Abstract
Phage therapy is now seen as a promising way to overcome the current global crisis in the spread of multidrug-resistant bacteria. However, phages are highly strain-specific, and in most cases one will have to isolate a new phage or search for a phage suitable for a therapeutic application in existing libraries. At an early stage of the isolation process, rapid screening techniques are needed to identify and type potential virulent phages. Here, we propose a simple PCR approach to differentiate between two families of virulent Staphylococcus phages (Herelleviridae and Rountreeviridae) and eleven genera of virulent Klebsiella phages (Przondovirus, Taipeivirus, Drulisvirus, Webervirus, Jiaodavirus, Sugarlandvirus, Slopekvirus, Jedunavirus, Marfavirus, Mydovirus and Yonseivirus). This assay includes a thorough search of a dataset comprising S. aureus (n = 269) and K. pneumoniae (n = 480) phage genomes available in the NCBI RefSeq/GenBank database for specific genes that are highly conserved at the taxonomic group level. The selected primers showed high sensitivity and specificity for both isolated DNA and crude phage lysates, which permits circumventing DNA purification protocols. Our approach can be extended and applied to any group of phages, given the large number of available genomes in the databases.
1. Introduction
The discovery of antibiotics marked a revolution in healthcare, changing the treatment of infectious diseases for many years to come. However, the emergence and rapid spread of multidrug-resistant (MDR) bacteria has become one of the major threats to public health in the twenty-first century. Based on a comprehensive assessment of the global burden of antimicrobial resistance (AMR), in 2019 AMR was shown to be the third leading cause of death [1]. The usefulness of antibiotics is waning, and it is possible that by the year 2050, MDR infections will be the leading cause of death worldwide [2]. Moreover, the following species pose the greatest danger, among which the proportion of MDR strains exceeds 50%: Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Staphylococcus spp., Enterococcus spp., Acinetobacter baumannii, and Pseudomonas aeruginosa [1].
One of the most promising approaches for overcoming the current global crisis of antimicrobial resistance is the usage of (bacterio)phages (viruses that kill bacteria) to treat infections caused by MDR strains. Phage therapy has been shown to be safe and effective against a wide range of bacterial pathogens in both preclinical and clinical studies [3,4,5,6]. However, due to the high specificity of phage–bacteria interactions, in most cases it is necessary to isolate a new phage or to search for a suitable one in existing phage libraries [7].
At the same time, it is important to emphasize that phages are living antimicrobials, which imposes particular requirements on their therapeutic use; specifically, only virulent phages (the reproduction of these bacteriophages always leads to cell lysis; unable to integrate into the genome of an infected bacterium) can be used clinically [8,9]. Whole-genome sequencing (WGS) is gradually introduced to confirm the virulent nature of the phage: knowing the entire phage nucleotide sequence makes it possible to check the genome for the presence of recombination or transposition gene modules (responsible for recombination and/or integration of the phage genome into the host cell genome), toxins, antibiotic resistance genes, and virulence factors [9,10]. Furthermore, the ongoing systematization of information about taxa suitable for therapeutic needs is a result of the active improvement of virus taxonomy, aided by the widespread use of WGS.
A universal hierarchical taxonomic system is being maintained and developed by the International Committee on Taxonomy of Viruses (ICTV). Genomic characteristics are now acknowledged to be the fundamental component of taxonomic classification [11,12]. Several approaches, such as computing genome-wide sequence similarities (ViPTree) [13], network-based genome taxonomy (vConTACT2) [14], and many others, are proposed for viral taxonomic classification. Although WGS-based methods are becoming more and more efficient, they remain rather time-consuming and labor-intensive, especially in the case of emergency selection of individualized phage cocktails. Therefore, the development of a cheaper and faster approach for primary screening is an urgent task for typing existing and de novo isolated phages.
To date, several approaches for phage typing have been proposed. Although alternative methods have been described [15,16], the majority of the studies on phage typing rely on the presence of molecular markers (signature genes) [17,18,19,20]. Furthermore, a significant portion of existing methods is devoted to the typing of temperate bacteriophages [19,20,21,22], which is connected with the need to type bacterial strains. Significantly fewer studies are concerned with the typing of virulent phages; such typing schemes have been proposed only for Salmonella enterica, Escherichia coli and Erwinia amylovora [17].
Here, we propose a novel PCR-based typing scheme for S. aureus and K. pneumoniae phages. To develop the scheme, a search for taxon-specific orthologous gene families was conducted on a dataset comprising n = 749 phage genomes. This assay led to the selection of n = 13 primer pairs that could identify two families of virulent Staphylococcus phages (Herelleviridae and Rountreeviridae) and 11 genera of virulent Klebsiella phages (Przondovirus, Taipeivirus, Drulisvirus, Webervirus, Jiaodavirus, Sugarlandvirus, Slopekvirus, Jedunavirus, Marfavirus, Mydovirus and Yonseivirus). The assessment of sensitivity and specificity of bacteriophages and bacteria from laboratory collections showed that the scheme is applicable both for isolated DNA and for crude phage lysates.
2. Results
2.1. Sample Collection and Phylogenetic Analysis
To develop a typing scheme, all available S. aureus (n = 269) and K. pneumoniae (n = 480) phage genomes in the NCBI RefSeq/GenBank database were used (Table S1A,B). Out of these, ~53% of Staphylococcus phages (n = 142) and ~25% of Klebsiella phages (n = 123) have been classified by the ICTV (Figure 1).
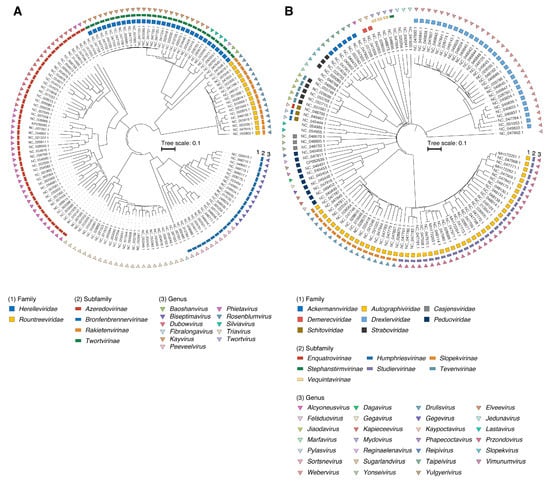
Figure 1.
Midpoint-rooted phylogenetic tree of (A) S. aureus (n = 123) and (B) K. pneumoniae (n = 142) phages based on protein distances between genomes. The three external rings indicate the family, subfamily and genus rank, respectively.
Using taxonomy derived from the ICTV database and the vConTACT2 network-based viral classification tool [14], we were able to assign taxonomy to most of the Staphylococcus and Klebsiella phage genomes retrieved from the NCBI RefSeq/GenBank database (Figures S1 and S2). In total, Staphylococcus phages belonged to 2 families, 4 subfamilies, and 11 genera. Meanwhile, a greater diversity of taxonomic groups was found among Klebsiella phages: 8 families, 7 subfamilies, and 27 genera (Table 1). For the n = 18 Staphylococcus and n = 178 Klebsiella phage genomes, genus-level classification was not assigned.

Table 1.
Taxonomy of S. aureus and K. pneumoniae bacteriophages.
2.2. Identification of Signature Genes Suitable for PCR Typing
We used the following criteria to search for taxon-specific groups of genes suitable for the development of a PCR typing scheme: ≥2 genomes at the genus level, the gene of interest is only present within members of the operational taxonomic unit (OTU), amino acid sequence identity threshold > 60% and average length > 400 bp. From 0 to 207 specific genes were identified for each OTU (Table 1 and Table S2). Phylogenetic trees inferred from protein distances between genomes and trees derived from pangenome analysis based on pairwise distance matrix from gene presence/absence data were found to be largely concordant and showed a similar clustering of OTUs (Figures S3 and S4).
Within the Staphylococcus phages, we were unable to identify specific genes that would allow us to accurately differentiate the Kayvirus, Biseptimavirus, Peeveelvirus and Fibralongavirus genera from the other most-closely related ones. However, at the Herelleviridae family level, we found n = 8 family-specific genes suitable for typing.
In contrast, for Klebsiella phages, we found specific genes for each genus, but not for families. Four genes were found to be orthologous for the Jiaodavirus and Slopekvirus genera of the Straboviridae family, but they were also found in members of the Marfavirus genus. Moreover, n = 12 genes were found to be common between the Jiaodavirus and Marfavirus genera, which indicates the close relationship between them.
2.3. PCR-Based Typing Scheme for Rapid Phage Classification
Genes were selected for typing if they allowed us to clearly distinguish among genera of virulent phages that have at least five representatives per genus (Table 1). Therefore, primers for K. pneumoniae phages were selected for the following genus-specific genes: internal virion protein D (Przondovirus), putative tail tube-associated baseplate protein (Taipeivirus), capsid protein (Drulisvirus), putative major head subunit precursor (Webervirus), alpha-glucosyl-transferase (Jiaodavirus), putative replicative DNA helicase (Sugarlandvirus), putative baseplate hub (Slopekvirus), virion structural protein (Jedunavirus), hypothetical protein (Marfavirus), putative helicase (Mydovirus) and DNA polymerase I (Yonseivirus). In Staphylococcus phages, for the Rountreeviridae and Herelleviridae families, primers were selected for the gene family encoding the resolvase and major capsid protein (Table 2, all alignments are available via Figshare).
Table 2.
Primer sets for taxon differentiation.
The sensitivity of the selected primer pairs was evaluated both on bacteriophage DNA isolated using phenol–chloroform extraction and directly on crude phage lysates. As a result of serial dilutions of the matrix, it was found that the limit of detection of PCR primers corresponds to a phage DNA concentration ranging from 150 to 1.5 pg/μL. Amplification of lysates was successful at a minimal concentration of 104–105 plaque-forming units per milliliter (PFU/mL) (Figure 2, Table S3).

Figure 2.
Polymerase chain reaction results for the S. aureus and K. pneumoniae bacteriophages. Lane 1: negative control; lanes 2–7: serially diluted bacteriophage DNA (15 ng/μL–0.15 pg/μL) as template; lane 8: DNA ladder (the ladder is composed of ten chromatography-purified individual DNA fragments (in base pairs): 1000, 900, 800, 700, 600, 500, 400, 300, 200, 100); lanes 9–14: serially diluted phage lysates (109–104 PFU/mL) as template.
Specificity of the primers was tested on non-target phage genera from the laboratory collection (n = 21) (Table S3). No nonspecific products were detected after the amplification of phages. Additionally, bacterial strains of S. aureus (n = 21) and K. pneumoniae (n = 16) were tested These strains were either hosts of the studied phages or were effectively lysed, and were considered as potential hosts. Only several reactions with K. pneumoniae strains yielded non-specific results, which appeared as faint bands that were different in size from positive controls. These results were later regarded as negative.
3. Discussion
Phages are one of the most abundant and diverse entities of the biosphere; according to some estimates, their number reaches about 1031 particles [23]. This wide diversity makes it possible to isolate a specific phage that can lyse almost any of the existing infectious bacterial strains. However, only virulent phages are suitable for phage therapy, and it is not possible to reliably determine the type of phage–bacteria interaction solely on the basis of plaque morphology. Thus, the suitability of a phage for therapy can only be established after acquiring its complete genome via WGS or other additional experiments [24].
In the case of personalized phage therapy, the most important aspect is the issue of time spent on the selection of a suitable phage, and therefore a pre-WGS screening that would be able to exclude temperate phages from the analysis would be greatly beneficial. It has been shown that phages belonging to the same taxonomic unit (genus, family) are characterized by the same type of interaction with the cell (virulent or temperate) [25], which makes it possible to take the taxonomic affiliation of phages as a basis for a rapid preliminary screening.
In addition to characterizing the type of phage-bacteria interaction, taxonomic affiliation makes it possible to determine the approximate size of the phage genome [26,27], which is also important for WGS analysis. Since the size of phage genomes varies greatly, this must be taken into account at the DNA library preparation step in order to achieve optimal coverage and high sequencing quality [27].
To create a PCR typing system, we limited ourselves to S. aureus and K. pneumoniae phages. Both bacterial species cause a wide range of infectious diseases and belong to the so-called ESKAPE (Enterococcus faecium, S. aureus, K. pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) group of pathogens, characterized by the high resistance of clinical strains to antimicrobial agents, which makes them important potential targets for phage therapy [1,4]. On the other hand, phages of these species are often well-characterized and a sufficient number of cases of their successful application have been described [4].
Staphylococcus phages are grouped into three major families: Herelleviridae (former Myoviridae), Rountreeviridae (former Podoviridae), and unclassified family (former Siphoviridae). The Klebsiella phages are classified into Ackermannviridae (former Myoviridae), Autographiviridae (former Podoviridae), Casjensviridae (former Siphoviridae), Demerecviridae (former Siphoviridae), Drexlerviridae (former Siphoviridae), Peduoviridae (former Myoviridae), Schitoviridae (former Podoviridae) and Straboviridae (former Myoviridae) families, as well as unclassified genera (formerly belonging to the families Myoviridae and Podoviridae) (Figure 1, Table 1). All known virulent S. aureus phages belong to the Herelleviridae and Rountreeviridae families [28], while temperate bacteriophages are not found in these families. Among the K. pneumoniae phages, n = 20 genera were found, comprising exclusively virulent phages. The aforementioned corroborates that the type of life cycle closely correlates with the taxonomic affiliation of phages.
Thirteen primer pairs were selected based on the gene alignments of the virulent S. aureus and K. pneumoniae phages (Table 2). In a number of cases, the genes chosen in this study, such as major capsid protein and DNA polymerase, match those previously proposed for phage typing and classification [17,29,30]. These genes play a key role in the physiology of phages and are widely distributed among various families. As previously established, their use as target genes for PCR typing schemes allows viruses to be reliably differentiated irrespective of the mosaic nature of their genome, presumably due to limited horizontal gene transfer [31]. This study made it possible to significantly expand the list of conservative genes specific to certain taxonomic units, which can be used in further phylogenetic studies (Table S2). For example, we have found eight specific genes for the family Herelleviridae, which are part of the previously described core genes of this family [32].
In silico, the selected primers showed the ability to detect all currently known S. aureus phages (Herelleviridae and Rountreeviridae families) associated with a virulent lifestyle. In the case of K. pneumoniae phages, the proposed PCR scheme is able to differentiate n = 11 from n = 20 genera of virulent bacteriophages. The vast majority of the studied virulent K. pneumoniae phages (92.4%) belong to phages from the aforementioned eleven genera.
Testing of the thirteen PCR schemes was carried out on a laboratory collection of phages, the genomes of which were not used for the primer design. Results obtained confirmed that the selected loci are conservative, and the primers have good specificity. Moreover, the limit of detection of the scheme was found to be rather high, which allows it to be used both when working with isolated DNA and with lysates when searching for a specific phage.
The proposed PCR assay has a number of limitations that must be taken into account. Although Sugarlandvirus, Slopekvirus, Jedunavirus, and Marfavirus primers showed high specificity in silico, they were not tested in vitro due to the absence of these phages in our laboratory collection. Moreover, for some primer systems, an experimental confirmation of sensitivity and specificity was carried out on a single phage, so sensitivity limits require further confirmation. Finally, it is necessary to consider the fact that several samples of K. pneumoniae showed nonspecific products, but we believe this issue can be overcome by introducing an additional control in the form of phage-free bacterial genomic DNA.
4. Materials and Methods
4.1. Genome Sampling and Annotation
A dataset comprising n = 749 complete phage genomes was retrieved from the NCBI RefSeq/GenBank database using ncbi-acc-download v.0.2.8 (https://github.com/kblin/ncbi-acc-download; accessed on 7 June 2022). Gene prediction and annotation were carried out using Prokka v.1.14.6 [33], supplemented with the pVOG [34], PHROG v.3 [35] and Caudovirales (http://s3.climb.ac.uk/ADM_share/Caudovirales.tar.gz; accessed on 7 June 2022) databases.
4.2. Phylogenetic Analysis
Trees based on pairwise distances between phage genomes were inferred using a standalone version of ViPTree v.1.1.2 [13]. For 265 genomes, the official taxonomy was derived from the ICTV Master Species List 2021.v1 (https://talk.ictvonline.org/files/master-species-lists/m/msl/13425/download; accessed on 7 June 2022). Phage genomes that were not present in the ICTV database were classified using vConTACT2 v.0.9.22 with default parameters using the “ProkaryoticViralRefSeq211-Merged” database [14] to obtain a taxonomic affiliation at the family or genus rank. Furthermore, a phage’s lifestyle (virulent or temperate) was predicted from conserved protein domains using BACPHLIP v.0.9.6 [10]. All trees were midpoint-rooted and visualized in RStudio v.2022.02.3 with R v.4.1.2 using ggtree v.3.2.1 [36], ggtreeExtra v.1.4.2 [37], ggplot2 v.3.3.5 [38], ggnewscale v.0.4.7 (https://zenodo.org/record/6385112; accessed on 7 June 2022), ggsci v.2.9 (https://github.com/nanxstats/ggsci; accessed on 7 June 2022), ggstar v.1.0.3 (https://github.com/xiangpin/ggstar; accessed on 7 June 2022), qualpalr v.0.4.3 [39] and here v.1.0.1 [40].
4.3. Pangenome Analysis and Homologues Identification
The resulting annotations in GFF3 format derived using Prokka were used for pangenome construction. Pangenome analyses were carried out using PIRATE v.1.0.4 [41] with amino acid identity thresholds of 30, 40, 50, 60, 70, 80, 90 and 95. SeqKit v.2.2.0 [42] and Seqtk v.1.3-r106 (https://github.com/lh3/seqtk; accessed on 7 June 2022) were used for handling sequences in FASTA format.
4.4. Primer Design
Genes were selected for potential use in the typing system for each operational taxonomic unit Orthologous genes with an identity of >60% and average length of >400 bp were used for further analysis. Orthologous gene family alignment was performed for each pair of genes using MAFFT v.7.490 [43] under default parameters. Primers were designed using BioEdit v.7.0.5.3 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html; accessed on 7 June 2022) and OLIGO v.6.31 (Molecular Biology Insights Inc., Cascade, CO, USA). The NCBI Primer-BLAST online tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/; accessed on 7 June 2022) was used to assess primer pair specificity [44]. The primers used in this study and PCR product lengths are listed in Table 1.
4.5. PCR Verification
Validation of the PCR typing assays was carried out on a laboratory collection of bacteriophages, including S. aureus (n = 6) bacteriophages from the families Herelleviridae and Rountreeviridae and K. pneumoniae (n = 15) bacteriophages from seven different genera (Przondovirus, Taipeivirus, Drulisvirus, Webervirus, Jiaodavirus, Mydovirus and Yonseivirus) (Table S3). A standard phenol–chloroform extraction protocol was used for phage DNA isolation [45]. DNA concentration was determined using a Qubit 2.0 spectrophotometer (Life Technologies, Darmstadt, Germany).
The standard PCR was carried out in 25 µL of the reaction mixture (66 mM Tris–HCl (pH 9.0), 16.6 mM (NH4)2SO4, 2.5 mM MgCl2, 250 µM of each dNTP, 1 U of Taq DNA polymerase (Promega, Madison, WI, USA) and 5 pmol of each primer). About 10 ng of phage DNA was used as a template for PCR. PCR was performed in a DNA Engine Tetrad (MJ Research, Inc., Saint-Bruno-de-Montarville, Quebec, Canada) at 96 °C for 2 min, followed by 35 cycles of 30 s at 95 °C, 20 s at 56 °C for Staphylococcus phage primers and 60 °C for Klebsiella phage primers, and 40 s at 72 °C. Amplification products were analyzed by 2% agarose gel electrophoresis followed by ethidium bromide staining.
The limit of detection of the PCR system was determined by serial dilutions of DNA template (15 ng/μL–0.15 pg/μL). In addition, crude phage lysates without preliminary purification, obtained by growing bacteriophages on their host strains in lysogeny broth (LB) medium, were used as a matrix. For PCR, 5 μL of lysates containing from 109 to 104 PFU/mL were taken.
PCR specificity was evaluated by cross-testing the selected primers on bacteriophages from other genera (Table S3). DNA from bacterial host strains and strains from the laboratory collection (S. aureus (n = 21) belonging to 21 multi-locus sequencing typing (MLST) sequence types (STs) and K. pneumoniae (n = 16) belonging to 16 MLST STs) were also used for testing. MLST was performed based on standard schemes [https://pubmlst.org/; accessed on 7 June 2022]. The primers for the arcC and rpoB housekeeping genes from S. aureus and K. pneumoniae, respectively, were used as positive controls.
5. Conclusions
The introduction of phage therapy in clinical practice has led to the development of criteria that a potential phage preparation must meet. The initial stage in the creation of such preparations, however, is currently rather empirical in nature, with phages that effectively lyse a certain pathogenic strain of a certain bacterial species being selected during screening.
In this work, we used generalized genomic data to create a PCR typing scheme for S. aureus and K. pneumoniae phages. This scheme could be used for a preliminary rapid screening of phages. The approach described here can be applied to other bacterial species, which will ultimately facilitate the selection of phages for further study and their application for therapeutic purposes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24054483/s1.
Author Contributions
Conceptualization, M.K., R.G. and E.S.; methodology, M.K., D.B., M.M. and E.S.; software, D.B.; validation, M.K. and E.S.; formal analysis, D.B.; investigation, M.K., D.B., M.M., N.K. and R.G.; data curation, M.K. and E.S.; writing the main manuscript text, M.K., D.B., R.G. and E.S.; visualization, D.B.; supervision, E.S.; project administration, E.S.; funding acquisition, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (research project no. 22-15-00443, https://rscf.ru/en/project/22-15-00443/).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Annotated genomes, sequence alignments, phylogenetic trees, and supplementary data are available at FigShare (https://doi.org/10.6084/m9.figshare.20337603.v1, accessed on 7 June 2022). The custom code underlying the analysis and scripts for generating the figures are available at GitHub (https://github.com/dbespiatykh/Kornienko_et_al_2022_phagePCRtyping.git, accessed on 7 June 2022).
Acknowledgments
We thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Lopukhin Federal Research and Clinical Center of Physical-Chemical Medicine of Federal Medical Biological Agency for the use the equipment.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Review on Antimicrobial Resistance. In Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; HM Government: London, UK, 2014.
- Leitner, L.; Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Chkonia, I.; Rigvava, S.; Chkhotua, A.; Changashvili, G.; McCallin, S.; Schneider, M.P.; et al. Intravesical bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: A randomised, placebo-controlled, double-blind clinical trial. Lancet Infect. Dis. 2021, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Suh, G.A.; Lodise, T.P.; Tamma, P.D.; Knisely, J.M.; Alexander, J.; Aslam, S.; Barton, K.D.; Bizzell, E.; Totten, K.M.C.; Campbell, J.L.; et al. Considerations for the Use of Phage Therapy in Clinical Practice. Antimicrob. Agents Chemother. 2022, 66, e0207121. [Google Scholar] [CrossRef] [PubMed]
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R.; Khalid, A.; Venturini, C.; Chard, R.; Morales, S.; et al. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 465–472. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Loh, B.; Leptihn, S. A Call For a Multidisciplinary Future of Phage Therapy to Combat Multi-drug Resistant Bacterial Infections. Infect. Microbes Dis. 2020, 2, 1–2. [Google Scholar] [CrossRef]
- Cui, Z.; Guo, X.; Feng, T.; Li, L. Exploring the whole standard operating procedure for phage therapy in clinical practice. J. Transl. Med. 2019, 17, 373. [Google Scholar] [CrossRef]
- Harper, D.R. Criteria for Selecting Suitable Infectious Diseases for Phage Therapy. Viruses 2018, 10, 177. [Google Scholar] [CrossRef]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Varsani, A.; Tong, Y.; Simmonds, P.; Sabanadzovic, S.; Rubino, L.; Roux, S.; Munoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on taxonomic classification of uncultivated viruses. Curr. Opin. Virol. 2021, 51, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, A.; Dahan, M.; Giesbertz, A.; Nilsson, A.; Nyberg, L.K.; Weinhold, E.; Ambjornsson, T.; Westerlund, F.; Ebenstein, Y. Bacteriophage strain typing by rapid single molecule analysis. Nucleic Acids Res. 2015, 43, e117. [Google Scholar] [CrossRef] [PubMed]
- Stverakova, D.; Sedo, O.; Benesik, M.; Zdrahal, Z.; Doskar, J.; Pantucek, R. Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry. Viruses 2018, 10, 176. [Google Scholar] [CrossRef]
- Born, Y.; Knecht, L.E.; Eigenmann, M.; Bolliger, M.; Klumpp, J.; Fieseler, L. A major-capsid-protein-based multiplex PCR assay for rapid identification of selected virulent bacteriophage types. Arch. Virol. 2019, 164, 819–830. [Google Scholar] [CrossRef]
- Moisan, M.; Moineau, S. Multilocus sequence typing scheme for the characterization of 936- like phages infecting Lactococcus lactis. Appl. Environ. Microbiol. 2012, 78, 4646–4653. [Google Scholar] [CrossRef]
- Doria, F.; Napoli, C.; Costantini, A.; Berta, G.; Saiz, J.C.; Garcia-Moruno, E. Development of a new method for detection and identification of Oenococcus oeni bacteriophages based on endolysin gene sequence and randomly amplified polymorphic DNA. Appl. Environ. Microbiol. 2013, 79, 4799–4805. [Google Scholar] [CrossRef]
- Kahankova, J.; Pantucek, R.; Goerke, C.; Ruzickova, V.; Holochova, P.; Doskar, J. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ. Microbiol. 2010, 12, 2527–2538. [Google Scholar] [CrossRef]
- Ko, D.S.; Seong, W.J.; Kim, D.; Kim, E.K.; Kim, N.H.; Lee, C.Y.; Kim, J.H.; Kwon, H.J. Molecular prophage typing of Staphylococcus aureus isolates from bovine mastitis. J. Vet. Sci. 2018, 19, 771–781. [Google Scholar] [CrossRef]
- Sanchini, A.; Del Grosso, M.; Villa, L.; Ammendolia, M.G.; Superti, F.; Monaco, M.; Pantosti, A. Typing of Panton-Valentine leukocidin-encoding phages carried by methicillin-susceptible and methicillin-resistant Staphylococcus aureus from Italy. Clin. Microbiol. Infect. 2014, 20, O840–O846. [Google Scholar] [CrossRef]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Keen, E.C.; Adhya, S.L.; Wormser, G.P. Phage Therapy: Current Research and Applications. Clin. Infect. Dis. 2015, 61, 141. [Google Scholar] [CrossRef]
- Grose, J.H.; Casjens, S.R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae. Virology 2014, 468–470, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Philipson, C.W.; Voegtly, L.J.; Lueder, M.R.; Long, K.A.; Rice, G.K.; Frey, K.G.; Biswas, B.; Cer, R.Z.; Hamilton, T.; Bishop-Lilly, K.A. Characterizing Phage Genomes for Therapeutic Applications. Viruses 2018, 10, 188. [Google Scholar] [CrossRef]
- Russell, D.A. Sequencing, Assembling, and Finishing Complete Bacteriophage Genomes. Methods Mol. Biol. 2018, 1681, 109–125. [Google Scholar] [PubMed]
- Deghorain, M.; Van Melderen, L. The staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef]
- Henry, M.; Bobay, L.-M.; Chevallereau, A.; Saussereau, E.; Ceyssens, P.-J.; Debarbieux, L. The Search for Therapeutic Bacteriophages Uncovers One New Subfamily and Two New Genera of Pseudomonas-Infecting Myoviridae. PLoS ONE 2015, 10, e0117163. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Ackermann, H.-W.; Anany, H.; Blasdel, B.; Connerton, I.F.; Goulding, D.; Griffiths, M.W.; Hooton, S.P.; Kutter, E.M.; Kropinski, A.M.; et al. A suggested new bacteriophage genus: “Viunalikevirus”. Arch. Virol. 2012, 157, 2035–2046. [Google Scholar] [CrossRef]
- Smith, K.C.; Castro-Nallar, E.; Fisher, J.N.; Breakwell, D.P.; Grose, J.H.; Burnett, S.H. Phage cluster relationships identified through single gene analysis. BMC Genom. 2013, 14, 410. [Google Scholar] [CrossRef]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.P.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Analysis of Spounaviruses as a Case Study for the Overdue Reclassification of Tailed Phages. Syst. Biol. 2020, 69, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Perez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genomics Bioinforma. 2021, 3, lqab067. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinforma. 2020, 69, e96. [Google Scholar] [CrossRef]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. ggtreeExtra: Compact Visualization of Richly Annotated Phylogenetic Data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Larsson, J. Qualpalr: Automatic Generation of Qualitative Color Palettes. R Package Version 0.4.3. Available online: https://CRAN.R-project.org/package=qualpalr (accessed on 20 July 2022).
- Muller, K. Here: A Simpler Way to Find Your Files. R Package Version 1.0.1. Available online: https://CRAN.R-project.org/package=here (accessed on 20 July 2022).
- Bayliss, S.C.; Thorpe, H.A.; Coyle, N.M.; Sheppard, S.K.; Feil, E.J. PIRATE: A fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 2019, 8, giz119. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual 2012 Edition (Final); Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).