
DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways



Abstract
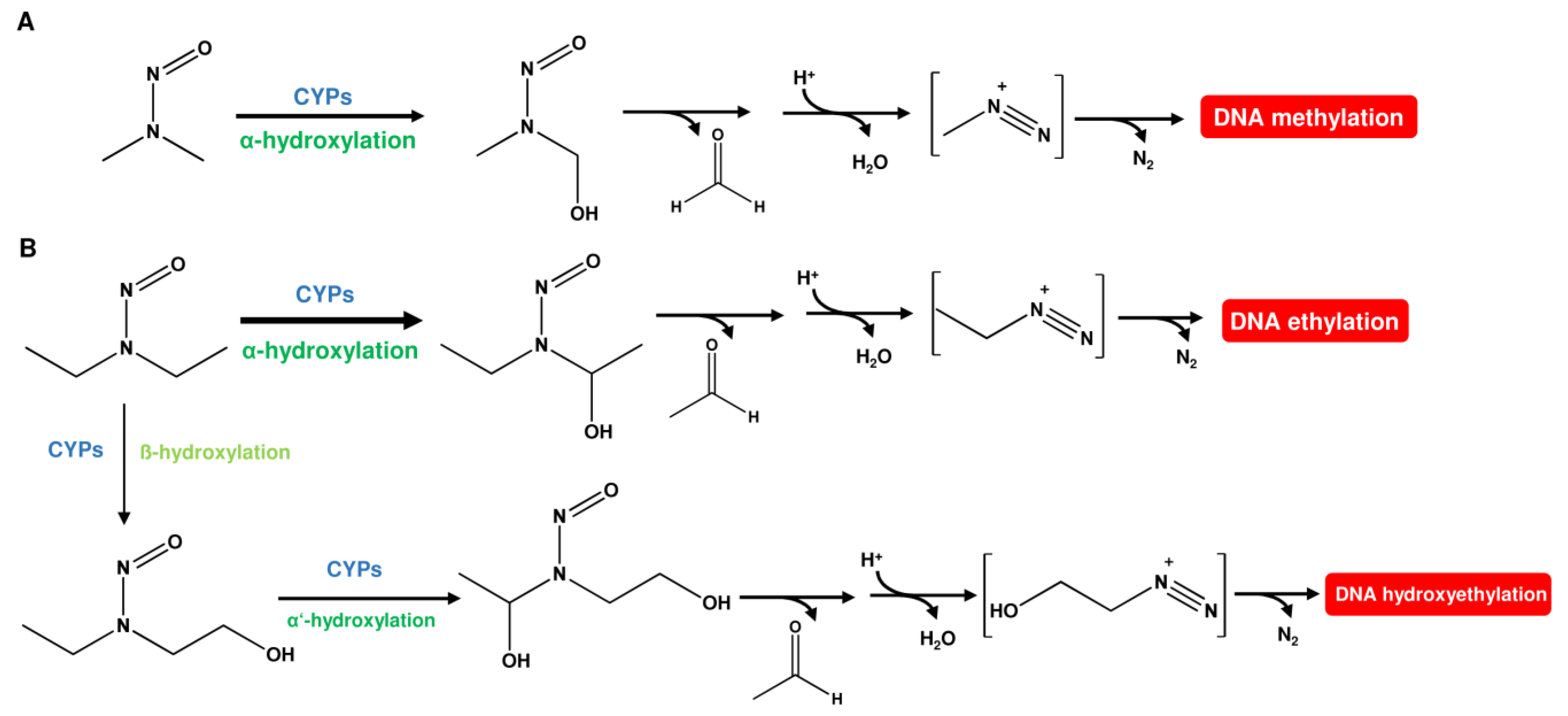
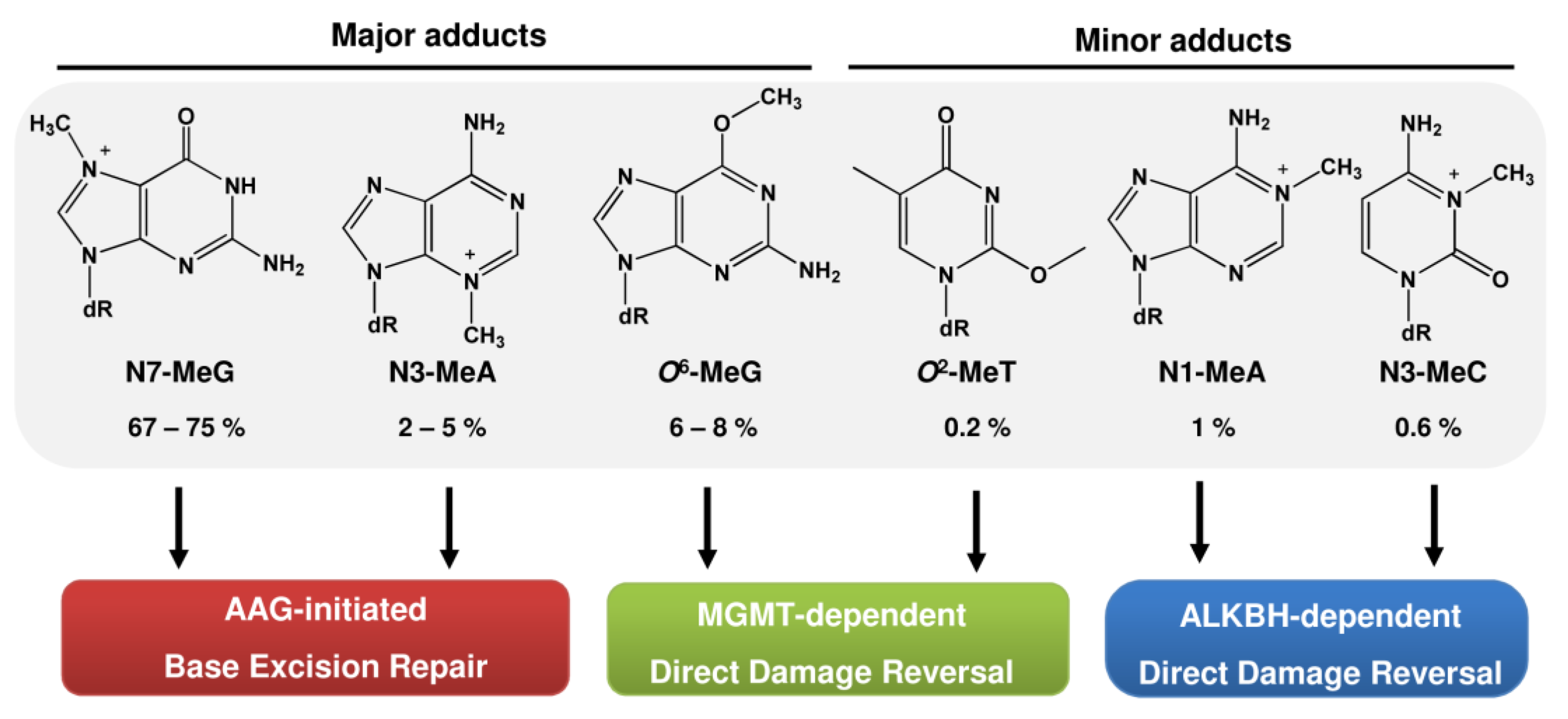
1. Sources of DNA Alkylation Damage
1.1. Nitrosamines (N-Nitrosamines)
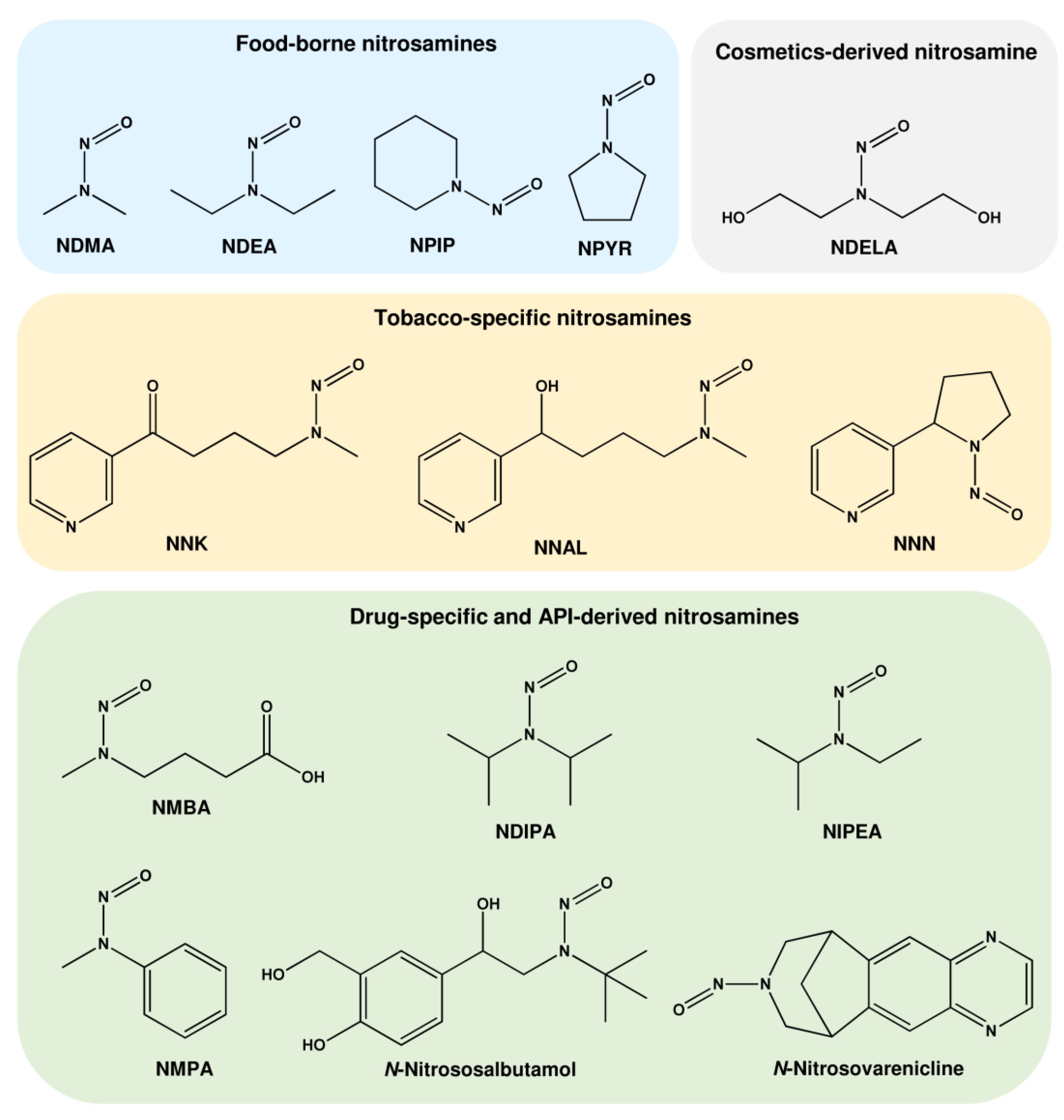
1.1.1. N-Nitrosamines as Contaminants in Food

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitrosamines | Abbreviation | Major DNA Alkylation Adducts | Sources |
|---|---|---|---|
| N-nitrosodimethylamine | NDMA | N7-MeG, N3-MeA O6-MeG, O2-MeT, O4-MeT | Food, drugs, tobacco smoke |
| N-nitrosodiethylamine | NDEA | N7-EtG, N3-EtA, O6-EtG, O2-EtT, O4-EtT | Food, drugs |
| N-nitrosopiperidine | NPIP | 7-(2-oxopropyl)-N1,N2-etheno-G, N2-(3,4,5,6-tetrahydro-2H-pyran-2-yl)-2‘-G | Food |
| N-nitrosopyrrolidine | NPYR | N7,8-ButanoG, N7-(4-Oxobutyl)-G, O4-(4-OH-Butyl)-T, and others | Food |
| N-nitrosodiethanolamine | NDELA | O6-OHEtG and others; glyoxal adducts | Cosmetics |
| N-nitroso-N-methyl-4-aminobutanoic acid | NMBA | unknown | Drugs |
| N-nitrosodiisopropylamine | NDIPA | unknown | Drugs |
| N-nitrosoethylisopropylamine | NEIPA | unknown | Drugs |
| N-nitrosomethylphenylamine | NMPA | unknown | Drugs |
| N-nitrosovarenicline | - | unknown | Drugs |
| N-nitrososalbutamol | - | unknown | Drugs |
| 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; (nicotine-derived nitrosamine ketone) | NNK | N7-MeG, N3-MeA, N3-MeG, O6-MeG, O4-MeG, O6-pobG | Tobacco smoke |
| 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol; (nicotine-derived nitrosamine alcohol) | NNAL | N7-MeG, N3-MeA, N3-MeG, O6-MeG, O4-MeG, O6-pobG | Tobacco smoke |
| N‘-nitrosonornicotine | NNN | O6-pobG | Tobacco smoke |
| N’-nitrosoanabasine | NAB | unknown | Tobacco smoke |
| N’-nitrosoanatabine | NAT | unknown | Tobacco smoke |
1.1.2. N-Nitrosamines as Impurities in Cosmetics

1.1.3. N-Nitrosamines Formed by Smoking

1.1.4. N-Nitrosamines as Impurities in Drugs
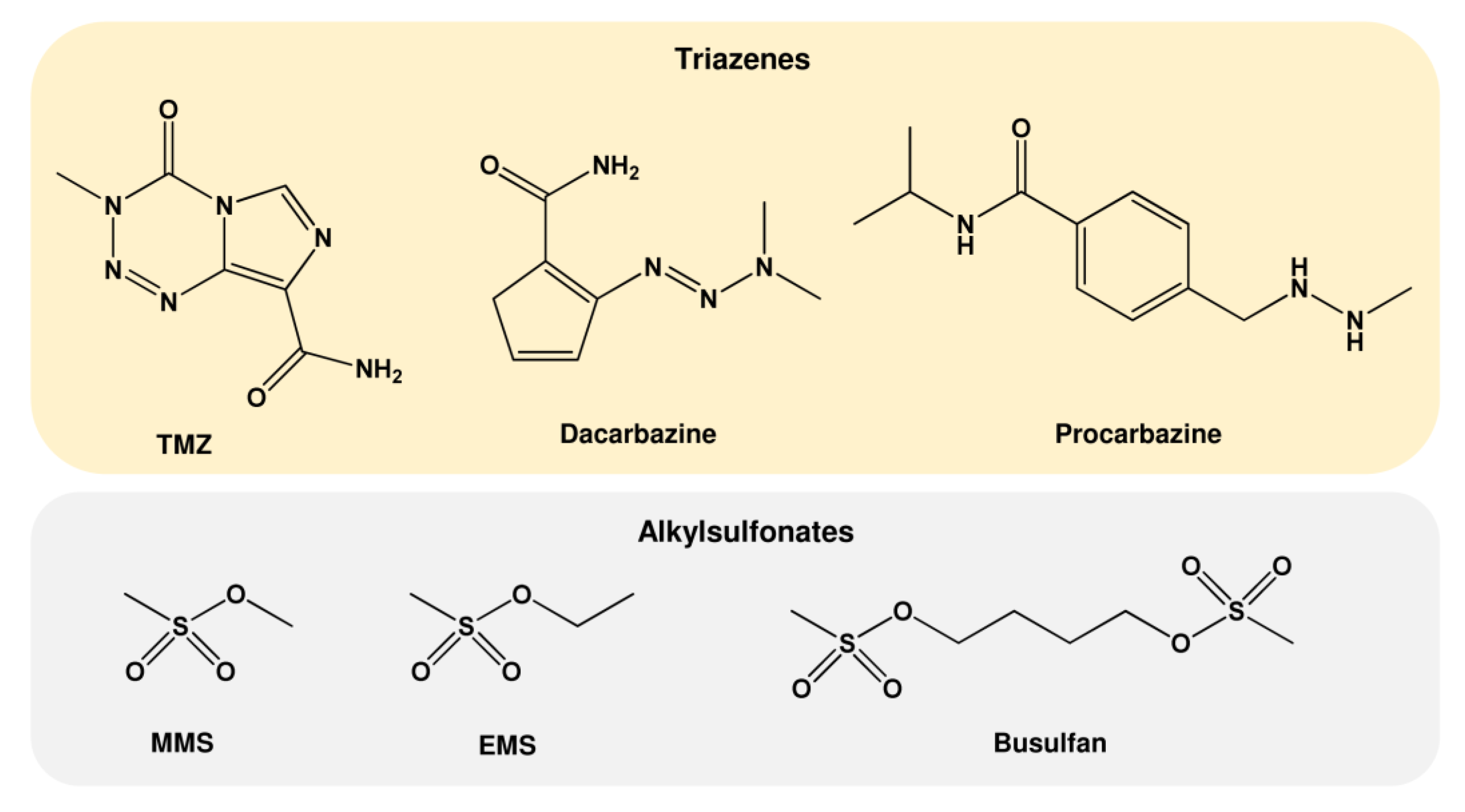
1.2. Nitrosamides (N-nitrosamides)

1.3. Further Alkylating Agents
| Further Alkylating Agents | Abbreviations | Major DNA Alkylation Adducts | Sources |
|---|---|---|---|
| Triazenes | |||
| N-Isopropyl-4-(2-methylhydrazinomethyl)benzamid | Procarbazine | N7-MeG, N3-MeA N3-MeG, O6-MeG | Anticancer drug |
| 5-(3,3-Dimethyl-1-triazenyl)imidazol-4-carboxamid | Dacarbazine | N7-MeG, N3-MeA N3-MeG, O6-MeG | Anticancer drug |
| 4-Methyl-5-oxo-2,3,4,6,8-pentazabicyclo [4.3.0]nona-2,7,9-trien-9-carboxamid | TMZ | N7-MeG, N3-MeA N3-MeG, O6-MeG | Anticancer drug |
| Alkylsulfonates Ethyl methanesulfonate Methyl methanesulfonate 4-methylsulfonyloxybutyl methanesulfonate | EMS MMS Busulfan | N7-MeG, N3-MeA N7-EtG, N3-EtA N7-MeG, N3-MeA | Basic research Basic research Anticancer drug |
2. Repair of DNA Alkylation Damage
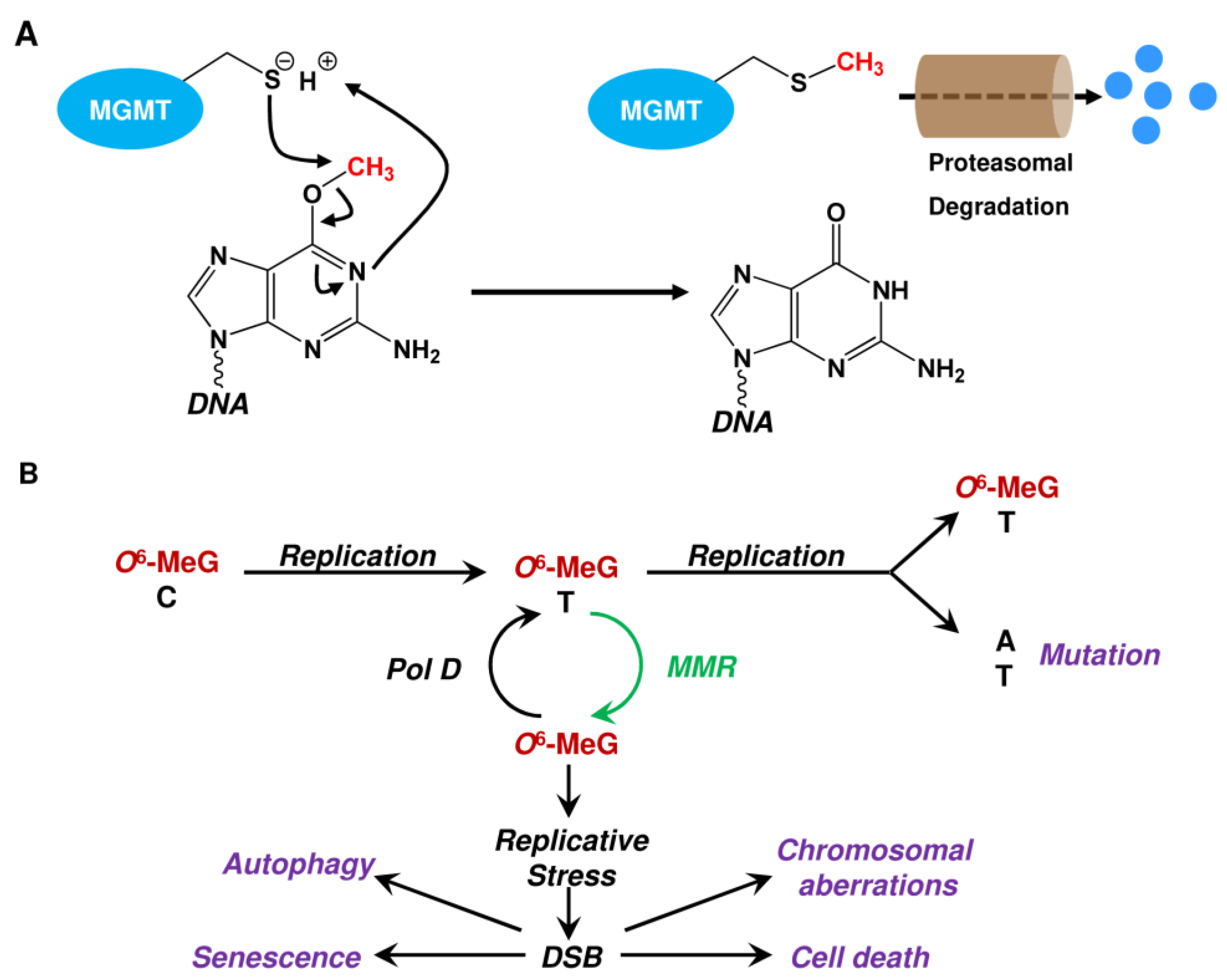
2.1. Repair by MGMT

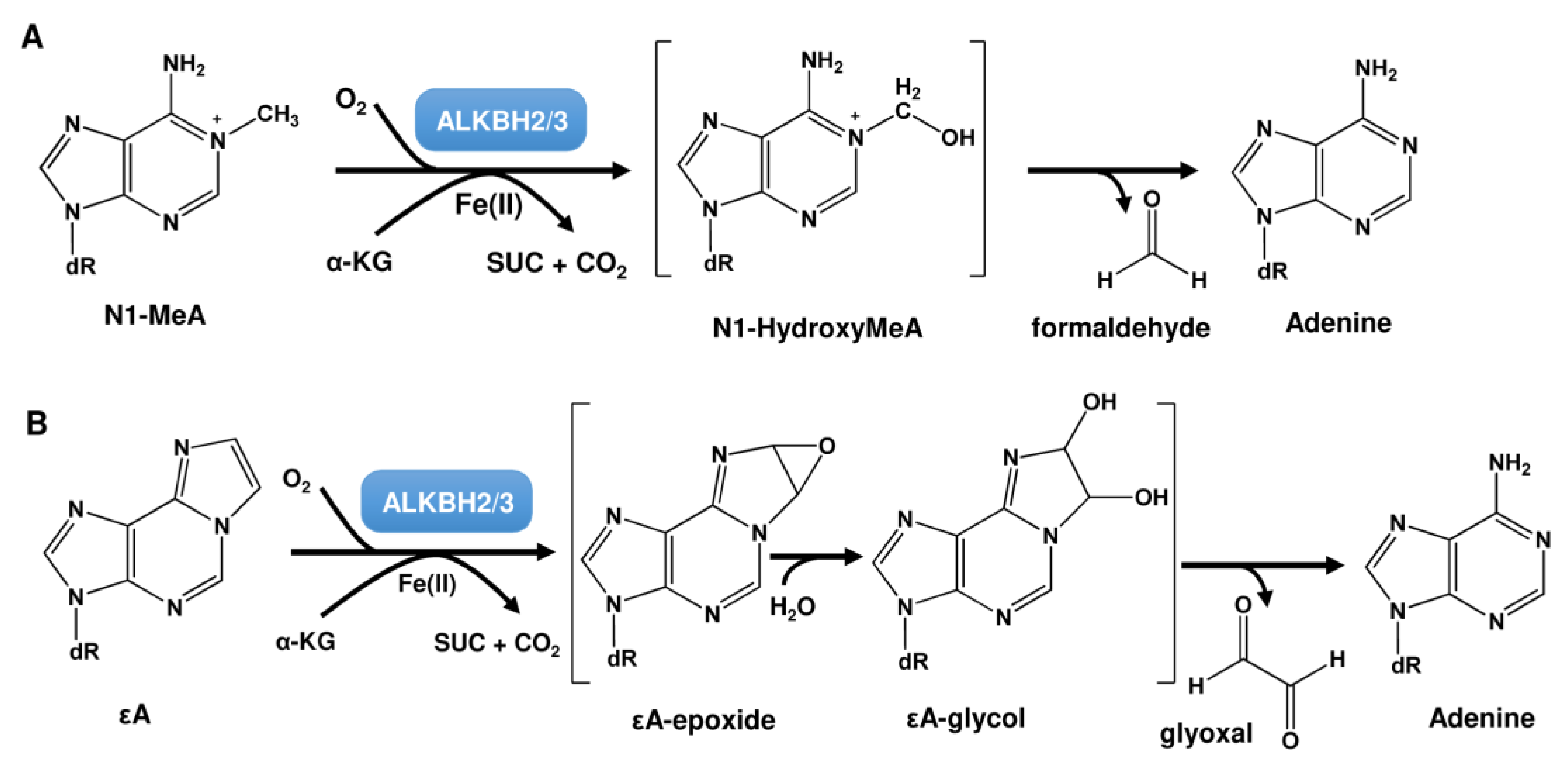
2.2. Repair by the ALKBH Family

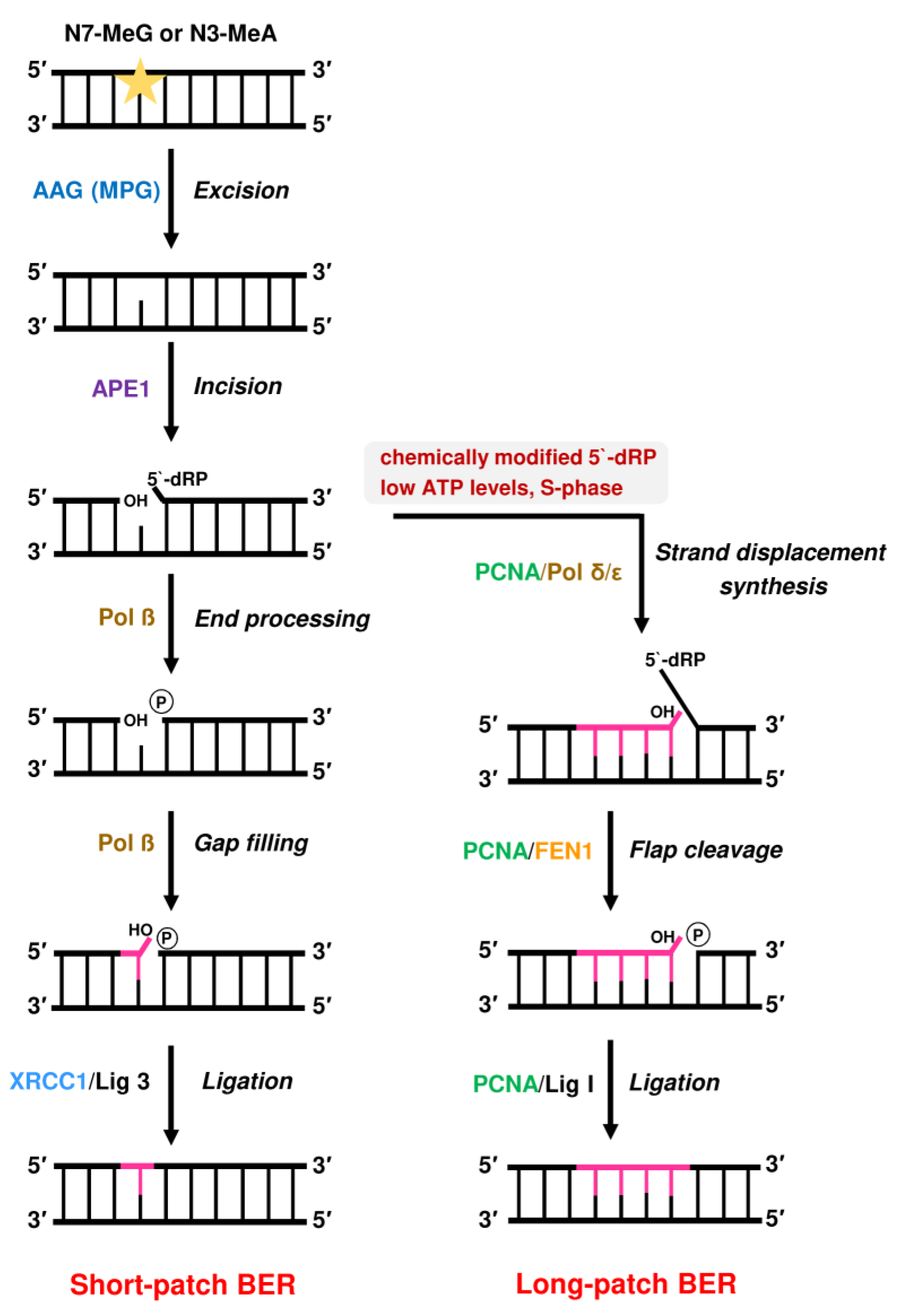
2.3. Repair by Base Excision Repair (BER) Initiated by AAG and Role of PARP-1

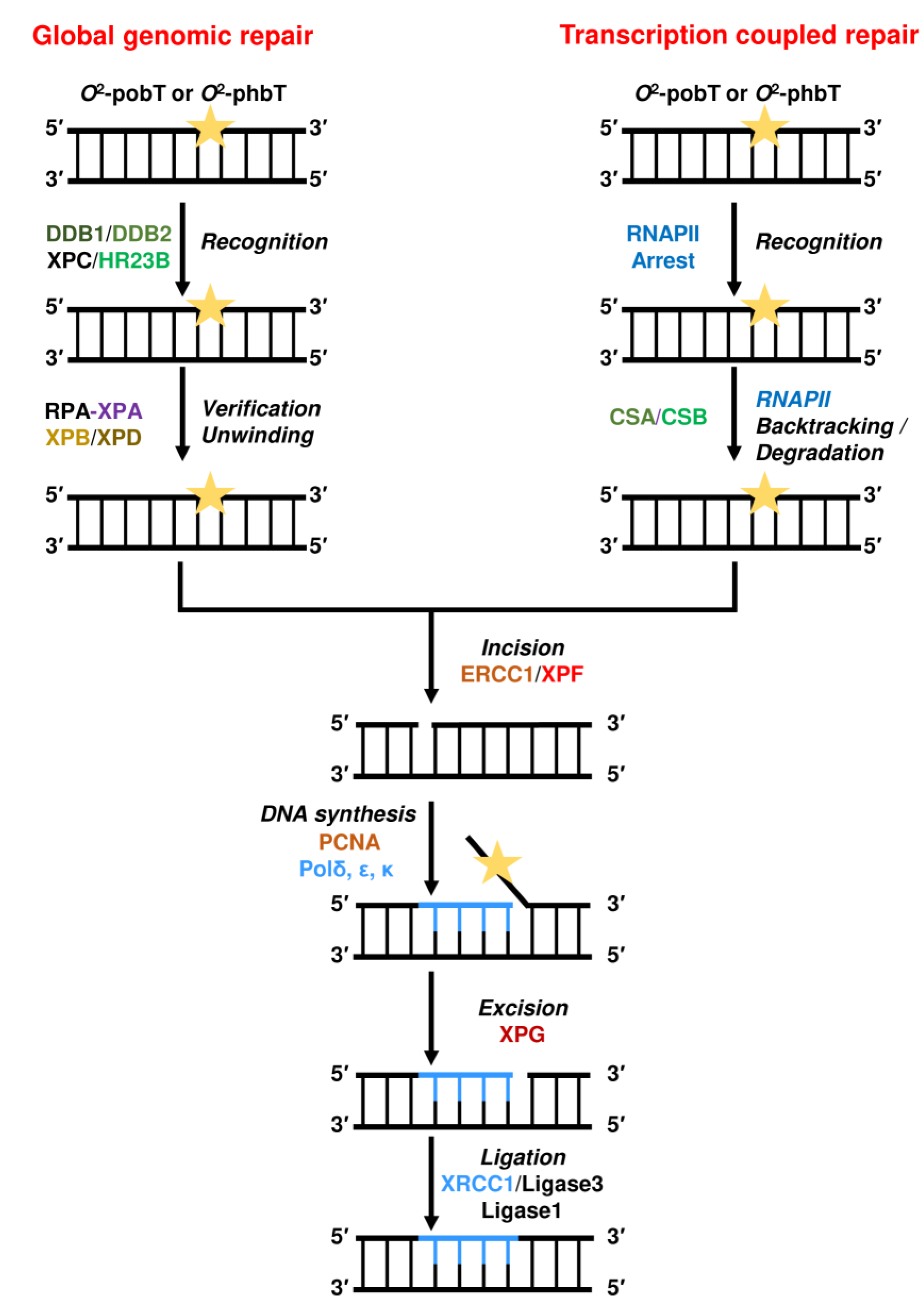
2.4. Repair by Nucleotide Excision Repair (NER)

2.5. Bypass by Translesion Synthesis (TLS)

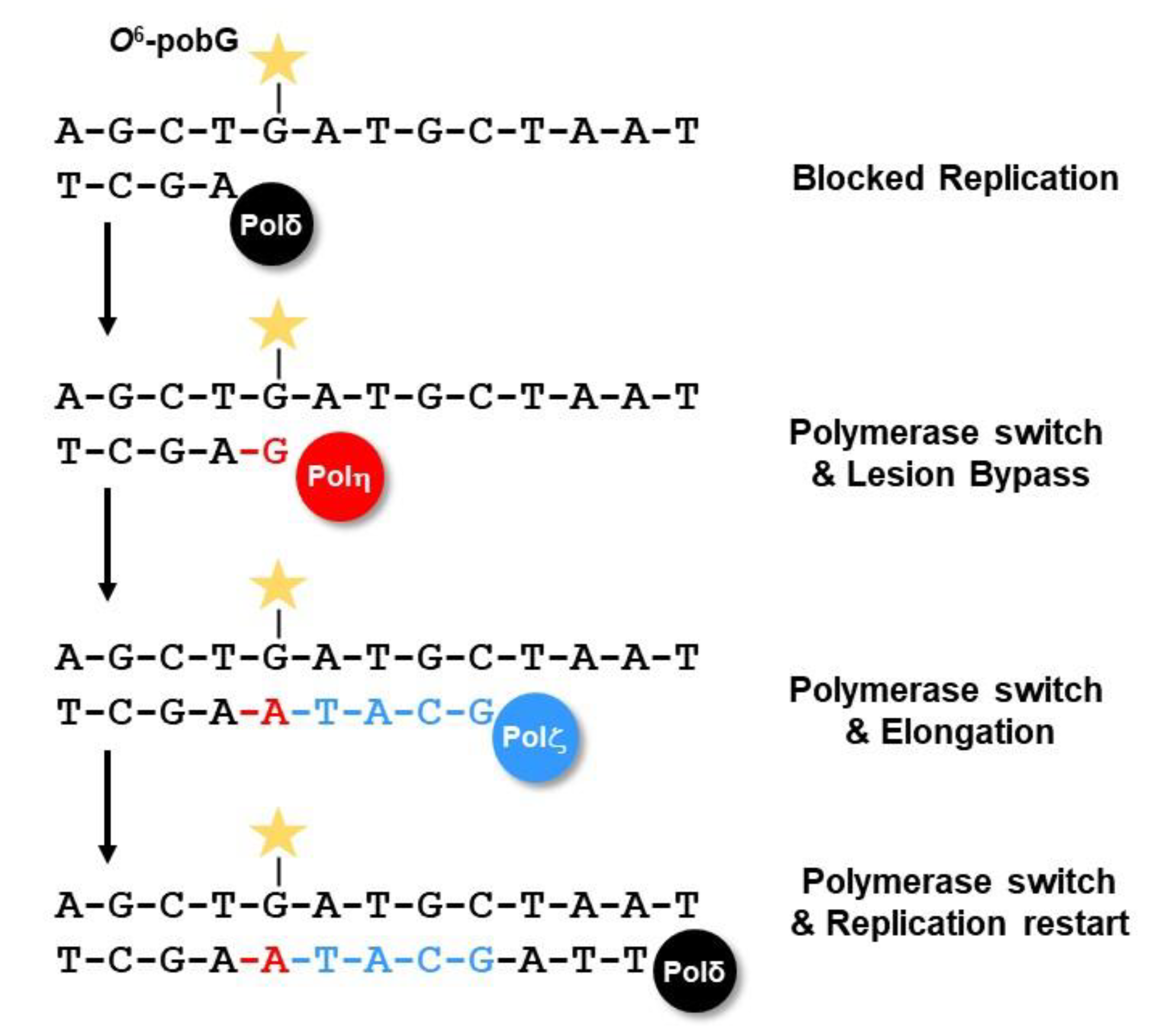
2.5.1. Bypass of O6-alkyl-dG Lesions
2.5.2. Bypass of O2/O4-alkyl-dT Lesions
2.5.3. Bypass of N-alkyl-dG Lesions
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAG | alkyladenine glycosylase |
| ALKBH | AlkB homolog |
| AP | apurinic/apyrimidinic |
| APE1 | AP endonuclease 1 |
| BER | base excision repair |
| CPDs | cyclobutane pyrimidine dimers |
| CS | Cockayne syndrome |
| CYP450 | cytochrome P450 |
| dRP | deoxyribose-5-phosphate |
| DSB | DNA double-strand break |
| ERCC1 | excision repair cross-complementing 1 |
| EXO1 | exonuclease 1 |
| FEN1 | flap endonuclease 1 |
| GG-NER | global genome NER |
| HR | homologous recombination |
| IR | ionizing radiation |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MMR | mismatch repair |
| MPG | N-methylpurine-DNA glycosylase (alias AAG) |
| NER | nucleotide excision repair |
| OGG1 | 8-oxoguanine DNA glycosylase |
| PAR | poly(ADP-ribose) |
| PARP-1 | poly(ADP-ribose) polymerase-1 |
| PCNA | proliferating cellular nuclear antigen |
| Pol | DNA polymerase |
| RFC | replication factor C |
| RNAP | RNA polymerase |
| RPA | replication protein A |
| SSB | DNA single-strand break |
| TC-NER | transcription-coupled NER |
| TFIIH | transcription factor II H |
| TLS | translesion synthesis |
| TTD | trichothiodystrophy |
| XP | xeroderma pigmentosum |
| XRCC1 | X-ray repair cross-complementing protein 1 |
| 6-4PP | pyrimidine-(6,4)-pyrimidone photoproduct |
References
- Druckrey, H.; Preussmann, R.; Ivankovic, S.; Schmahl, D. Organotropic carcinogenic effects of 65 various N-nitroso- compounds on BD rats. Z. Fur Krebsforsch. 1967, 69, 103–201. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Loechler, E.L.; Green, C.L.; Essigmann, J.M. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. USA 1984, 81, 6271–6275. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.E.; Corrigan, J.J.; Armijo, A.L.; Nazari, I.S.; Kohale, I.N.; Torous, D.K.; Avlasevich, S.L.; Croy, R.G.; Wadduwage, D.N.; Carrasco, S.E.; et al. Excision of mutagenic replication-blocking lesions suppresses cancer but promotes cytotoxicity and lethality in nitrosamine-exposed mice. Cell Rep. 2021, 34, 108864. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Taverna, P.; Sedgwick, B. Generation of an endogenous DNA-methylating agent by nitrosation in E. coli. J. Bacteriol. 1996, 178, 5105–5111. [Google Scholar] [CrossRef]
- Barrows, L.R.; Magee, P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351. [Google Scholar] [CrossRef]
- Shuker, D.E.; Margison, G.P. Nitrosated glycine derivatives as a potential source of O6-methylguanine in DNA. Cancer Res 1997, 57, 366–369. [Google Scholar]
- Busby, W.F., Jr.; Shuker, D.E.; Charnley, G.; Newberne, P.M.; Tannenbaum, S.R.; Wogan, G.N. Carcinogenicity in rats of the nitrosated bile acid conjugates N-nitrosoglycocholic acid and N-nitrosotaurocholic acid. Cancer Res. 1985, 45, 1367–1371. [Google Scholar]
- Beard, J.C.; Swager, T.M. An Organic Chemist’s Guide to N-Nitrosamines: Their Structure, Reactivity, and Role as Contaminants. J. Org. Chem. 2021, 86, 2037–2057. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Approaches to cancer prevention based on an understanding of N-nitrosamine carcinogenesis. Proc. Soc. Exp. Biol. Medicine. Soc. Exp. Biol. Med. 1997, 216, 181–191. [Google Scholar] [CrossRef]
- IARC. IARC Monographs on the Identification of Carcinogenic Hazards to Humans. Available online: https://monographs.iarc.who.int/list-of-classifications (accessed on 15 February 2023).
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res. 1999, 443, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Gushgari, A.J.; Halden, R.U. Critical review of major sources of human exposure to N-nitrosamines. Chemosphere 2018, 210, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Behsnilian, D.; Butz, P.; Greiner, R.; Lautenschlaeger, R. Process-induced undesirable compounds: Chances of non-thermal approaches. Meat Sci. 2014, 98, 392–403. [Google Scholar] [CrossRef]
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.H.; Bailey, N.; Bandaletova, T.; Bowman, R.; Cross, A.J.; Pollock, J.; Shuker, D.E.; Bingham, S.A. Red meat enhances the colonic formation of the DNA adduct O6-carboxymethyl guanine: Implications for colorectal cancer risk. Cancer Res. 2006, 66, 1859–1865. [Google Scholar] [CrossRef]
- Joosen, A.M.; Kuhnle, G.G.; Aspinall, S.M.; Barrow, T.M.; Lecommandeur, E.; Azqueta, A.; Collins, A.R.; Bingham, S.A. Effect of processed and red meat on endogenous nitrosation and DNA damage. Carcinogenesis 2009, 30, 1402–1407. [Google Scholar] [CrossRef]
- Santarelli, R.L.; Vendeuvre, J.L.; Naud, N.; Tache, S.; Gueraud, F.; Viau, M.; Genot, C.; Corpet, D.E.; Pierre, F.H. Meat processing and colon carcinogenesis: Cooked, nitrite-treated, and oxidized high-heme cured meat promotes mucin-depleted foci in rats. Cancer Prev. Res. 2010, 3, 852–864. [Google Scholar] [CrossRef]
- Seiwert, N.; Adam, J.; Steinberg, P.; Wirtz, S.; Schwerdtle, T.; Adams-Quack, P.; Hovelmeyer, N.; Kaina, B.; Foersch, S.; Fahrer, J. Chronic intestinal inflammation drives colorectal tumor formation triggered by dietary heme iron in vivo. Arch. Toxicol. 2021, 95, 2507–2522. [Google Scholar] [CrossRef]
- Yamazaki, H.; Inui, Y.; Yun, C.H.; Guengerich, F.P.; Shimada, T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 1992, 13, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Den Engelse, L.; Menkveld, G.J.; De Brij, R.J.; Tates, A.D. Formation and stability of alkylated pyrimidines and purines (including imidazole ring-opened 7-alkylguanine) and alkylphosphotriesters in liver DNA of adult rats treated with ethylnitrosourea or dimethylnitrosamine. Carcinogenesis 1986, 7, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Camus, A.M.; Geneste, O.; Honkakoski, P.; Bereziat, J.C.; Henderson, C.J.; Wolf, C.R.; Bartsch, H.; Lang, M.A. High variability of nitrosamine metabolism among individuals: Role of cytochromes P450 2A6 and 2E1 in the dealkylation of N-nitrosodimethylamine and N-nitrosodiethylamine in mice and humans. Mol. Carcinog. 1993, 7, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Verna, L.; Whysner, J.; Williams, G.M. N-nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef] [PubMed]
- von Hofe, E.; Kleihues, P.; Keefer, L.K. Extent of DNA 2-hydroxyethylation by N-nitrosomethylethylamine and N-nitrosodiethylamine in vivo. Carcinogenesis 1986, 7, 1335–1337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.; Hecht, S.S. Metabolic Activation and DNA Interactions of Carcinogenic N-Nitrosamines to Which Humans Are Commonly Exposed. Int. J. Mol. Sci. 2022, 23, 4559. [Google Scholar] [CrossRef]
- Schothorst, R.C.; Somers, H.H. Determination of N-nitrosodiethanolamine in cosmetic products by LC-MS-MS. Anal. Bioanal. Chem. 2005, 381, 681–685. [Google Scholar] [CrossRef]
- Liu, Y.; Glatt, H. Mutagenicity of N-nitrosodiethanolamine in a V79-derived cell line expressing two human biotransformation enzymes. Mutat. Res. 2008, 643, 64–69. [Google Scholar] [CrossRef]
- Loeppky, R.N.; Goelzer, P. Microsome-mediated oxidation of N-nitrosodiethanolamine (NDELA), a bident carcinogen. Chem. Res. Toxicol. 2002, 15, 457–469. [Google Scholar] [CrossRef]
- Loeppky, R.N.; Ye, Q.; Goelzer, P.; Chen, Y. DNA adducts from N-nitrosodiethanolamine and related beta-oxidized nitrosamines in vivo: (32)P-postlabeling methods for glyoxal- and O(6)-hydroxyethyldeoxyguanosine adducts. Chem. Res. Toxicol. 2002, 15, 470–482. [Google Scholar] [CrossRef]
- Hecht, S.S. Progress and challenges in selected areas of tobacco carcinogenesis. Chem. Res. Toxicol. 2008, 21, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Chen, C.B.; Hirota, N.; Ornaf, R.M.; Tso, T.C.; Hoffmann, D. Tobacco-specific nitrosamines: Formation from nicotine in vitro and during tobacco curing and carcinogenicity in strain A mice. J. Natl. Cancer Inst. 1978, 60, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Rivenson, A.; Braley, J.; DiBello, J.; Adams, J.D.; Hoffmann, D. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986, 46, 4162–4166. [Google Scholar] [PubMed]
- Hecht, S.S.; Hoffmann, D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis 1988, 9, 875–884. [Google Scholar] [CrossRef] [PubMed]
- DeVore, N.M.; Scott, E.E. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J. Biol. Chem. 2012, 287, 26576–26585. [Google Scholar] [CrossRef]
- Wong, H.L.; Zhang, X.; Zhang, Q.Y.; Gu, J.; Ding, X.; Hecht, S.S.; Murphy, S.E. Metabolic activation of the tobacco carcinogen 4-(methylnitrosamino)-(3-pyridyl)-1-butanone by cytochrome P450 2A13 in human fetal nasal microsomes. Chem. Res. Toxicol. 2005, 18, 913–918. [Google Scholar] [CrossRef]
- Jalas, J.R.; Ding, X.; Murphy, S.E. Comparative metabolism of the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol by rat cytochrome P450 2A3 and human cytochrome P450 2A13. Drug Metab. Dispos. Biol. Fate Chem. 2003, 31, 1199–1202. [Google Scholar] [CrossRef]
- Li, Y.; Hecht, S.S. Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. Int. J. Mol. Sci. 2022, 23, 5109. [Google Scholar] [CrossRef]
- Lao, Y.; Villalta, P.W.; Sturla, S.J.; Wang, M.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 674–682. [Google Scholar] [CrossRef]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N’-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Trushin, N.; Castonguay, A.; Rivenson, A. Comparative tumorigenicity and DNA methylation in F344 rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N-nitrosodimethylamine. Cancer Res. 1986, 46, 498–502. [Google Scholar] [PubMed]
- Trushin, N.; Rivenson, A.; Hecht, S.S. Evidence supporting the role of DNA pyridyloxobutylation in rat nasal carcinogenesis by tobacco-specific nitrosamines. Cancer Res. 1994, 54, 1205–1211. [Google Scholar] [PubMed]
- Peterson, L.A.; Hecht, S.S. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991, 51, 5557–5564. [Google Scholar] [PubMed]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- FDA. Laboratory Analysis of Valsartan Products. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/laboratory-analysis-valsartan-products (accessed on 21 December 2022).
- EMA. EMA Update on Metformin Diabetes Medicines. Available online: https://www.ema.europa.eu/en/news/ema-update-metformin-diabetes-medicines (accessed on 21 December 2022).
- Keire, D.A.; Bream, R.; Wollein, U.; Schmaler-Ripcke, J.; Burchardt, A.; Conti, M.; Zmyslowski, A.; Keizers, P.; Morin, J.; Poh, J.; et al. International Regulatory Collaboration on the Analysis of Nitrosamines in Metformin-Containing Medicines. AAPS J. 2022, 24, 56. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline ICH M7 (R1)—Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk; EMA: London, UK, 2015. [Google Scholar]
- Tuesuwan, B.; Vongsutilers, V. Nitrosamine Contamination in Pharmaceuticals: Threat, Impact, and Control. J. Pharm. Sci. 2021, 110, 3118–3128. [Google Scholar] [CrossRef]
- FDA. Control of Nitrosamine Impurities in Human Drugs. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs (accessed on 21 December 2022).
- FDA. Pfizer Expands Voluntary Nationwide Recall to include All Lots of CHANTIX® (Varenicline) Tablets Due to N-Nitroso Varenicline Content. Available online: https://www.fda.gov/safety/recalls-market-withdrawals-safety-alerts/pfizer-expands-voluntary-nationwide-recall-include-all-lots-chantixr-varenicline-tablets-due-n (accessed on 21 December 2022).
- HSA. Recall of Ventolin 2mg Tablets. Available online: https://www.hsa.gov.sg/announcements/product-recall/recall-of-ventolin-2mg-tablets (accessed on 21 December 2022).
- Skipper, H.E.; Schabel, F.M., Jr.; Trader, M.W.; Thomson, J.R. Experimental evaluation of potential anticancer agents. VI. Anatomical distribution of leukemic cells and failure of chemotherapy. Cancer Res. 1961, 21, 1154–1164. [Google Scholar]
- Brulikova, L.; Hlavac, J.; Hradil, P. DNA interstrand cross-linking agents and their chemotherapeutic potential. Curr. Med. Chem. 2012, 19, 364–385. [Google Scholar] [CrossRef]
- Ludlum, D.B. DNA alkylation by the haloethylnitrosoureas: Nature of modifications produced and their enzymatic repair or removal. Mutat. Res. 1990, 233, 117–126. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Glaab, W.E.; Tindall, K.R.; Skopek, T.R. Specificity of mutations induced by methyl methanesulfonate in mismatch repair-deficient human cancer cell lines. Mutat. Res. 1999, 427, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, B.; Spurr, N.; Karran, P. cDNA cloning and chromosomal assignment of the human O6-methylguanine-DNA methyltransferase. cDNA expression in E. coli and gene expression in human cells. J. Biol. Chem. 1990, 265, 9563–9569. [Google Scholar] [CrossRef] [PubMed]
- Sassanfar, M.; Dosanjh, M.K.; Essigmann, J.M.; Samson, L. Relative efficiencies of the bacterial, yeast, and human DNA methyltransferases for the repair of O6-methylguanine and O4-methylthymine. Suggestive evidence for O4-methylthymine repair by eukaryotic methyltransferases. J. Biol. Chem. 1991, 266, 2767–2771. [Google Scholar] [CrossRef]
- Koike, G.; Maki, H.; Takeya, H.; Hayakawa, H.; Sekiguchi, M. Purification, structure, and biochemical properties of human O6-methylguanine-DNA methyltransferase. J. Biol. Chem. 1990, 265, 14754–14762. [Google Scholar] [CrossRef]
- Zak, P.; Kleibl, K.; Laval, F. Repair of O6-methylguanine and O4-methylthymine by the human and rat O6-methylguanine-DNA methyltransferases. J. Biol. Chem. 1994, 269, 730–733. [Google Scholar] [CrossRef]
- Pegg, A.E. Mammalian O6-alkylguanine-DNA alkyltransferase: Regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar] [PubMed]
- Pegg, A.E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Peterson, L.A.; Liu, X.K.; Hecht, S.S. Pyridyloxobutyl DNA adducts inhibit the repair of O6-methylguanine. Cancer Res. 1993, 53, 2780–2785. [Google Scholar]
- Wang, L.; Spratt, T.E.; Liu, X.K.; Hecht, S.S.; Pegg, A.E.; Peterson, L.A. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1997, 10, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Spratt, T.E.; Pegg, A.E.; Peterson, L.A. Synthesis of DNA oligonucleotides containing site-specifically incorporated O6-[4-oxo-4-(3-pyridyl)butyl]guanine and their reaction with O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1999, 12, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.M.; Kenney, P.M.; Peterson, L.A. The pyridyloxobutyl DNA adduct, O6-[4-oxo-4-(3-pyridyl)butyl]guanine, is detected in tissues from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-treated A/J mice. Chem. Res. Toxicol. 2003, 16, 1–6. [Google Scholar] [CrossRef]
- Pauly, G.T.; Peterson, L.A.; Moschel, R.C. Mutagenesis by O(6)-[4-oxo-4-(3-pyridyl)butyl]guanine in E. coli and human cells. Chem. Res. Toxicol. 2002, 15, 165–169. [Google Scholar] [CrossRef]
- Kleihues, P.; Margison, G.P. Carcinogenicity of N-methyl-N-nitrosourea: Possible role of excision repair of O6-methylguanine from DNA. J. Natl. Cancer Inst. 1974, 53, 1839–1841. [Google Scholar]
- Becker, K.; Dosch, J.; Gregel, C.M.; Martin, B.A.; Kaina, B. Targeted expression of human O(6)-methylguanine-DNA methyltransferase (MGMT) in transgenic mice protects against tumor initiation in two-stage skin carcinogenesis. Cancer Res. 1996, 56, 3244–3249. [Google Scholar]
- Becker, K.; Gregel, C.; Fricke, C.; Komitowski, D.; Dosch, J.; Kaina, B. DNA repair protein MGMT protects against N-methyl-N-nitrosourea-induced conversion of benign into malignant tumors. Carcinogenesis 2003, 24, 541–546. [Google Scholar] [CrossRef]
- Becker, K.; Gregel, C.M.; Kaina, B. The DNA repair protein O6-methylguanine-DNA methyltransferase protects against skin tumor formation induced by antineoplastic chloroethylnitrosourea. Cancer Res. 1997, 57, 3335–3338. [Google Scholar]
- Nakatsuru, Y.; Matsukuma, S.; Nemoto, N.; Sugano, H.; Sekiguchi, M.; Ishikawa, T. O6-methylguanine-DNA methyltransferase protects against nitrosamine-induced hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 6468–6472. [Google Scholar] [CrossRef]
- Sakumi, K.; Shiraishi, A.; Shimizu, S.; Tsuzuki, T.; Ishikawa, T.; Sekiguchi, M. Methylnitrosourea-induced tumorigenesis in MGMT gene knockout mice. Cancer Res. 1997, 57, 2415–2418. [Google Scholar] [PubMed]
- Liu, L.; Qin, X.; Gerson, S.L. Reduced lung tumorigenesis in human methylguanine DNA--methyltransferase transgenic mice achieved by expression of transgene within the target cell. Carcinogenesis 1999, 20, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Dumenco, L.L.; Allay, E.; Norton, K.; Gerson, S.L. The prevention of thymic lymphomas in transgenic mice by human O6-alkylguanine-DNA alkyltransferase. Science 1993, 259, 219–222. [Google Scholar] [CrossRef]
- Liu, L.; Allay, E.; Dumenco, L.L.; Gerson, S.L. Rapid repair of O6-methylguanine-DNA adducts protects transgenic mice from N-methylnitrosourea-induced thymic lymphomas. Cancer Res. 1994, 54, 4648–4652. [Google Scholar] [PubMed]
- Wirtz, S.; Nagel, G.; Eshkind, L.; Neurath, M.F.; Samson, L.D.; Kaina, B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis 2010, 31, 2111–2117. [Google Scholar] [CrossRef]
- Bugni, J.M.; Meira, L.B.; Samson, L.D. Alkylation-induced colon tumorigenesis in mice deficient in the Mgmt and Msh6 proteins. Oncogene 2009, 28, 734–741. [Google Scholar] [CrossRef]
- Fahrer, J.; Frisch, J.; Nagel, G.; Kraus, A.; Dörsam, B.; Thomas, A.D.; Reissig, S.; Waisman, A.; Kaina, B. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis 2015, 36, 1235–1244. [Google Scholar] [CrossRef]
- Kraus, A.; McKeague, M.; Seiwert, N.; Nagel, G.; Geisen, S.M.; Ziegler, N.; Trantakis, I.A.; Kaina, B.; Thomas, A.D.; Sturla, S.J.; et al. Immunological and mass spectrometry-based approaches to determine thresholds of the mutagenic DNA adduct O(6)-methylguanine in vivo. Arch. Toxicol. 2019, 93, 559–572. [Google Scholar] [CrossRef]
- Sandercock, L.E.; Hahn, J.N.; Li, L.; Luchman, H.A.; Giesbrecht, J.L.; Peterson, L.A.; Jirik, F.R. Mgmt deficiency alters the in vivo mutational spectrum of tissues exposed to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Carcinogenesis 2008, 29, 866–874. [Google Scholar] [CrossRef]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in resistance to alkylating anticancer drugs. Int. J. Clin. Pharmacol. Ther. 2002, 40, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Kawate, H.; Sakumi, K.; Tsuzuki, T.; Nakatsuru, Y.; Ishikawa, T.; Takahashi, S.; Takano, H.; Noda, T.; Sekiguchi, M. Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc. Natl. Acad. Sci. USA 1998, 95, 5116–5120. [Google Scholar] [CrossRef]
- Eadie, J.S.; Conrad, M.; Toorchen, D.; Topal, M.D. Mechanism of mutagenesis by O6-methylguanine. Nature 1984, 308, 201–203. [Google Scholar] [CrossRef]
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.H.; Reardon, J.T.; Sancar, A.; Lilley, D.M.J.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447. [Google Scholar] [CrossRef]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef]
- Ochs, K.; Kaina, B. Apoptosis induced by DNA damage O6-methylguanine is Bcl-2 and caspase-9/3 regulated and Fas/caspase-8 independent. Cancer Res. 2000, 60, 5815–5824. [Google Scholar]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Stratenwerth, B.; Geisen, S.M.; He, Y.; Beltzig, L.; Sturla, S.J.; Kaina, B. Molecular Dosimetry of Temozolomide: Quantification of Critical Lesions, Correlation to Cell Death Responses, and Threshold Doses. Mol. Cancer Ther. 2021, 20, 1789–1799. [Google Scholar] [CrossRef]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair 2009, 8, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Gotzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, C.; Tatsch, L.; Brandstetter Vilar, J.; Rasenberger, B.; Beltzig, L.; Kaina, B.; Tomicic, M.T.; Christmann, M. Targeting c-IAP1, c-IAP2, and Bcl-2 Eliminates Senescent Glioblastoma Cells Following Temozolomide Treatment. Cancers 2021, 13, 3585. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Povey, A.C.; Kaina, B.; Santibanez Koref, M.F. Variability and regulation of O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003, 24, 625–635. [Google Scholar] [CrossRef]
- Povey, A.C.; Margison, G.P.; Santibanez-Koref, M.F. Lung cancer risk and variation in MGMT activity and sequence. DNA Repair 2007, 6, 1134–1144. [Google Scholar] [CrossRef]
- Grombacher, T.; Eichhorn, U.; Kaina, B. p53 is involved in regulation of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) by DNA damaging agents. Oncogene 1998, 17, 845–851. [Google Scholar] [CrossRef]
- Harris, L.C.; Remack, J.S.; Houghton, P.J.; Brent, T.P. Wild-type p53 suppresses transcription of the human O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1996, 56, 2029–2032. [Google Scholar]
- Blough, M.D.; Zlatescu, M.C.; Cairncross, J.G. O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res. 2007, 67, 580–584. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Shou, J.; Mullapudi, S.R.; Lang, F.F., Jr.; Rao, J.S.; Ali-Osman, F. Enforced expression of wild-type p53 curtails the transcription of the O(6)-methylguanine-DNA methyltransferase gene in human tumor cells and enhances their sensitivity to alkylating agents. Clin. Cancer Res. 2001, 7, 1398–1409. [Google Scholar] [PubMed]
- Bocangel, D.; Sengupta, S.; Mitra, S.; Bhakat, K.K. p53-Mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer. Res. 2009, 29, 3741–3750. [Google Scholar] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef]
- Fritz, G.; Tano, K.; Mitra, S.; Kaina, B. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol. Cell. Biol. 1991, 11, 4660–4668. [Google Scholar]
- Grombacher, T.; Kaina, B. Constitutive expression and inducibility of O6-methylguanine-DNA methyltransferase and N-methylpurine-DNA glycosylase in rat liver cells exhibiting different status of differentiation. Biochim. Et Biophys. Acta 1995, 1270, 63–72. [Google Scholar] [CrossRef]
- Grombacher, T.; Mitra, S.; Kaina, B. Induction of the alkyltransferase (MGMT) gene by DNA damaging agents and the glucocorticoid dexamethasone and comparison with the response of base excision repair genes. Carcinogenesis 1996, 17, 2329–2336. [Google Scholar] [CrossRef]
- Rafferty, J.A.; Clarke, A.R.; Sellappan, D.; Koref, M.S.; Frayling, I.M.; Margison, G.P. Induction of murine O6-alkylguanine-DNA-alkyltransferase in response to ionising radiation is p53 gene dose dependent. Oncogene 1996, 12, 693–697. [Google Scholar]
- Boldogh, I.; Ramana, C.V.; Chen, Z.; Biswas, T.; Hazra, T.K.; Grosch, S.; Grombacher, T.; Mitra, S.; Kaina, B. Regulation of expression of the DNA repair gene O6-methylguanine-DNA methyltransferase via protein kinase C-mediated signaling. Cancer Res. 1998, 58, 3950–3956. [Google Scholar]
- Aasland, D.; Reich, T.R.; Tomicic, M.T.; Switzeny, O.J.; Kaina, B.; Christmann, M. Repair gene O(6)-methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J. Neurochem. 2018, 144, 139–151. [Google Scholar] [CrossRef]
- Costello, J.F.; Futscher, B.W.; Kroes, R.A.; Pieper, R.O. Methylation-related chromatin structure is associated with exclusion of transcription factors from and suppressed expression of the O-6-methylguanine DNA methyltransferase gene in human glioma cell lines. Mol. Cell. Biol. 1994, 14, 6515–6521. [Google Scholar]
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.C.; Potter, P.M.; Tano, K.; Shiota, S.; Mitra, S.; Brent, T.P. Characterization of the promoter region of the human O6-methylguanine-DNA methyltransferase gene. Nucleic Acids Res. 1991, 19, 6163–6167. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; von Wronski, M.A.; Brent, T.P. Localization of methylation sites in the human O6-methylguanine-DNA methyltransferase promoter: Correlation with gene suppression. Carcinogenesis 1995, 16, 1385–1390. [Google Scholar] [CrossRef]
- Qian, X.C.; Brent, T.P. Methylation hot spots in the 5’ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997, 57, 3672–3677. [Google Scholar] [PubMed]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Et Biophys. Acta 2011, 1816, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Janssen, K.; Eichhorn-Grombacher, U.; Schlink, K.; Nitzsche, S.; Oesch, F.; Kaina, B. Long-time expression of DNA repair enzymes MGMT and APE in human peripheral blood mononuclear cells. Arch. Toxicol. 2001, 75, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.; Haugen, A.; Myrnes, B.; Guddal, P.H. Repair of premutagenic DNA lesions in human fetal tissues: Evidence for low levels of O6-methylguanine-DNA methyltransferase and uracil-DNA glycosylase activity in some tissues. Carcinogenesis 1983, 4, 1559–1564. [Google Scholar] [CrossRef]
- Briegert, M.; Enk, A.H.; Kaina, B. Change in expression of MGMT during maturation of human monocytes into dendritic cells. DNA Repair 2007, 6, 1255–1263. [Google Scholar] [CrossRef]
- Christmann, M.; Nagel, G.; Horn, S.; Krahn, U.; Wiewrodt, D.; Sommer, C.; Kaina, B. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: A comparative study on astrocytoma and glioblastoma. Int. J. Cancer. J. Int. Du Cancer 2010, 127, 2106–2118. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Vilar, J.B.; Christmann, M.; Tomicic, M.T. Alterations in Molecular Profiles Affecting Glioblastoma Resistance to Radiochemotherapy: Where Does the Good Go? Cancers 2022, 14, 2416. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Velu, C.S.; Smith, Q.R.; Bhat, G.J.; Srivenugopal, K.S. Increased expression of the MGMT repair protein mediated by cysteine prodrugs and chemopreventative natural products in human lymphocytes and tumor cell lines. Carcinogenesis 2007, 28, 378–389. [Google Scholar] [CrossRef]
- Huber, W.W.; Scharf, G.; Nagel, G.; Prustomersky, S.; Schulte-Hermann, R.; Kaina, B. Coffee and its chemopreventive components Kahweol and Cafestol increase the activity of O6-methylguanine-DNA methyltransferase in rat liver--comparison with phase II xenobiotic metabolism. Mutat. Res. 2003, 522, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014, 35, 692–702. [Google Scholar] [CrossRef]
- Göder, A.; Nagel, G.; Kraus, A.; Dörsam, B.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic acid inhibits the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) and triggers its depletion in colorectal cancer cells with concomitant autophagy induction. Carcinogenesis 2015, 36, 817–831. [Google Scholar] [CrossRef]
- Tsai, C.K.; Huang, L.C.; Wu, Y.P.; Kan, I.Y.; Hueng, D.Y. SNAP reverses temozolomide resistance in human glioblastoma multiforme cells through down-regulation of MGMT. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 14171–14184. [Google Scholar] [CrossRef]
- Kuo, C.C.; Liu, J.F.; Shiah, H.S.; Ma, L.C.; Chang, J.Y. Tamoxifen accelerates proteasomal degradation of O6-methylguanine DNA methyltransferase in human cancer cells. Int. J. Cancer. J. Int. Du Cancer 2007, 121, 2293–2300. [Google Scholar] [CrossRef]
- Samson, L.; Cairns, J. A new pathway for DNA repair in E. coli. Nature 1977, 267, 281–283. [Google Scholar] [CrossRef]
- Falnes, P.O.; Johansen, R.F.; Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in E. coli. Nature 2002, 419, 178–182. [Google Scholar] [CrossRef]
- Trewick, S.C.; Henshaw, T.F.; Hausinger, R.P.; Lindahl, T.; Sedgwick, B. Oxidative demethylation by E. coli AlkB directly reverts DNA base damage. Nature 2002, 419, 174–178. [Google Scholar] [CrossRef]
- Delaney, J.C.; Essigmann, J.M. Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB E. coli. Proc. Natl. Acad. Sci. USA 2004, 101, 14051–14056. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Tang, Q.; Bian, K.; Humulock, Z.T.; Yang, X.; Jost, M.; Drennan, C.L.; Essigmann, J.M.; Li, D. Adaptive Response Enzyme AlkB Preferentially Repairs 1-Methylguanine and 3-Methylthymine Adducts in Double-Stranded DNA. Chem. Res. Toxicol. 2016, 29, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.C.; Smeester, L.; Wong, C.; Frick, L.E.; Taghizadeh, K.; Wishnok, J.S.; Drennan, C.L.; Samson, L.D.; Essigmann, J.M. AlkB reverses etheno DNA lesions caused by lipid oxidation in vitro and in vivo. Nat. Struct. Mol. Biol. 2005, 12, 855–860. [Google Scholar] [CrossRef]
- Mishina, Y.; Yang, C.G.; He, C. Direct repair of the exocyclic DNA adduct 1,N6-ethenoadenine by the DNA repair AlkB proteins. J. Am. Chem. Soc. 2005, 127, 14594–14595. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.E.; Delaney, J.C.; Wong, C.; Drennan, C.L.; Essigmann, J.M. Alleviation of 1,N6-ethanoadenine genotoxicity by the E. coli adaptive response protein AlkB. Proc. Natl. Acad. Sci. USA 2007, 104, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Bates, P.A.; Paik, J.; Jacobs, S.C.; Lindahl, T. Repair of alkylated DNA: Recent advances. DNA Repair 2007, 6, 429–442. [Google Scholar] [CrossRef]
- Aas, P.A.; Otterlei, M.; Falnes, P.O.; Vagbo, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjoras, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 421, 859–863. [Google Scholar] [CrossRef]
- Duncan, T.; Trewick, S.C.; Koivisto, P.; Bates, P.A.; Lindahl, T.; Sedgwick, B. Reversal of DNA alkylation damage by two human dioxygenases. Proc. Natl. Acad. Sci. USA 2002, 99, 16660–16665. [Google Scholar] [CrossRef]
- Qi, R.; Bian, K.; Chen, F.; Tang, Q.; Zhou, X.; Li, D. Sequence Dependent Repair of 1,N(6)-Ethenoadenine by DNA Repair Enzymes ALKBH2, ALKBH3, and AlkB. Molecules 2021, 26, 5285. [Google Scholar] [CrossRef]
- Fu, D.; Samson, L.D. Direct repair of 3,N(4)-ethenocytosine by the human ALKBH2 dioxygenase is blocked by the AAG/MPG glycosylase. DNA Repair 2012, 11, 46–52. [Google Scholar] [CrossRef]
- Zdzalik, D.; Domanska, A.; Prorok, P.; Kosicki, K.; van den Born, E.; Falnes, P.O.; Rizzo, C.J.; Guengerich, F.P.; Tudek, B. Differential repair of etheno-DNA adducts by bacterial and human AlkB proteins. DNA Repair 2015, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, P.; Robins, P.; Lindahl, T.; Sedgwick, B. Demethylation of 3-methylthymine in DNA by bacterial and human DNA dioxygenases. J. Biol. Chem. 2004, 279, 40470–40474. [Google Scholar] [CrossRef] [PubMed]
- Falnes, P.O. Repair of 3-methylthymine and 1-methylguanine lesions by bacterial and human AlkB proteins. Nucleic Acids Res. 2004, 32, 6260–6267. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.; Lenz, S.A.P.; Tang, Q.; Chen, F.; Qi, R.; Jost, M.; Drennan, C.L.; Essigmann, J.M.; Wetmore, S.D.; Li, D. DNA repair enzymes ALKBH2, ALKBH3, and AlkB oxidize 5-methylcytosine to 5-hydroxymethylcytosine, 5-formylcytosine and 5-carboxylcytosine in vitro. Nucleic Acids Res. 2019, 47, 5522–5529. [Google Scholar] [CrossRef]
- Nay, S.L.; Lee, D.H.; Bates, S.E.; O’Connor, T.R. Alkbh2 protects against lethality and mutation in primary mouse embryonic fibroblasts. DNA Repair 2012, 11, 502–510. [Google Scholar] [CrossRef]
- Ringvoll, J.; Nordstrand, L.M.; Vagbo, C.B.; Talstad, V.; Reite, K.; Aas, P.A.; Lauritzen, K.H.; Liabakk, N.B.; Bjork, A.; Doughty, R.W.; et al. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 2006, 25, 2189–2198. [Google Scholar] [CrossRef]
- Calvo, J.A.; Meira, L.B.; Lee, C.Y.; Moroski-Erkul, C.A.; Abolhassani, N.; Taghizadeh, K.; Eichinger, L.W.; Muthupalani, S.; Nordstrand, L.M.; Klungland, A.; et al. DNA repair is indispensable for survival after acute inflammation. J. Clin. Investig. 2012, 122, 2680–2689. [Google Scholar] [CrossRef]
- Gilljam, K.M.; Feyzi, E.; Aas, P.A.; Sousa, M.M.; Muller, R.; Vagbo, C.B.; Catterall, T.C.; Liabakk, N.B.; Slupphaug, G.; Drablos, F.; et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. J. Cell Biol. 2009, 186, 645–654. [Google Scholar] [CrossRef]
- Fu, D.; Samson, L.D.; Hubscher, U.; van Loon, B. The interaction between ALKBH2 DNA repair enzyme and PCNA is direct, mediated by the hydrophobic pocket of PCNA and perturbed in naturally-occurring ALKBH2 variants. DNA Repair 2015, 35, 13–18. [Google Scholar] [CrossRef]
- Mohan, M.; Akula, D.; Dhillon, A.; Goyal, A.; Anindya, R. Human RAD51 paralogue RAD51C fosters repair of alkylated DNA by interacting with the ALKBH3 demethylase. Nucleic Acids Res. 2019, 47, 11729–11745. [Google Scholar] [CrossRef]
- Brickner, J.R.; Townley, B.A.; Mosammaparast, N. Intersections between transcription-coupled repair and alkylation damage reversal. DNA Repair 2019, 81, 102663. [Google Scholar] [CrossRef] [PubMed]
- Soll, J.M.; Brickner, J.R.; Mudge, M.C.; Mosammaparast, N. RNA ligase-like domain in activating signal cointegrator 1 complex subunit 1 (ASCC1) regulates ASCC complex function during alkylation damage. J. Biol. Chem. 2018, 293, 13524–13533. [Google Scholar] [CrossRef]
- Brickner, J.R.; Soll, J.M.; Lombardi, P.M.; Vagbo, C.B.; Mudge, M.C.; Oyeniran, C.; Rabe, R.; Jackson, J.; Sullender, M.E.; Blazosky, E.; et al. A ubiquitin-dependent signalling axis specific for ALKBH-mediated DNA dealkylation repair. Nature 2017, 551, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Dango, S.; Mosammaparast, N.; Sowa, M.E.; Xiong, L.J.; Wu, F.; Park, K.; Rubin, M.; Gygi, S.; Harper, J.W.; Shi, Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol. Cell 2011, 44, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.C.; Prestegarden, L.; Grudic, A.; Hegi, M.E.; Tysnes, B.B.; Bjerkvig, R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro-Oncol. 2013, 15, 269–278. [Google Scholar] [CrossRef]
- Stefansson, O.A.; Hermanowicz, S.; van der Horst, J.; Hilmarsdottir, H.; Staszczak, Z.; Jonasson, J.G.; Tryggvadottir, L.; Gudjonsson, T.; Sigurdsson, S. CpG promoter methylation of the ALKBH3 alkylation repair gene in breast cancer. BMC Cancer 2017, 17, 469. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Tran, T.Q.; Ishak Gabra, M.B.; Lowman, X.H.; Yang, Y.; Reid, M.A.; Pan, M.; O’Connor, T.R.; Kong, M. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017, 15, e2002810. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Engelward, B.P.; Weeda, G.; Wyatt, M.D.; Broekhof, J.L.; de Wit, J.; Donker, I.; Allan, J.M.; Gold, B.; Hoeijmakers, J.H.; Samson, L.D. Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 13087–13092. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Ellenberger, T. Dissecting the broad substrate specificity of human 3-methyladenine-DNA glycosylase. J. Biol. Chem. 2004, 279, 9750–9757. [Google Scholar] [CrossRef]
- Lee, C.Y.; Delaney, J.C.; Kartalou, M.; Lingaraju, G.M.; Maor-Shoshani, A.; Essigmann, J.M.; Samson, L.D. Recognition and processing of a new repertoire of DNA substrates by human 3-methyladenine DNA glycosylase (AAG). Biochemistry 2009, 48, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Lin, C.R. Noninvasive measurement of smoking-associated N(3)-ethyladenine and N(7)-ethylguanine in human salivary DNA by stable isotope dilution nanoflow liquid chromatography-nanospray ionization tandem mass spectrometry. Toxicol. Lett. 2014, 225, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Gates, K.S.; Nooner, T.; Dutta, S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol. 2004, 17, 839–856. [Google Scholar] [CrossRef]
- Engelward, B.P.; Dreslin, A.; Christensen, J.; Huszar, D.; Kurahara, C.; Samson, L. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. EMBO J. 1996, 15, 945–952. [Google Scholar] [CrossRef]
- Elder, R.H.; Jansen, J.G.; Weeks, R.J.; Willington, M.A.; Deans, B.; Watson, A.J.; Mynett, K.J.; Bailey, J.A.; Cooper, D.P.; Rafferty, J.A.; et al. Alkylpurine-DNA-N-glycosylase knockout mice show increased susceptibility to induction of mutations by methyl methanesulfonate. Mol. Cell. Biol. 1998, 18, 5828–5837. [Google Scholar] [CrossRef]
- Encell, L.; Shuker, D.E.; Foiles, P.G.; Gold, B. The in vitro methylation of DNA by a minor groove binding methyl sulfonate ester. Chem. Res. Toxicol. 1996, 9, 563–567. [Google Scholar] [CrossRef]
- Engelward, B.P.; Allan, J.M.; Dreslin, A.J.; Kelly, J.D.; Wu, M.M.; Gold, B.; Samson, L.D. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. J. Biol. Chem. 1998, 273, 5412–5418. [Google Scholar] [CrossRef]
- Smith, S.A.; Engelward, B.P. In vivo repair of methylation damage in Aag 3-methyladenine DNA glycosylase null mouse cells. Nucleic Acids Res. 2000, 28, 3294–3300. [Google Scholar] [CrossRef]
- Coquerelle, T.; Dosch, J.; Kaina, B. Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents--a case of imbalanced DNA repair. Mutat. Res. 1995, 336, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.L.; Seo, Y.R.; Smith, M.L.; Kelley, M.R. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003, 63, 608–615. [Google Scholar] [PubMed]
- Trivedi, R.N.; Almeida, K.H.; Fornsaglio, J.L.; Schamus, S.; Sobol, R.W. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005, 65, 6394–6400. [Google Scholar] [CrossRef]
- Tang, J.B.; Svilar, D.; Trivedi, R.N.; Wang, X.H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-Oncol. 2011, 13, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Löbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef]
- Crosbie, P.A.; Watson, A.J.; Agius, R.; Barber, P.V.; Margison, G.P.; Povey, A.C. Elevated N3-methylpurine-DNA glycosylase DNA repair activity is associated with lung cancer. Mutat. Res. 2012, 732, 43–46. [Google Scholar] [CrossRef]
- Calvo, J.A.; Moroski-Erkul, C.A.; Lake, A.; Eichinger, L.W.; Shah, D.; Jhun, I.; Limsirichai, P.; Bronson, R.T.; Christiani, D.C.; Meira, L.B.; et al. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLoS Genet. 2013, 9, e1003413. [Google Scholar] [CrossRef]
- Chakravarti, D.; Ibeanu, G.C.; Tano, K.; Mitra, S. Cloning and expression in E. coli of a human cDNA encoding the DNA repair protein N-methylpurine-DNA glycosylase. J. Biol. Chem. 1991, 266, 15710–15715. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Schaich, M.A.; Zhong, Z.; van Loon, B.; Watkins, S.C.; Van Houten, B. Cooperative interaction between AAG and UV-DDB in the removal of modified bases. Nucleic Acids Res. 2022, 50, 12856–12871. [Google Scholar] [CrossRef]
- Demple, B.; Sung, J.S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Wilson, S.H. Structure and mechanism of DNA polymerase Beta. Chem. Rev. 2006, 106, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.K.; Berg, B.J.; Prasad, R.; Molina, J.T.; Beard, W.A.; Tomkinson, A.E.; Wilson, S.H. Mammalian abasic site base excision repair. Identification of the reaction sequence and rate-determining steps. J. Biol. Chem. 1998, 273, 21203–21209. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, E.; Taylor, R.; Cevasco, M.; Abbondandolo, A.; Caldecott, K.; Frosina, G. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J. Biol. Chem. 1997, 272, 23970–23975. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.M.; Wickstead, B.; Cronin, S.; Caldecott, K.W. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol. CB 1998, 8, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Dianova, I.I.; Sleeth, K.M.; Allinson, S.L.; Parsons, J.L.; Breslin, C.; Caldecott, K.W.; Dianov, G.L. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res. 2004, 32, 2550–2555. [Google Scholar] [CrossRef]
- Sleeth, K.M.; Robson, R.L.; Dianov, G.L. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry 2004, 43, 12924–12930. [Google Scholar] [CrossRef]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: Reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar] [CrossRef]
- Petermann, E.; Ziegler, M.; Oei, S.L. ATP-dependent selection between single nucleotide and long patch base excision repair. DNA Repair 2003, 2, 1101–1114. [Google Scholar] [CrossRef]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef]
- Stucki, M.; Pascucci, B.; Parlanti, E.; Fortini, P.; Wilson, S.H.; Hubscher, U.; Dogliotti, E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene 1998, 17, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.S.; McKenna, A.E.; Motycka, T.A.; Matsumoto, Y.; Tomkinson, A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. CB 2000, 10, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Ström, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 286, 10690–10701. [Google Scholar] [CrossRef]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef]
- Mangerich, A.; Bürkle, A. Pleiotropic cellular functions of PARP1 in longevity and aging: Genome maintenance meets inflammation. Oxidative Med. Cell. Longev. 2012, 2012, 321653. [Google Scholar] [CrossRef]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar] [CrossRef]
- Teloni, F.; Altmeyer, M. Readers of poly(ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2016, 44, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Bürkle, A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007, 35, e143. [Google Scholar] [CrossRef] [PubMed]
- Breslin, C.; Hornyak, P.; Ridley, A.; Rulten, S.L.; Hanzlikova, H.; Oliver, A.W.; Caldecott, K.W. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015, 43, 6934–6944. [Google Scholar] [CrossRef]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef] [PubMed]
- de Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef]
- Trucco, C.; Oliver, F.J.; de Murcia, G.; Menissier-de Murcia, J. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998, 26, 2644–2649. [Google Scholar] [CrossRef]
- Masutani, M.; Nozaki, T.; Nakamoto, K.; Nakagama, H.; Suzuki, H.; Kusuoka, O.; Tsutsumi, M.; Sugimura, T. The response of Parp knockout mice against DNA damaging agents. Mutat. Res. 2000, 462, 159–166. [Google Scholar] [CrossRef]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef]
- Shibata, A.; Kamada, N.; Masumura, K.; Nohmi, T.; Kobayashi, S.; Teraoka, H.; Nakagama, H.; Sugimura, T.; Suzuki, H.; Masutani, M. Parp-1 deficiency causes an increase of deletion mutations and insertions/rearrangements in vivo after treatment with an alkylating agent. Oncogene 2005, 24, 1328–1337. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Masutani, M.; Nozaki, T.; Kusuoka, O.; Tsujiuchi, T.; Nakagama, H.; Suzuki, H.; Konishi, Y.; Sugimura, T. Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout mice to nitrosamine carcinogenicity. Carcinogenesis 2001, 22, 1–3. [Google Scholar] [CrossRef]
- Nozaki, T.; Fujihara, H.; Watanabe, M.; Tsutsumi, M.; Nakamoto, K.; Kusuoka, O.; Kamada, N.; Suzuki, H.; Nakagama, H.; Sugimura, T.; et al. Parp-1 deficiency implicated in colon and liver tumorigenesis induced by azoxymethane. Cancer Sci. 2003, 94, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Corrigan, J.J.; Fake, K.R.; Calvo, J.A.; Samson, L.D. PARP inhibitors protect against sex- and AAG-dependent alkylation-induced neural degeneration. Oncotarget 2017, 8, 68707–68720. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagne, J.P.; Barbi de Moura, M.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Ke, Y.; Zeng, X.; Gao, J.; Ba, X.; Wang, R. The key players of parthanatos: Opportunities for targeting multiple levels in the therapy of parthanatos-based pathogenesis. Cell. Mol. Life Sci. CMLS 2022, 79, 60. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Haince, J.F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354, aad6872. [Google Scholar] [CrossRef]
- Yang, M.; Wang, C.; Zhou, M.; Bao, L.; Wang, Y.; Kumar, A.; Xing, C.; Luo, W.; Wang, Y. KDM6B promotes PARthanatos via suppression of O6-methylguanine DNA methyltransferase repair and sustained checkpoint response. Nucleic Acids Res. 2022, 50, 6313–6331. [Google Scholar] [CrossRef]
- Christmann, M.; Tomicic, M.T.; Roos, W.P.; Kaina, B. Mechanisms of human DNA repair: An update. Toxicology 2003, 193, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, T.; Roos, W.P.; Kramer, O.H.; Strik, H.M.; Kaina, B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim. Et Biophys. Acta. Rev. Cancer 2017, 1868, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Muniandy, P.A.; Liu, J.; Majumdar, A.; Liu, S.T.; Seidman, M.M. DNA interstrand crosslink repair in mammalian cells: Step by step. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 23–49. [Google Scholar] [CrossRef]
- Kusumoto, R.; Masutani, C.; Sugasawa, K.; Iwai, S.; Araki, M.; Uchida, A.; Mizukoshi, T.; Hanaoka, F. Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutat. Res. 2001, 485, 219–227. [Google Scholar] [CrossRef]
- Tang, J.; Chu, G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 2002, 1, 601–616. [Google Scholar] [CrossRef]
- Evans, E.; Moggs, J.G.; Hwang, J.R.; Egly, J.M.; Wood, R.D. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997, 16, 6559–6573. [Google Scholar] [CrossRef]
- Krasikova, Y.; Rechkunova, N.; Lavrik, O. Nucleotide Excision Repair: From Molecular Defects to Neurological Abnormalities. Int. J. Mol. Sci. 2021, 22, 6220. [Google Scholar] [CrossRef]
- Giannattasio, M.; Follonier, C.; Tourriere, H.; Puddu, F.; Lazzaro, F.; Pasero, P.; Lopes, M.; Plevani, P.; Muzi-Falconi, M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol. Cell 2010, 40, 50–62. [Google Scholar] [CrossRef]
- Svejstrup, J.Q. Rescue of arrested RNA polymerase II complexes. J. Cell Sci. 2003, 116, 447–451. [Google Scholar] [CrossRef]
- Mijal, R.S.; Thomson, N.M.; Fleischer, N.L.; Pauly, G.T.; Moschel, R.C.; Kanugula, S.; Fang, Q.; Pegg, A.E.; Peterson, L.A. The repair of the tobacco specific nitrosamine derived adduct O6-[4-Oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem. Res. Toxicol. 2004, 17, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Urban, A.M.; Upadhyaya, P.; Cao, Q.; Peterson, L.A. Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem. Res. Toxicol. 2012, 25, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Perdigao, J.; Pegg, A.E.; Lao, Y.; Hecht, S.S.; Lindgren, B.R.; Reardon, J.T.; Sancar, A.; Wattenberg, E.V.; Peterson, L.A. The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2009, 22, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Kotandeniya, D.; Murphy, D.; Yan, S.; Park, S.; Seneviratne, U.; Koopmeiners, J.S.; Pegg, A.; Kanugula, S.; Kassie, F.; Tretyakova, N. Kinetics of O(6)-pyridyloxobutyl-2’-deoxyguanosine repair by human O(6)-alkylguanine DNA alkyltransferase. Biochemistry 2013, 52, 4075–4088. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Wang, Y. Liquid Chromatography-Tandem Mass Spectrometry for the Quantification of Tobacco-Specific Nitrosamine-Induced DNA Adducts in Mammalian Cells. Anal. Chem. 2017, 89, 9124–9130. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; Johnson, C.S.; Kovi, R.C.; James-Yi, S.A.; O’Sullivan, M.G.; Wang, M.; Le, C.T.; Khariwala, S.S.; Upadhyaya, P.; Hecht, S.S. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis 2014, 35, 2798–2806. [Google Scholar] [CrossRef]
- Carlson, E.S.; Upadhyaya, P.; Villalta, P.W.; Ma, B.; Hecht, S.S. Analysis and Identification of 2’-Deoxyadenosine-Derived Adducts in Lung and Liver DNA of F-344 Rats Treated with the Tobacco-Specific Carcinogen 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone and Enantiomers of its Metabolite 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018, 31, 358–370. [Google Scholar] [CrossRef]
- Stepanov, I.; Hecht, S.S. Mitochondrial DNA adducts in the lung and liver of F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and (S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 406–414. [Google Scholar] [CrossRef]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Stepanov, I.; Hecht, S.S. Pyridylhydroxybutyl and pyridyloxobutyl DNA phosphate adduct formation in rats treated chronically with enantiomers of the tobacco-specific nitrosamine metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Mutagenesis 2017, 32, 561–570. [Google Scholar] [CrossRef]
- Ma, B.; Villalta, P.W.; Zarth, A.T.; Kotandeniya, D.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Comprehensive High-Resolution Mass Spectrometric Analysis of DNA Phosphate Adducts Formed by the Tobacco-Specific Lung Carcinogen 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2015, 28, 2151–2159. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Bedard, L.L.; Massey, T.E. Repair of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA pyridyloxobutylation by nucleotide excision repair. Cancer Lett. 2008, 260, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Massey, T.E. In vivo treatment with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces organ-specific alterations in in vitro repair of DNA pyridyloxobutylation. Mutat. Res. 2009, 663, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Leng, J.; Tan, Y.; Price, N.E.; Wang, Y. Quantification of DNA Lesions Induced by 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol in Mammalian Cells. Chem. Res. Toxicol. 2019, 32, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.W.; Sledziewska-Gojska, E.; Hirani-Hojatti, S. In vivo effect of DNA repair on the transition frequency produced from a single O6-methyl- or O6-n-butyl-guanine in a T:G base pair. Mol. Gen. Genet. 1988, 213, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.W.; Sledziewska-Gojska, E.; Hirani-Hojatti, S.; Borowy-Borowski, H. uvrA and recA mutations inhibit a site-specific transition produced by a single O6-methylguanine in gene G of bacteriophage phi X174. Proc. Natl. Acad. Sci. USA 1985, 82, 7173–7177. [Google Scholar] [CrossRef]
- Boyle, J.M.; Margison, G.P.; Saffhill, R. Evidence for the excision repair of O6-n-butyldeoxyguanosine in human cells. Carcinogenesis 1986, 7, 1987–1990. [Google Scholar] [CrossRef]
- Boyle, J.M.; Saffhill, R.; Margison, G.P.; Fox, M. A comparison of cell survival, mutation and persistence of putative promutagenic lesions in Chinese hamster cells exposed to BNU or MNU. Carcinogenesis 1986, 7, 1981–1985. [Google Scholar] [CrossRef]
- Bol, S.A.; van Steeg, H.; van Oostrom, C.T.; Tates, A.D.; Vrieling, H.; de Groot, A.J.; Mullenders, L.H.; van Zeeland, A.A.; Jansen, J.G. Nucleotide excision repair modulates the cytotoxic and mutagenic effects of N-n-butyl-N-nitrosourea in cultured mammalian cells as well as in mouse splenocytes in vivo. Mutagenesis 1999, 14, 317–322. [Google Scholar] [CrossRef]
- Du, H.; Wang, P.; Li, L.; Wang, Y. Repair and translesion synthesis of O(6)-alkylguanine DNA lesions in human cells. J. Biol. Chem. 2019, 294, 11144–11153. [Google Scholar] [CrossRef]
- Bronstein, S.M.; Skopek, T.R.; Swenberg, J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992, 52, 2008–2011. [Google Scholar] [PubMed]
- Kostka, T.; Empl, M.T.; Seiwert, N.; Geisen, S.M.; Hoffmann, P.; Adam, J.; Seeger, B.; Shay, J.W.; Christmann, M.; Sturla, S.J.; et al. Repair of O6-carboxymethylguanine adducts by O6-methylguanine-DNA methyltransferase in human colon epithelial cells. Carcinogenesis 2021, 42, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, C.M.N.; Escher, N.A.; Kim, H.S.; Geisen, S.M.; Fontana, G.A.; Yeo, J.E.; Scharer, O.D.; Sturla, S.J. A combination of direct reversion and nucleotide excision repair counters the mutagenic effects of DNA carboxymethylation. DNA Repair 2022, 110, 103262. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.L.; Fairhurst, N.; Challis, B.C.; Shuker, D.E. Synthesis, characterization, and immunochemical detection of O6-(carboxymethyl)-2’-deoxyguanosine: A DNA adduct formed by nitrosated glycine derivatives. Chem. Res. Toxicol. 1997, 10, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Geisen, S.M.; Aloisi, C.M.N.; Huber, S.M.; Sandell, E.S.; Escher, N.A.; Sturla, S.J. Direct Alkylation of Deoxyguanosine by Azaserine Leads to O(6)-Carboxymethyldeoxyguanosine. Chem. Res. Toxicol. 2021, 34, 1518–1529. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Aasland, D.; Naumann, S.C.; Meise, R.; Barckhausen, C.; Kaina, B.; Christmann, M. Translesion polymerase eta is upregulated by cancer therapeutics and confers anticancer drug resistance. Cancer Res. 2014, 74, 5585–5596. [Google Scholar] [CrossRef]
- Roos, W.P.; Tsaalbi-Shtylik, A.; Tsaryk, R.; Guvercin, F.; de Wind, N.; Kaina, B. The translesion polymerase Rev3L in the tolerance of alkylating anticancer drugs. Mol. Pharmacol. 2009, 76, 927–934. [Google Scholar] [CrossRef]
- Hanisch, D.; Krumm, A.; Diehl, T.; Stork, C.M.; Dejung, M.; Butter, F.; Kim, E.; Brenner, W.; Fritz, G.; Hofmann, T.G.; et al. Class I HDAC overexpression promotes temozolomide resistance in glioma cells by regulating RAD18 expression. Cell Death Dis. 2022, 13, 293. [Google Scholar] [CrossRef]
- Kaszubowski, J.D.; Trakselis, M.A. Beyond the Lesion: Back to High Fidelity DNA Synthesis. Front. Mol. Biosci. 2021, 8, 811540. [Google Scholar] [CrossRef]
- Groth, P.; Auslander, S.; Majumder, M.M.; Schultz, N.; Johansson, F.; Petermann, E.; Helleday, T. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol. 2010, 402, 70–82. [Google Scholar] [CrossRef]
- Voigt, J.M.; Topal, M.D. O6-methylguanine-induced replication blocks. Carcinogenesis 1995, 16, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Prakash, S.; Prakash, L. Replication past O(6)-methylguanine by yeast and human DNA polymerase eta. Mol. Cell. Biol. 2000, 20, 8001–8007. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perrino, F.W.; Blans, P.; Harvey, S.; Gelhaus, S.L.; McGrath, C.; Akman, S.A.; Jenkins, G.S.; LaCourse, W.R.; Fishbein, J.C. The N2-ethylguanine and the O6-ethyl- and O6-methylguanine lesions in DNA: Contrasting responses from the “bypass” DNA polymerase eta and the replicative DNA polymerase alpha. Chem. Res. Toxicol. 2003, 16, 1616–1623. [Google Scholar] [CrossRef]
- Räz, M.H.; Dexter, H.R.; Millington, C.L.; van Loon, B.; Williams, D.M.; Sturla, S.J. Bypass of Mutagenic O(6)-Carboxymethylguanine DNA Adducts by Human Y- and B-Family Polymerases. Chem. Res. Toxicol. 2016, 29, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Choi, J.Y.; Guengerich, F.P. Kinetic analysis of translesion synthesis opposite bulky N2- and O6-alkylguanine DNA adducts by human DNA polymerase REV1. J. Biol. Chem. 2008, 283, 23645–23655. [Google Scholar] [CrossRef]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef]
- Grevatt, P.C.; Solomon, J.J.; Bhanot, O.S. In vitro mispairing specificity of O2-ethylthymidine. Biochemistry 1992, 31, 4181–4188. [Google Scholar] [CrossRef]
- Williams, N.L.; Wang, P.; Wang, Y. Replicative Bypass of O(2)-Alkylthymidine Lesions in Vitro. Chem. Res. Toxicol. 2016, 29, 1755–1761. [Google Scholar] [CrossRef]
- Wu, J.; Li, L.; Wang, P.; You, C.; Williams, N.L.; Wang, Y. Translesion synthesis of O4-alkylthymidine lesions in human cells. Nucleic Acids Res. 2016, 44, 9256–9265. [Google Scholar] [CrossRef][Green Version]
- Du, H.; Leng, J.; Wang, P.; Li, L.; Wang, Y. Impact of tobacco-specific nitrosamine-derived DNA adducts on the efficiency and fidelity of DNA replication in human cells. J. Biol. Chem. 2018, 293, 11100–11108. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Yoon, J.H.; Roy Choudhury, J.; Prakash, L.; Prakash, S. Genetic Control of Replication through N1-methyladenine in Human Cells. J. Biol. Chem. 2015, 290, 29794–29800. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Basu, D.; Choudhury, J.R.; Prakash, S.; Prakash, L. DNA polymerase lambda promotes error-free replication through Watson-Crick impairing N1-methyl-deoxyadenosine adduct in conjunction with DNA polymerase zeta. J. Biol. Chem. 2021, 297, 100868. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Roy Choudhury, J.; Park, J.; Prakash, S.; Prakash, L. Translesion synthesis DNA polymerases promote error-free replication through the minor-groove DNA adduct 3-deaza-3-methyladenine. J. Biol. Chem. 2017, 292, 18682–18688. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Guengerich, F.P. Adduct size limits efficient and error-free bypass across bulky N2-guanine DNA lesions by human DNA polymerase eta. J. Mol. Biol. 2005, 352, 72–90. [Google Scholar] [CrossRef]
- Choi, J.Y.; Guengerich, F.P. Kinetic evidence for inefficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase iota. J. Biol. Chem. 2006, 281, 12315–12324. [Google Scholar] [CrossRef]
- Choi, J.Y.; Angel, K.C.; Guengerich, F.P. Translesion synthesis across bulky N2-alkyl guanine DNA adducts by human DNA polymerase kappa. J. Biol. Chem. 2006, 281, 21062–21072. [Google Scholar] [CrossRef]
- Choi, J.Y.; Zang, H.; Angel, K.C.; Kozekov, I.D.; Goodenough, A.K.; Rizzo, C.J.; Guengerich, F.P. Translesion synthesis across 1,N2-ethenoguanine by human DNA polymerases. Chem. Res. Toxicol. 2006, 19, 879–886. [Google Scholar] [CrossRef]
- Yoon, J.H.; Johnson, R.E.; Prakash, L.; Prakash, S. DNA polymerase theta accomplishes translesion synthesis opposite 1,N(6)-ethenodeoxyadenosine with a remarkably high fidelity in human cells. Genes Dev. 2019, 33, 282–287. [Google Scholar] [CrossRef]



| N-nitrosamides | Abbreviations | Major DNA Alkylation Adducts | Sources |
|---|---|---|---|
| N-nitrosoureas | |||
| N-methyl-N-nitrosourea | MNU | N7-MeG, N3-MeA, N3-MeG, O6-MeG | Anticancer drug, Basic research |
| N-ethyl-N-nitrosourea | ENU | N7-EtG, N3-EtA, N3-EtG, O6-EtG | Basic research |
| N-(methylnitrosocarbamoyl)-α-D-glucosamine (Streptozocin) | N7-MeG, N3-MeA, N3-MeG, O6-MeG | Anticancer drug | |
| 1,3-Bis(2-chloroethyl)-1-nitrosourea; Carmustine | BCNU | N7-ClEtG, O6-ClEtG, N1,O6-EthenoG, G-C and G-G crosslinks | Anticancer drug |
| 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea; Lomustine | CCNU | N7-ClEtG, O6-ClEtG, N1,O6-EthenoG, G-C and G-G crosslinks | Anticancer drug |
| N-nitrosoguanidines | |||
| N-methyl-N’-nitro-N-nitrosoguanidine | MNNG | N7-MeG, N3-MeA N3-MeG, O6-MeG | Basic research |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahrer, J.; Christmann, M. DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. Int. J. Mol. Sci. 2023, 24, 4684. https://doi.org/10.3390/ijms24054684
Fahrer J, Christmann M. DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. International Journal of Molecular Sciences. 2023; 24(5):4684. https://doi.org/10.3390/ijms24054684
Chicago/Turabian StyleFahrer, Jörg, and Markus Christmann. 2023. "DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways" International Journal of Molecular Sciences 24, no. 5: 4684. https://doi.org/10.3390/ijms24054684
APA StyleFahrer, J., & Christmann, M. (2023). DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. International Journal of Molecular Sciences, 24(5), 4684. https://doi.org/10.3390/ijms24054684