Cancer Stem Cells: Heterogeneity, Microenvironment and Related Therapeutic Strategies. Cell Biochem. Funct. 2010, 28, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Klein-Soyer, C.; Hemmendinger, S.; Cazenave, J.P. Culture of Human Vascular Endothelial Cells on a Positively Charged Polystyrene Surface, Primaria: Comparison with Fibronectin-Coated Tissue Culture Grade Polystyrene. Biomaterials 1989, 10, 85–90. [Google Scholar] [CrossRef]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma Cell Motility Is Associated with Reduced Transcription of Proapoptotic and Proliferation Genes: A CDNA Microarray Analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef]
- Holland, J.; Hersh, L.; Bryhan, M.; Onyiriuka, E.; Ziegler, L. Culture of Human Vascular Endothelial Cells on an RGD-Containing Synthetic Peptide Attached to a Starch-Coated Polystyrene Surface: Comparison with Fibronectin-Coated Tissue Grade Polystyrene. Biomaterials 1996, 17, 2147–2156. [Google Scholar] [CrossRef]
- Inda, M.d.M.; Bonavia, R.; Seoane, J. Glioblastoma Multiforme: A Look Inside Its Heterogeneous Nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Retallack, H.; di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika Virus Cell Tropism in the Developing Human Brain and Inhibition by Azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [Green Version]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.F.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.v.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin Avβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Jurado, K.A. Playing Favorites: Integrin Avβ5 Mediates Preferential Zika Infection of Neural Stem Cells. Cell Stem Cell 2020, 26, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mao, Y.; Li, Q.; Qiu, Z.; Li, J.; Li, X.; Liang, W.; Xu, M.; Li, A.; Cai, X.; et al. Efficient Gene Transfer to Kidney Using a Lentiviral Vector Pseudotyped with Zika Virus Envelope Glycoprotein. Hum. Gene. 2022, 33, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Jiménez, F.; Pérez-Olais, J.H.; Raymond, C.; King, B.J.; McClure, C.P.; Urbanowicz, R.A.; Ball, J.K. Challenges on the Development of a Pseudotyping Assay for Zika Glycoproteins. J. Med. Microbiol. 2021, 70, 001413. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Popova, E.; Göttlinger, H.G. Nef Can Enhance the Infectivity of Receptor-Pseudotyped Human Immunodeficiency Virus Type 1 Particles. J. Virol. 2008, 82, 10811–10819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.P.; Hsieh, S.C.; King, C.C.; Wang, W.K. Characterization of Retrovirus-Based Reporter Viruses Pseudotyped with the Precursor Membrane and Envelope Glycoproteins of Four Serotypes of Dengue Viruses. Virology 2007, 368, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Ketloy, C.; Keelapang, P.; Prompetchara, E.; Suphatrakul, A.; Puttikhunt, C.; Kasinrerk, W.; Konishi, E.; Sittisombut, N.; Ruxrungtham, K. Strategies to Improve the Immunogenicity of PrM+E Dengue Virus Type-2 DNA Vaccine. Asian Pac. J. Allergy Immunol. 2017, 35, 11–19. [Google Scholar] [CrossRef]
- Op De Beeck, A.; Rouillé, Y.; Caron, M.; Duvet, S.; Dubuisson, J. The Transmembrane Domains of the PrM and E Proteins of Yellow Fever Virus Are Endoplasmic Reticulum Localization Signals. J. Virol. 2004, 78, 12591–12602. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, T.A.; Li, L.; Park, E. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 2017, 33, 369–390. [Google Scholar] [CrossRef]
- Juan, T.; Fürthauer, M. Biogenesis and Function of ESCRT-Dependent Extracellular Vesicles. Semin. Cell Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef]
- Bressan, R.B.; Dewari, P.S.; Kalantzaki, M.; Gangoso, E.; Matjusaitis, M.; Garcia-Diaz, C.; Blin, C.; Grant, V.; Bulstrode, H.; Gogolok, S.; et al. Efficient CRISPR/Cas9-Assisted Gene Targeting Enables Rapid and Precise Genetic Manipulation of Mammalian Neural Stem Cells. Development 2017, 144, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, C.M.; Ding, Y.; Hoellerbauer, P.; Davis, R.J.; Basom, R.; Girard, E.J.; Lee, E.; Corrin, P.; Hart, T.; Bolouri, H.; et al. Genome-Wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 2015, 13, 2425–2439. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.; Ohgaki, H.; Wiestler, O.; Kleihues, P.; Ellison, D. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacheny, A.; Ben, W.; Mostapha, E.; Mellies, N.; De Longueville, F. Reliable and Robust Animal-Component-Free HMSC-BM Expansion on Ready-to-Use Eppendorf CCCadvanced ® FN1 Motifs Surface. Eppendorf 2020, 390, 1–12. [Google Scholar]
- Bellis, S.L. Advantages of RGD Peptides for Directing Cell Association with Biomaterials. Biomaterials 2011, 32, 4205–4210. [Google Scholar] [CrossRef] [Green Version]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD Modified Polymers: Biomaterials for Stimulated Cell Adhesion and Beyond. Biomaterials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | MGMT- Promotor 1 | MIB-1 2 | GFAP 3 | Pretherapy | Sample Date | Isolated Tumor Cells Growth Rate 4 p53 S100 | ||
|---|---|---|---|---|---|---|---|---|
| P-01 | not methylated | >40% | positive | none | 6/21 | ++6 | <10% | <10% |
| P-02 | hypermethylation | 20% | positive | none | 6/21 | + | nd | nd |
| P-03 | not methylated | 12% | positive | Stupp 5 | 6/21 | ++ | nd | nd |
| P-04 | not methylated | >30% | positive | mitoxantrone | 7/21 | + | nd | nd |
| P-05 | not methylated | 30% | positive | none | 7/21 | +++6 | <10% | <10% |
| P-06 | hypermethylation | 30% | positive | Stupp | 7/21 | - | nd | nd |
| P-07 | not methylated | 30% | positive | Stupp | 7/21 | + | nd | nd |
| P-08 | hypermethylation | 10% | positive | Stupp | 7/21 | + | nd | nd |
| P-09 | hypermethylation | 40% | positive | Stupp | 8/21 | ++++6 | 80% | <10% |
| P-10 | hypermethylation | 40% | positive | Stupp | 8/21 | +++6 | 50% | <10% |
| P-11 | not methylated | >30% | positive | none | 9/21 | +++ | nd | nd |
| P-12 | hypermethylation | 20% | positive | none | 9/21 | ++++6 | <10% | <10% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pöhlking, C.; Beier, S.; Formanski, J.P.; Friese, M.; Schreiber, M.; Schwalbe, B. Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1. Int. J. Mol. Sci. 2023, 24, 4467. https://doi.org/10.3390/ijms24054467
Pöhlking C, Beier S, Formanski JP, Friese M, Schreiber M, Schwalbe B. Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1. International Journal of Molecular Sciences. 2023; 24(5):4467. https://doi.org/10.3390/ijms24054467
Chicago/Turabian StylePöhlking, Celine, Sebastian Beier, Jan Patrick Formanski, Michael Friese, Michael Schreiber, and Birco Schwalbe. 2023. "Isolation of Cells from Glioblastoma Multiforme Grade 4 Tumors for Infection with Zika Virus prME and ME Pseudotyped HIV-1" International Journal of Molecular Sciences 24, no. 5: 4467. https://doi.org/10.3390/ijms24054467