


Transfer of Proteins from Cultured Human Adipose to Blood Cells and Induction of Anabolic Phenotype Are Controlled by Serum, Insulin and Sulfonylurea Drugs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
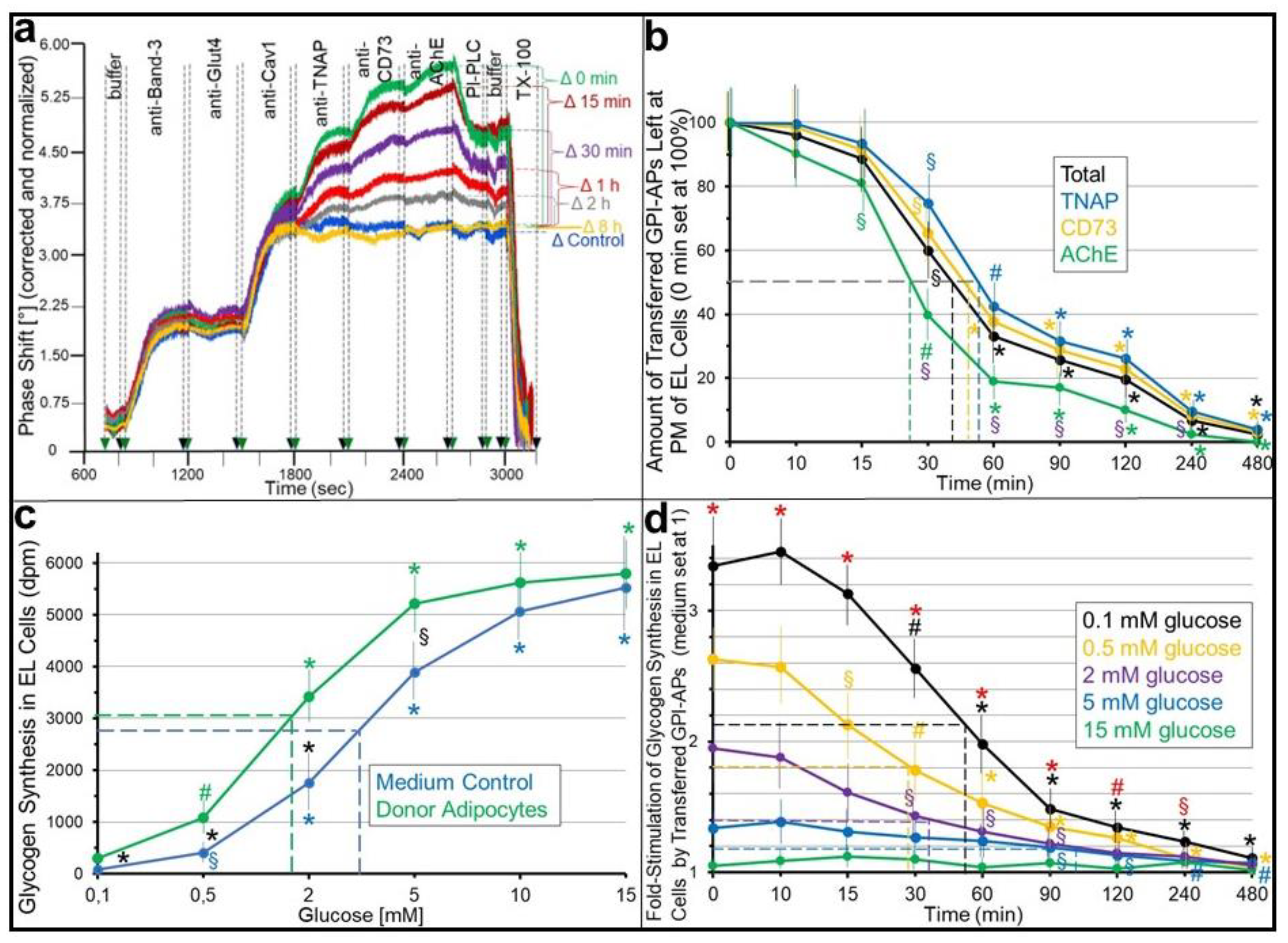
2.1. Stimulation of Basal Glycogen Synthesis upon Transfer of GPI-APs Depends on Their Localization at the PMs of the Acceptor ELCs
2.2. Insulin and Antidiabetic SUs Inhibit Both GPI-AP Transfer to and Transfer-Induced Glycogen Synthesis in Acceptor ELCs
2.3. Insulin and SU inhibition of GPI-AP Transfer and Transfer-Induced Glycogen Synthesis Is Controlled by Serum
2.4. Full-Length GPI-APs Displaced from Serum Proteins by PIG(-Proteins) Are Transferred to and Stimulate Glycogen Synthesis in Acceptor Cells
3. Discussion
3.1. Residence at PMs of Transferred GPI-APs as a Prerequisite for the Induction of Anabolic Effects
3.2. “Indirect” Transfer of GPI-APs and Its Control by Insulin and SUs
3.3. Contribution of the “Indirect” Intercellular Transfer of GPI-APs to Insulin and SU Action
3.4. The Interplay between the “Indirect” and the “Direct” Modes of GPI-AP Transfer
4. Materials and Methods
4.1. Ethical Approval
4.2. Transwell Co-Culture of Human Adipocytes and GPI-Deficient ELCs
4.3. Assay of GPI-AP Transfer
4.4. Assay of Glycogen Synthesis
4.5. siRNA Transfection of Human Adipocytes
4.6. Immobilization of Anti-GPLD1 Antibody at the Chip Surface
4.7. Statistical Analysis
4.8. Miscellaneous
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haldar, K.; Ferguson, M.A.J.; Cross, G.A.M. Acylation of a Plasmodium falciparum merozoite surface antigen via sn-1,2-diacylglycerol. J. Biol. Chem. 1985, 260, 4969–4974. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.J.; Homans, S.W.; Dwek, R.A.; Rademacher, T.W. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science 1988, 239, 753–759. [Google Scholar] [CrossRef]
- UniProt, C. 2015 UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Eisenhaber, B.; Bork, P.; Eisenhaber, B. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: Analysis of protein sequence data from complete genomes. Protein Eng. 2001, 14, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, S.; Rodriguez-Gallardo, S.; Sabido-Bozo, S.; Muniz, M. Endoplasmic reticulum export of GPI-anchored proteins. Int. J. Mol. Sci. 2019, 20, 3506. [Google Scholar] [CrossRef] [Green Version]
- Yi-Shi, L.; Fujita, M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 2020, 48, 1129–1138. [Google Scholar]
- Kinoshita, T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020, 10, 190290. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.G.; Kasai, R.S.; Hirosawa, K.M.; Nemoto, Y.L.; Ishibashi, M.; Miwa, Y.; Fujiwara, T.K.; Kusumi, A. Transient GPI-anchored protein homodimers are units for raft organization and function. Nat. Chem. Biol. 2012, 8, 774–783. [Google Scholar] [CrossRef]
- Lebreton, S.; Zurzolo, C.; Paladino, S. Organization of GPI-anchored proteins at the cell surface and its physiopathological relevance. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 403–419. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Goni, F.M. “Rafts”: A nickname for putative transient nanodomains. Chem. Phys. Lipids 2019, 218, 34–39. [Google Scholar] [CrossRef]
- Raghupathy, R.; Anilkumar, A.A.; Polley, A.; Singh, P.P.; Yadev, M.; Johnson, C.; Suryawanshi, S.; Saikam, V.; Sawant, S.D.; Panda, A.; et al. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell 2015, 161, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Nosjean, O.; Briolay, A.; Roux, B. Mammalian GPI proteins: Sorting, membrane residence and functions. Biochem. Biophys. Acta 1997, 1331, 153–186. [Google Scholar] [CrossRef]
- Fujihara, Y.; Ikawa, M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016, 57, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.A. The release of glycosylphosphatidylinositol-anchored proteins from the cell surface. Arch. Biochem. Biophys. 2018, 656, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. Glycosylphosphatidylinositol-Anchored Proteins and Their Release from Cells—From Phenomenon to Meaning, 1st ed.; Nova Science Publishers Inc.: New York, NY, USA, 2018; pp. 39–91. [Google Scholar]
- Incardona, J.P.; Rosenberry, T.L. Replacement of the glycoinositol phospholipid anchor of Drosophila acetylcholinesterase with a transmembrane domain does not alter sorting in neurons and epithelia but results in behavioral defects. Mol. Biol. Cell 1996, 7, 613–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemble, G.W.; Henis, Y.I.; White, J.M. GPI- and transmembrane-anchored influenza hemagglutinin differ in structure and receptor binding activity. J. Cell Biol. 1993, 122, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Guesdon, F.; Kaabi, Y.; Riley, A.H.; Wilkinson, I.R.; Gray, C.; James, D.C.; Artymiuk, P.J.; Sayers, J.R.; Ross, R.J. Expression of a glycosylphosphatidylinositol-anchored ligand, growth hormone, blocks receptor signaling. Biosci. Rep. 2012, 32, 653–660. [Google Scholar] [CrossRef]
- Djafarzadeh, R.; Sauter, M.; Notohamiprodjo, S.; Noessner, E.; Goyal, P.; Siess, W.; Wörnle, M.; Ribeiro, A.; Himmelein, S.; Sitter, T.; et al. Recombinant GPI-anchored TIMP-1 stimulates growth and migration of peritoneal mesothelial cells. PLoS ONE 2012, 4, e33963. [Google Scholar]
- Müller, G.; Bandlow, W. Glucose induces lipolytic cleavage of a glycolipidic plasma membrane anchor in yeast. J. Cell Biol. 1993, 122, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G.; Dearey, E.A.; Pünter, J. The sulphonylurea drug, glimepiride, stimulates release of glycosylphosphatidylinositol-anchored plasma membrane proteins from 3T3 adipocytes. Biochem. J. 1993, 289, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Dearey, E.A.; Korndörfer, A.; Bandlow, W. Stimulation of a glycosylphosphatidylinositol-specific phospholipase by insulin and the sulfonylurea, glimepiride, in rat adipocytes depends on increased glucose transport. J. Cell Biol. 1994, 126, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Bandlow, W.; Wied, S.; Müller, G. Glucose induces amphiphilic to hydrophilic conversion of a subset of glycosyl-phosphatidylinositol-anchored ectoproteins in yeast. Arch. Biochem. Biophys. 1995, 324, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R. The role of glycosyl-phosphoinositides in hormone action. J. Bioenerg. Biomembr. 1991, 23, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Romero, G.; Luttrell, L.; Rogol, A.; Zeller, K.; Hewlett, E.; Larner, J. Phosphatidylinositol-glycan anchors of membrane proteins: Potential precursors of insulin mediators. Science 1988, 240, 509–511. [Google Scholar] [CrossRef]
- Movahedi, S.; Hooper, N.M. Insulin stimulates the release of the glycosylphosphatidylinositol-anchored membrane dipeptidase from 3T3-L1 adipocytes through the action of a phospholipase C. Biochem. J. 1997, 326, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Ingham, V.; Williams, A.; Bate, C. Glimepiride reduces CD14 expression and cytokine secretion from macrophages. J. Neuroinflamm. 2014, 11, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmichi, M.; Decker, S.J.; Saltiel, A.R. Nerve growth factor stimulates the tyrosine phosphorylation of a 38-kDa protein that specifically associates with the src homology domain of phospholipase c-γ1. J. Biol. Chem. 1992, 267, 21601–21606. [Google Scholar] [CrossRef]
- Müller, G.A.; Müller, T.D. Biological role of the intercellular transfer of glycosylphosphatidylinositol-anchored proteins: Stimulation of lipid and glycogen synthesis. Int. J. Mol. Sci. 2022, 23, 7418. [Google Scholar] [CrossRef]
- Davitz, M.A.; Hereld, D.; Shak, S.; Krakow, J.; Englund, P.T.; Nussenzweig, V. A glycan-phosphatidylinositol-specific phospholipase D in human serum. Science 1987, 238, 81–84. [Google Scholar] [CrossRef]
- Li, J.-Y.; Hollfelder, K.; Huang, K.-S.; Low, M.G. Structural features of GPI-specific phospholipase D revealed by proteolytic fragmentation and Ca2+ binding studies. J. Biol. Chem. 1994, 269, 28963–28971. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Ussar, S.; Tschöp, M.H.; Müller, T.D. Age-dependent membrane release and degradation of full-length glycosylphosphatidylinositol-anchored proteins in rats. Mech. Ageing Dev. 2020, 190, 111307. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Lechner, A.; Tschöp, M.H.; Müller, T.D. Interaction of full-length glycosylphosphatidylinositol-anchored proteins with serum proteins and their translocation to cells in vitro depend on the (pre-)diabetic state in rats and humans. Biomedicines 2021, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Mohney, R.P.; Mutka, S.C.; Ravi, L.; Singleton, D.R.; Perry, G.; Tartakoff, A.M.; Medof, M.E. Derivation and characterization of glycoinositol-phospholipid anchor-defective human K562 cell clones. J. Biol. Chem. 1992, 267, 5272–5278. [Google Scholar] [CrossRef] [PubMed]
- Andrä, J.; Böhling, A.; Gronewold, T.M.A.; Schlecht, U.; Perpeet, M.; Gutsmann, T. Surface acoustic wave biosensor as a tool to study the interaction of antimicrobial peptides with phospholipid and lipopolysaccharide model membranes. Langmuir 2008, 24, 9148–9153. [Google Scholar] [CrossRef] [PubMed]
- Gronewold, T.M.A.; Glass, S.; Quandt, E.; Famulok, M. Monitoring complex formation in the blood-coagulation cascade using aptamer-coated SAW sensors. Biosens. Bioelectron. 2005, 20, 2044–2052. [Google Scholar] [CrossRef]
- Huang, K.; Park, S. Affinity purification of glycosylphosphatidylinositol-anchored proteins by alpha-toxin. In Glycosaminoglycans. Methods in Molecular Biology; Balagurunathan, K., Nakato, H., Desai, U., Saijoh, Y., Eds.; Humana Press: New York, NY, USA, 2022; Volume 2303. [Google Scholar]
- Lazar, D.F.; Knez, J.J.; Medof, M.E.; Cuatrecasas, P.; Saltiel, A.R. Stimulation of glycogen synthesis by insulin in human erythroleukemia cells requires the synthesis of glycosylphosphatidylinositol. Proc. Natl. Acad. Sci. USA 1994, 91, 9665–9669. [Google Scholar] [CrossRef] [Green Version]
- Davitz, M.A.; Low, M.G.; Nussenzweig, V. Release of decay-accelerating factor (DAF) from the cell membrane by phosphatidylinositol-specific phospholipase C (PIPLC). Selective modification of a complement regulatory protein. J. Exp. Med. 1986, 163, 1150–1161. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.W.; Low, M.G. Biosynthesis of glycosylphosphatidylinositol-anchored human placental alkaline phosphatase: Evidence for a phospholipase C-sensitive precursor and its post-attachment conversion into a phospholipase C-resistant form. Biochem. J. 1994, 301, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Fu, H.; He, B.; Wang, D.; Qin, M.; Yang, D.; Liu, D.; Song, G.; Shi, Y.; Zhang, H.; et al. Rho GTPases in A549 and Caco-2 cells dominating the endocytic pathways of nanocarbons with different morphologies. Int. J. Nanomed. 2018, 13, 4391–4404. [Google Scholar] [CrossRef] [Green Version]
- Lakhan, S.E.; Sabharanjak, S.; De, A. Endocytosis of glycosylphosphatidylinositol-anchored proteins. J. Biomed. Sci. 2009, 16, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadda, R.; Howes, M.T.; Plowman, S.J.; Hancock, J.F.; Parton, R.G.; Mayor, S. Cholesterol-sensitive Cdc42 activation regulates actin polymerization for endocytosis via the GEEC pathway. Traffic 2007, 8, 702–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.P.A.; Boucrot, E. Mechanisms of carrier formation during clathrin-independent endocytosis. Trends Cell Biol. 2018, 28, 188–200. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Bouquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, A.; Yip, C.; Paye, A.; Blacher, S.; Munaut, C.; Deroanne, C.; Noel, A.; Sounni, N.E. Dynamics of internalization and recycling of the prometastatic membrane type 4 matrix metalloproteinase (MT4-MMP) in breast cancer cells. FEBS J. 2016, 283, 704–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Chip-based sensing of the intercellular transfer of cell surface proteins: Regulation by the metabolic state. Biomedicines 2021, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Stralfors, P. Insulin second messengers. Bioessays 1997, 19, 327–335. [Google Scholar] [CrossRef]
- Sharom, F.J.; Radeva, G. GPI-anchored protein cleavage in the regulation of transmembrane signals. Subcell. Biochem. 2004, 37, 285–315. [Google Scholar] [PubMed]
- Varela-Nieto, I.; Leon, Y.; Caro, H.N. Cell signalling by inositol phosphoglycans from different species. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 1996, 115, 223–241. [Google Scholar] [CrossRef]
- Müller, G.; Schulz, A.; Wied, S.; Frick, W. Regulation of lipid raft proteins by glimepiride- and insulin-induced glycosylphosphatidylinositol-specific phospholipase C in rat adipocytes. Biochem. Pharmacol. 2005, 69, 761–780. [Google Scholar] [CrossRef]
- Arner, P.; Pettersson, A.; Mitchell, P.J.; Dunbar, J.D.; Kharitonenkov, A.; Ryden, M. FGF21 attenuates lipolysis in human adipocytes—A possible link to improved insulin sensitivity. FEBS Lett. 2008, 582, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- DeMarsilis, A.; Reddy, N.; Boutari, C.; Filippaios, A.; Sternthal, E.; Katsiki, N.; Mantzoros, C. Pharmacotherapy of type 2 diabetes: An update and future directions. Metabolism 2022, 137, 155332. [Google Scholar] [CrossRef]
- Agardh, C.D.; Björgell, P.; Nilsson-Ehle, P. The effects of tolbutamide on lipoproteins, lipoprotein lipase and hormone-sensitive lipase. Diabetes Res. Clin. Pract. 1999, 46, 99–108. [Google Scholar] [CrossRef]
- Müller, G.A.; Herling, A.W.; Stemmer, K.; Lechner, A.; Tschöp, M.H. Chip-based sensing for release of unprocessed cell surface proteins in vitro and in serum and its (patho)physiological relevance. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E212–E233. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Wied, S. The sulfonylurea drug, glimepiride, stimulates glucose transport, glucose transporter translocation, and dephosphorylation in insulin-resistant rat adipocytes in vitro. Diabetes 1993, 42, 1852–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocuzzi, E.; Szczotka, L.B.; Brodbeck, W.G.; Bardenstein, D.S.; Wei, T.; Medof, M.E. Tears contain the complement regulator CD59 as well as decay-accelerating factor (DAF). Clin. Exp. Immunol. 2001, 123, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Dvorakova, M.C. Future perspective of diabetic animal models. Endocrinol. Metab. Immune Dis.–Drug Targets 2020, 20, 25–38. [Google Scholar] [CrossRef]
- Wang, T.; Tang, X.; Hu, X.; Wang, J.; Chen, G. Reduction in the dietary VA status prevents type 2 diabetes and obesity in Zucker diabetic fatty rats. Biomolecules 2022, 12, 528. [Google Scholar] [CrossRef]
- Xia, C.; Zhang, X.; Cao, T.; Wang, J.; Li, C.; Yue, L.; Niu, K.; Shen, Y.; Ma, G.; Chen, F. Hepatic transcriptome analysis revealing the molecular pathogenesis of type 2 diabetes mellitus in Zucker Diabetic Fatty rats. Front. Endocrinol. 2020, 11, 565858. [Google Scholar] [CrossRef]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Hirose, S.; Ravi, L.; Hazra, S.V.; Medof, M.E. Assembly and deacylation of N-acetylglucosaminyl-plasmanylinositol in normal and affected paroxysmal nocturnal hemoglobinuria cells. Proc. Natl. Acad. Sci. USA 1991, 88, 3762–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G.A.; Tschöp, M.H.; Müller, T.D. Upregulated phospholipase D activity toward glycosylphosphatidylinositol-anchored proteins in micelle-like serum complexes in metabolically deranged rats and humans. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E462–E479. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Balatskaya, M.N.; Tkachuk, V.A. Glycosylphosphatidylinositol-anchored proteins as regulators of cortical cytoskeleton. Biochemistry 2016, 81, 636–650. [Google Scholar] [CrossRef]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.-A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic crosstalk: Cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014, 12, e1001832. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Geisen, K. Characterization of the molecular mode of action of the sulfonylurea, glimepiride, at adipocytes. Horm. Metab. Res. 1996, 28, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Satoh, Y.; Geisen, K. Extrapancreatic effects of sulfonylureas—A comparison between glimepiride and conventional sulfonylureas. Diabetes Res. Clin. Pract. 1995, 28, S115–S137. [Google Scholar] [CrossRef]
- Anderson, S.M.; Yu, G.; Giattina, M.; Miller, J.L. Intercellular transfer of glycosylphosphatidylinositol (GPI)-linked protein: Release and uptake of CD4-GPI from recombinant adeno-associated virus-transduced Hela cells. Proc. Nat. Acad. Sci. USA 1996, 93, 5894–5898. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.L.; Lisanti, M.P.; Rodriguez-Boulan, E.; Saltiel, A.R. Insulin-stimulated release of lipoprotein lipase by metabolism of its phosphatidylinositol anchor. Science 1988, 241, 1670–1672. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Proks, P.; Reimann, F.; Green, N.; Gribble, F.; Ashcroft, F. Sulfonylurea stimulation of insulin secretion. Diabetes 2002, 51 (Suppl. 3), S368–S376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamos, E.L.; Stein, S.A.; Davis, S.N. Sulfonylureas and meglitinides: Historical and contemporary issues. Panminerva Med. 2013, 55, 239–251. [Google Scholar] [PubMed]
- Prendergast, B.D. Glyburide and glipizide, second-generation oral sulfonylurea hypoglycemic agents. Clin. Pharm. 1984, 3, 473–485. [Google Scholar] [PubMed]
- Brietzke, S.A. Oral antihyperglycemic treatment options for type 2 diabetes mellitus. Med. Clin. North Am. 2015, 99, 87–106. [Google Scholar] [CrossRef]
- Kramer, W.; Müller, G.; Geisen, K. Characterization of the molecular mode of action of the sulfonylurea, glimepiride, at beta-cells. Horm. Metab. Res. 1996, 28, 464–468. [Google Scholar] [CrossRef]
- Kramer, W.; Müller, G.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Hartz, D.; Summ, H.-D. Differential interaction of glimepiride and glibenclamide with the beta-cell sulfonylurea receptor. II. Photoaffinity labeling of a 65 kDa protein by (3H)glimepiride. Biochim. Biophys. Acta 1994, 1191, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Schulz, A.; Hartz, D.; Dearey, E.-A.; Wetekam, E.-M.; Ökonomopulos, R.; Crecelius, A.; Wied, S.; Frick, W. Novel glimepiride derivatives with potential as double-edged swords against type II diabetes. Arch. Physiol. Biochem. 2010, 116, 3–20. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Wetekam, E.M.; Crecelius, A.; Unkelbach, A.; Pünter, J. Stimulation of glucose utilization in 3T3 adipocytes and rat diaphragms in vitro by the sulfonylureas, glimepiride and glibenclamide, is correlated with modulations of the cAMP regulatory cascade. Biochem. Pharmacol. 1994, 48, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Haupt, A.; Kausch, C.; Dahl, D.; Bachmann, O.; Stumvoll, M.; Häring, H.-U.; Matthaei, S. Effect of glimepiride on insulin-stimulated glycogen synthesis in cultured human skeletal muscle cells: A comparison to glibenclamide. Diabetes Care 2002, 25, 2129–2132. [Google Scholar] [CrossRef] [Green Version]
- Bähr, M.; von Holtey, M.; Müller, G.; Eckel, J. Direct stimulation of myocardial glucose transport and glucose transporter-1 (GLUT1) and GLUT4 protein expression by the sulfonylurea glimepiride. Endocrinology 1995, 136, 2547–2553. [Google Scholar] [CrossRef]
- Emini-Sadiku, M.; Car, N.; Begolli, L.; Blaslov, K.; Haliti, E.; Bahtiri, E. The differential influence of glimepiride and glibenclamide on insulin resistance and adiponectin levels in patients with type 2 diabetes. Endocr. J. 2019, 66, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Draeger, E. Clinical profile of glimepiride. Diabetes Res. Clin. Pract. 1995, 28, S139–S146. [Google Scholar] [CrossRef]
- Rosenstock, J.; Samols, E.; Muchmore, D.B.; Schneider, J. Glimepiride, a new once-daily sulfonylurea. A double-blind placebo-controlled study of NIDDM patients. Glimepiride study group. Diabetes Care 1996, 19, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Raptis, S.A.; Hatziagelaki, E.; Dimitriadis, G.; Draeger, K.E.; Pfeiffer, C.; Raptis, A.E. Comparative effects of glimepiride and glibenclamide on blood glucose, C-peptide and insulin concentrations in the fasting and postprandial state in normal man. Exp. Clin. Endocrinol. Diabetes 1999, 107, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Hirose, T.; Asakawa, H.; Itoh, Y.; Kamado, K.; Tokunaga, K.; Tomita, K.; Masuda, H.; Watanabe, N.; Namba, M. Efficacy of glimepiride in type 2 diabetic patients treated with glibenclamide. Diabetes Res. Clin. Pract. 2004, 66, S129–S132. [Google Scholar] [CrossRef] [PubMed]
- Hribal, M.L.; D’Alfonso, R.; Giovannone, B.; Lauro, D.; Liu, Y.Y.; Borboni, P.; Federici, M.; Lauro, R.; Sesti, G. The sulfonylurea glimepiride regulates intracellular routing of the insulin-receptor complexes through their interaction with specific protein kinase C isoforms. Mol. Pharmacol. 2001, 59, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Gao, Z.; He, C.; Xiang, R.; van Kuppevelt, T.H.; Belting, M.; Zhang, S. GRP75 upregulates clathrin-independent endocytosis through actin cytoskeleton reorganization mediated by the concurrent activation of Cdc42 and RhoA. Exp. Cell Res. 2016, 343, 223–236. [Google Scholar] [CrossRef]
- Guha, A.; Sriram, V.; Krishnan, K.S.; Mayor, S. shibire mutations reveal distinct dynamin-independent and -dependent endocytic pathways in primary cultures of Drosophila hemocytes. J. Cell Sci. 2003, 116, 3373–3386. [Google Scholar] [CrossRef] [Green Version]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Mol. Biol. Cell 2004, 15, 3542–3552. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.; Wetekam, E.; Jung, C.; Bandlow, W. Membrane association of lipoprotein lipase and a cAMP-binding ectoprotein in rat adipocytes. Endocrinology 1997, 33, 12149–12159. [Google Scholar] [CrossRef] [Green Version]
- Langtry, H.D.; Balfour, J.A. Glimepiride. A review of its use in the management of type 2 diabetes mellitus. Drugs 1998, 55, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Kaku, K.; Inoue, Y.; Kaneko, T. Extrapancreatic effects of sulfonylurea drugs. Diabetes Res. Clin. Pract. 1995, 28, S105–S108. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M. Insulin mediators revisited. Cell. Signal. 1989, 1, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Low, M.G.; Saltiel, A.R. Structural and functional roles of glycosylphosphatidylinositol in membranes. Science 1988, 239, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V. Lipid-derived mediators in insulin. Proc. Soc. Exp. Biol. Med. 1990, 195, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, G.N.; Pratt, J.C. Glycosylated phosphatidylinositol molecules as second messengers. Semin. Immunol. 1994, 6, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Frick, W.; Bauer, A.; Bauer, J.; Wied, S.; Müller, G. Structure-activity relationship of synthetic phosphoinositolglycans mimicking metabolic insulin action. Biochemistry 1998, 37, 13421–13436. [Google Scholar] [CrossRef]
- Müller, G.; Wied, S.; Piossek, C.; Bauer, A.; Bauer, J.; Frick, W. Convergence and divergence of the signaling pathways for insulin and phosphoinositolglycans. Mol. Med. 1998, 4, 299–323. [Google Scholar] [CrossRef] [Green Version]
- Larner, J.; Brautigam, D.L.; Thorner, M.O. D-chiro-inositol-glycans in insulin signaling and insulin resistance. Mol. Med. 2010, 16, 543–552. [Google Scholar] [CrossRef]
- Fonteles, M.C.; Huang, L.C.; Larner, J. Infusion of pH 2.0 D-chiro-inositol glycan insulin putative mediator normalizes plasma glucose in streptozotocin diabetic rats at a dose equivalent to insulin without inducing hypoglycaemia. Diabetologia 1996, 39, 731–734. [Google Scholar] [CrossRef]
- Shaskin, P.N.; Shashkina, E.F.; Fernqvist-Forbes, E.; Zhou, Y.P.; Grill, V.; Katz, A. Insulin mediators in man: Effects of glucose ingestion and insulin resistance. Diabetologia 1997, 40, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Deeg, M.A., Brass; Eric, P.B.; Rosenberry, T.L. Inositol glycan phosphate derived from human erythrocyte acetylcholinesterase glycolipid anchor and inositol cyclic 1,2-phosphate antagonize glucagon activation of glycogen phosphorylase. Diabetes 1993, 42, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Misek, D.E.; Saltiel, A.R. An inositol phosphate glycan derived from a Trypanosoma brucei glycosyl-phosphatidylinositol mimics some of the metabolic actions of insulin. J. Biol. Chem. 1992, 267, 16266–16276. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Wied, S.; Crecelius, A.; Kessler, A.; Eckel, J. Phosphoinositolglycan-peptides from yeast potently induce metabolic insulin actions in isolated rat adipocytes, cardiomyocytes and diaphragms. Endocrinology 1997, 138, 3459–3475. [Google Scholar] [CrossRef]
- Kessler, A.; Müller, G.; Wied, S.; Crecelius, A.; Eckel, J. Signalling pathways of an insulin-mimetic phosphoinositolglycan-peptide in muscle and adipose tissue. Biochem. J. 1998, 330, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G. The molecular mechanism of the insulin-mimetic/sensitizing activity of the antidiabetic sulfonylurea drug Amaryl (review). Mol. Med. 2000, 6, 907–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, G.; Welte, S. Caveolae are the targets for the insulin-independent blood glucose-decreasing activity of the sulfonylurea glimepiride. Diabetol. Prakt. 2001, 2 (Suppl. B), 45–55. [Google Scholar]
- Müller, G.; Welte, S. Lipid raft domains are the targets for the insulin-independent blood glucose-decreasing activity of the sulfonylurea glimepiride. Rec. Res. Develop. Endocrinol. 2002, 3, 401–423. [Google Scholar]
- Loureiro, Z.Y.; Solivan-Rivera, J.; Corvera, S. Adipocyte heterogeneity underlying tissue functions. Endocrinology 2022, 163, bqab138. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef]
- Cherkas, A.; Holota, S.; Mdzinarashvili, T.; Gabbianelli, R.; Zarkovic, N. Glucose as a major antioxidant: When, what for and why it fails? Antioxidants 2020, 9, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertheuil, N.; Chaput, B.; Menard, C.; Varin, A.; Laloze, J.; Watier, E.; Tarte, K. Adipose mesenchymal stromal cells: Definition, immunomodulatory properties, mechanical isolation and interest for plastic surgery. Ann. Chir. Plast. Esthet. 2019, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Manea, E. A step closer in defining glycosylphosphatidylinositol anchored proteins in health and glycosylation disorders. Mol. Genet. Metab. Rep. 2018, 16, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A. Membrane insertion and intracellular transfer of glycosylphosphatidylinositol-anchored proteins: Potential therapeutic applications. Arch. Physiol. Biochem. 2020, 126, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Bonfilio, R.; de Araujo, M.B.; Salgado, H.R.N. A review of analytical techniques for determination of glimepiride: Present and perspectives. Ther. Drug Monit. 2010, 32, 550–559. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, G.A.; Müller, T.D. Transfer of Proteins from Cultured Human Adipose to Blood Cells and Induction of Anabolic Phenotype Are Controlled by Serum, Insulin and Sulfonylurea Drugs. Int. J. Mol. Sci. 2023, 24, 4825. https://doi.org/10.3390/ijms24054825
Müller GA, Müller TD. Transfer of Proteins from Cultured Human Adipose to Blood Cells and Induction of Anabolic Phenotype Are Controlled by Serum, Insulin and Sulfonylurea Drugs. International Journal of Molecular Sciences. 2023; 24(5):4825. https://doi.org/10.3390/ijms24054825
Chicago/Turabian StyleMüller, Günter A., and Timo D. Müller. 2023. "Transfer of Proteins from Cultured Human Adipose to Blood Cells and Induction of Anabolic Phenotype Are Controlled by Serum, Insulin and Sulfonylurea Drugs" International Journal of Molecular Sciences 24, no. 5: 4825. https://doi.org/10.3390/ijms24054825
APA StyleMüller, G. A., & Müller, T. D. (2023). Transfer of Proteins from Cultured Human Adipose to Blood Cells and Induction of Anabolic Phenotype Are Controlled by Serum, Insulin and Sulfonylurea Drugs. International Journal of Molecular Sciences, 24(5), 4825. https://doi.org/10.3390/ijms24054825