
Combinatorial Blood Platelets-Derived circRNA and mRNA Signature for Early-Stage Lung Cancer Detection


 , , , , , , ,
, , , , , , ,  ,
,
Abstract
:
1. Introduction
2. Results
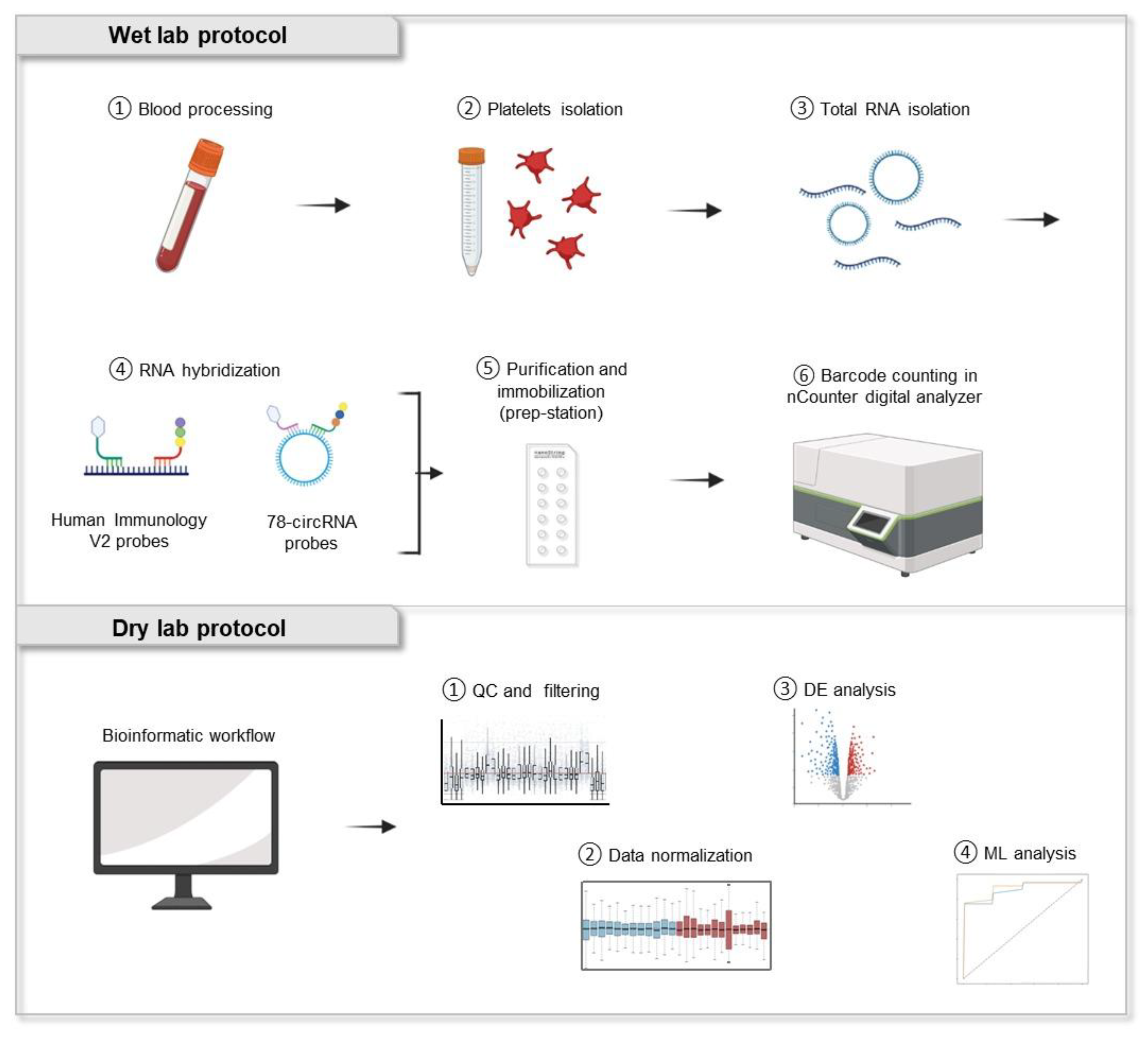
2.1. Analysis of Blood Platelet-Derived RNA Using NanoString nCounter Technology
2.2. Profiling mRNA and circRNA Derived from Lung Cancer Patients and Non-Cancer Individuals Using Human Immunology V2 Panel and 78-circRNA Custom Panel
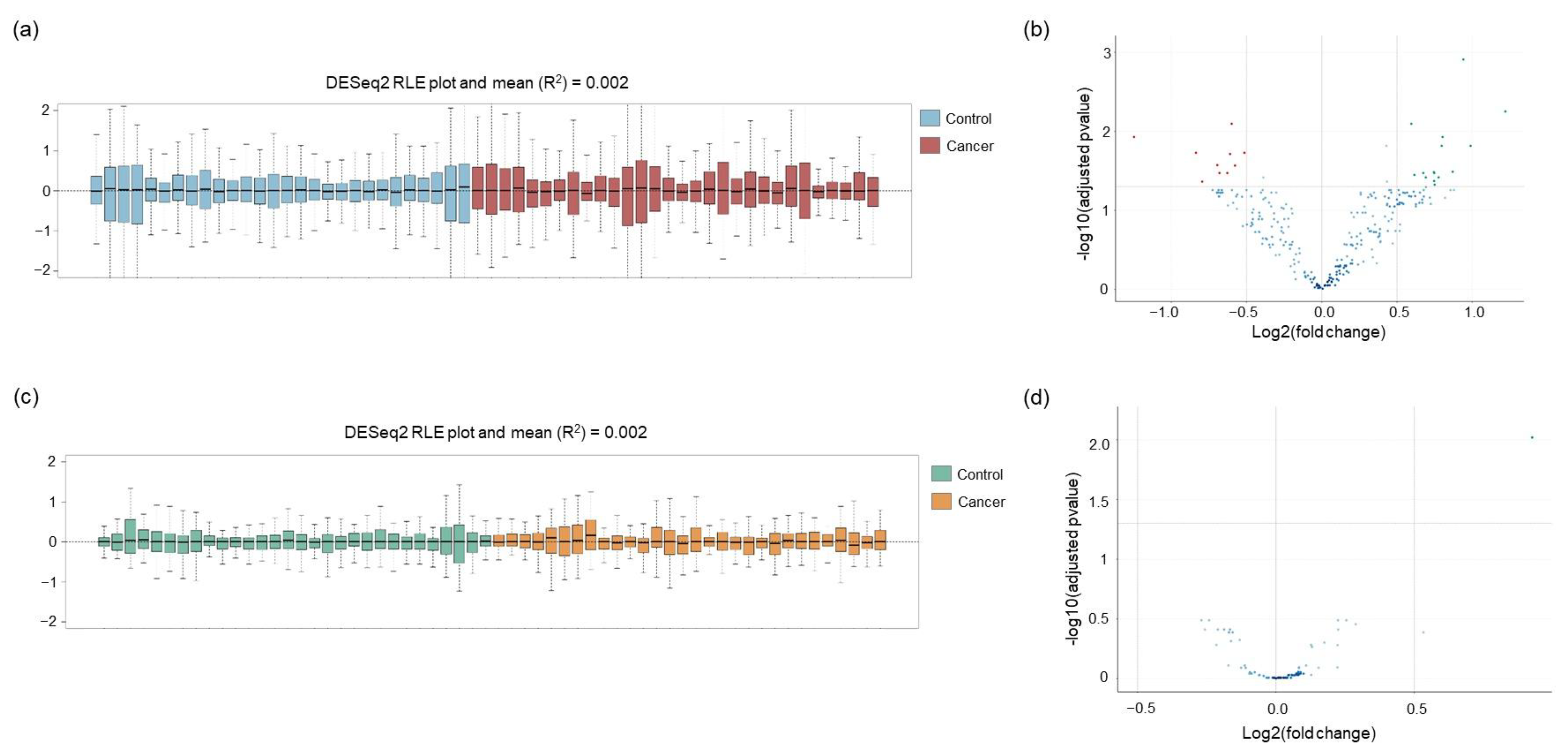
2.3. Normalization of the Raw Counts and Differential Gene Expression between Lung Cancer Patients and Non-Cancer Individuals
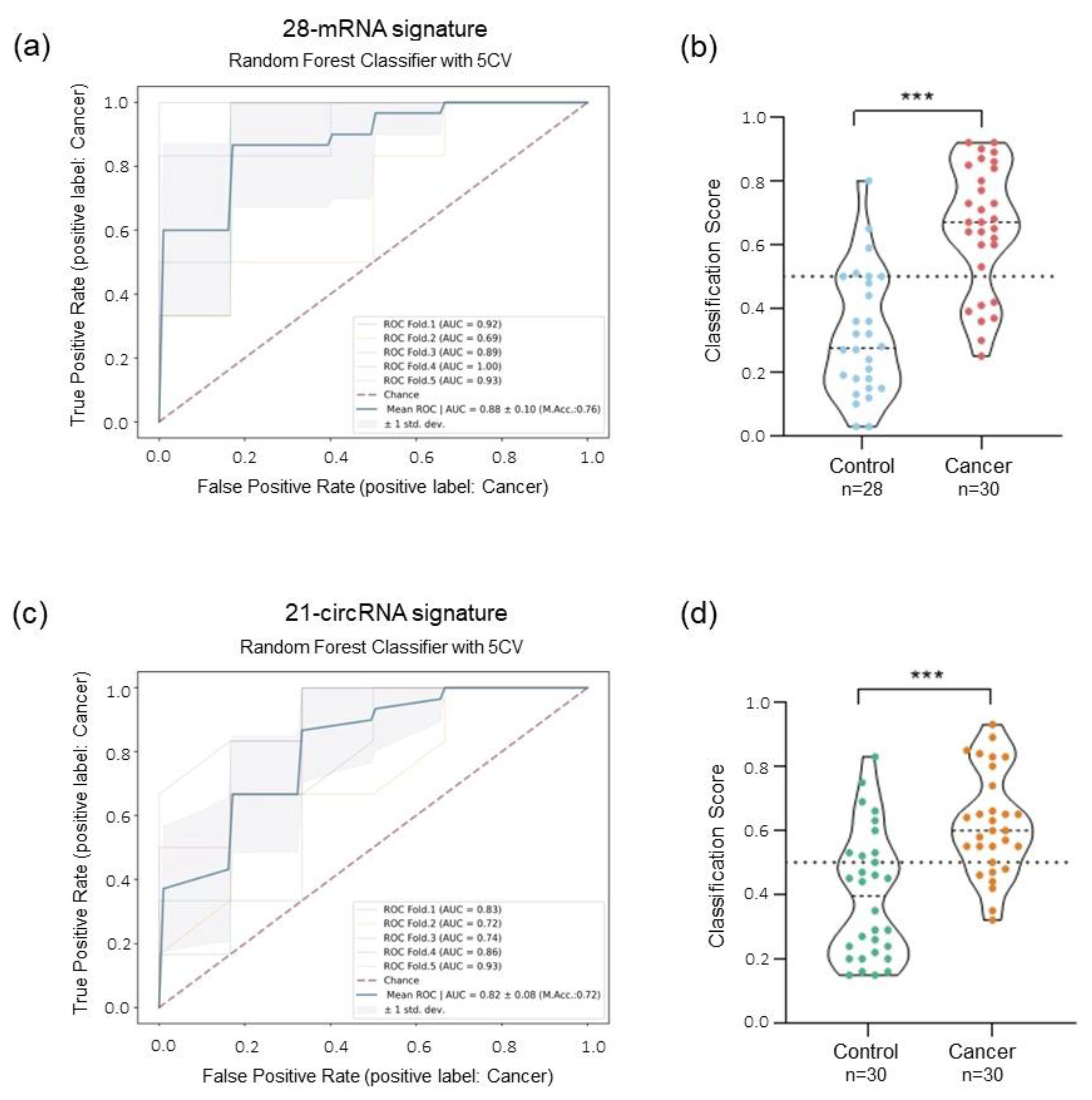
2.4. ML-Classifier Development and Performance for Detection of Lung Cancer Patients Using Human Immunology V2 Panel and 78-circRNA Custom Panel
2.5. Combinatorial Analysis: mRNA and circRNA Signature for the Detection of Lung Cancer Patients
2.6. Early-Stage Lung Cancer Detection Using Combinatorial Signature of mRNA and circRNA
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Population Study
4.2. Isolation of Blood Platelets
4.3. Total RNA Isolation
4.4. Gene Expression Analysis Using nCounter
4.5. Data Normalization and Differential Expression Analysis
4.6. Feature Selection and Classification Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC lung cancer staging project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM Classification for lung cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Jeon, D.S.; Kim, H.C.; Kim, S.H.; Kim, T.-J.; Kim, H.K.; Moon, M.H.; Beck, K.S.; Suh, Y.-G.; Song, C.; Ahn, J.S.; et al. Five-Year Overall Survival and Prognostic Factors in Patients with Lung Cancer: Results from the Korean Association of Lung Cancer Registry (KALC-R) 2015. Cancer Res. Treat. 2022, 55, 103–111. [Google Scholar] [CrossRef] [PubMed]
- SEER. SEER*Explorer: An Interactive Website for SEER Cancer Statistics. Surveillance Research Program, National Cancer Institute. Available online: https://seer.cancer.gov/explorer/ (accessed on 27 September 2021).
- Bracht, J.W.P.; Mayo-de-las-Casas, C.; Berenguer, J.; Karachaliou, N.; Rosell, R. The Present and Future of Liquid Biopsies in Non-Small Cell Lung Cancer: Combining Four Biosources for Diagnosis, Prognosis, Prediction, and Disease Monitoring. Curr. Oncol. Rep. 2018, 20, 70. [Google Scholar] [CrossRef]
- Antunes-Ferreira, M.; Koppers-Lalic, D.; Würdinger, T. Circulating platelets as liquid biopsy sources for cancer detection. Mol. Oncol. 2021, 15, 1727–1743. [Google Scholar] [CrossRef]
- Best, M.G.; Wesseling, P.; Wurdinger, T. Tumor-educated platelets as a noninvasive biomarker source for cancer detection and progression monitoring. Cancer Res. 2018, 78, 3407–3412. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, S.; Nilsson, R.J.; Wurdinger, T. Platelets and tumor-associated RNA transfer. Blood 2021, 137, 3181–3191. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Best, M.G.; Sol, N.; In ‘t Veld, S.G.J.G.; Vancura, A.; Muller, M.; Niemeijer, A.L.N.; Fejes, A.V.; Tjon Kon Fat, L.A.; Huis In ‘t Veld, A.E.; Leurs, C.; et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell 2017, 32, 238–252.e9. [Google Scholar] [CrossRef] [Green Version]
- Sol, N.; In ‘t Veld, S.G.J.G.; Vancura, A.; Tjerkstra, M.; Leurs, C.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Zwaan, K.; et al. Tumor-Educated Platelet RNA for the Detection and (Pseudo)progression Monitoring of Glioblastoma. Cell Rep. Med. 2020, 1, 100101. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; In’t Veld, S.G.J.G.; Dwarshuis, G.; Van Den Broek, D.; Sol, N.; Best, M.G.; Van Coevorden, F.; Haas, R.L.; Beijnen, J.H.; van Houdt, W.J.; et al. RNA-sequencing of tumor-educated platelets, a novel biomarker for blood-based sarcoma diagnostics. Cancers 2020, 12, 1372. [Google Scholar] [CrossRef]
- D’ambrosi, S.; Visser, A.; Antunes-Ferreira, M.; Poutsma, A.; Giannoukakos, S.; Sol, N.; Sabrkhany, S.; Bahce, I.; Kuijpers, M.J.E.; Oude Egbrink, M.G.A.; et al. The analysis of platelet-derived circRNA repertoire as potential diagnostic biomarker for non-small cell lung cancer. Cancers 2021, 13, 4644. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.Y.W.H.; Pa, D.; Donahue, A.D.D.; Sc, D. The production of blood platelets in lungs. J. Exp. Med. 1936, 6, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Mende, N.; Bastos, H.P.; Santoro, A.; Mahbubani, K.T.; Ciaurro, V.; Calderbank, E.F.; Londoño, M.Q.; Sham, K.; Mantica, G.; Morishima, T.; et al. Unique molecular and functional features of extramedullary hematopoietic stem and progenitor cell reservoirs in humans. Blood 2022, 139, 3387–3401. [Google Scholar] [CrossRef]
- Johnston, I.; Hayes, V.; Poncz, M. Threading an elephant through the eye of a needle: Where are platelets made? Cell Res. 2017, 27, 1079–1080. [Google Scholar] [CrossRef] [Green Version]
- Calverley, D.C.; Phang, T.L.; Choudhury, Q.G.; Gao, B.; Oton, A.B.; Weyant, M.J.; Geraci, M.W. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin. Transl. Sci. 2010, 3, 227–232. [Google Scholar] [CrossRef]
- Heinhuis, K.; In ’t Veld, S.; Dwarshuis, G.; van den Broek, D.; Sol, N.; Best, M.; Koenen, A.; Steeghs, N.; Coevorden, F.; Haas, R.; et al. RNA-Sequencing of Tumor-Educated Platelets, A Novel Biomarker for Blood Based Sarcoma Diagnostics. Eur. J. Surg. Oncol. 2020, 46, e7. [Google Scholar] [CrossRef]
- Best, M.G.; In ’t Veld, S.G.J.G.; Sol, N.; Wurdinger, T. RNA sequencing and swarm intelligence–enhanced classification algorithm development for blood-based disease diagnostics using spliced blood platelet RNA. Nat. Protoc. 2019, 14, 1206–1234. [Google Scholar] [CrossRef]
- In ’t Veld, S.G.J.G.; Arkani, M.; Post, E.; Antunes-Ferreira, M.; D’Ambrosi, S.; Vessies, D.C.L.; Vermunt, L.; Vancura, A.; Muller, M.; Niemeijer, A.L.N.; et al. Detection and localization of early- and late-stage cancers using platelet RNA. Cancer Cell 2022, 40, 999–1009.e6. [Google Scholar] [CrossRef] [PubMed]
- Mantini, G.; Meijer, L.L.; Glogovitis, I.; In ‘t Veld, S.G.J.G.; Paleckyte, R.; Capula, M.; Le Large, T.Y.S.; Morelli, L.; Pham, T.V.; Piersma, S.R.; et al. Omics analysis of educated platelets in cancer and benign disease of the pancreas. Cancers 2021, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Alhasan, A.A.; Izuogu, O.G.; Al-Balool, H.H.; Steyn, J.S.; Evans, A.; Colzani, M.; Ghevaert, C.; Mountford, J.C.; Marenah, L.; Elliott, D.J.; et al. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood 2016, 127, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Lee, S.K.; Sood, A.K. Circular RNAs in Cancer. Mol. Ther.-Nucleic Acids 2019, 16, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, S.K.; Horne, W.T.; Kolls, J.K. Research Techniques Made Simple: Methodology and Clinical Applications of RNA Sequencing. J. Investig. Dermatol. 2016, 136, e77–e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnant, M.; Filipits, M.; Greil, R.; Stoeger, H.; Rudas, M.; Bago-Horvath, Z.; Mlineritsch, B.; Kwasny, W.; Knauer, M.; Singer, C.; et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: Using the PAM50 Risk of Recurrence score in 1478 postmenopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann. Oncol. 2014, 25, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hannouf, M.B.; Zaric, G.S.; Blanchette, P.; Brezden-Masley, C.; Paulden, M.; McCabe, C.; Raphael, J.; Brackstone, M. Cost-effectiveness analysis of multigene expression profiling assays to guide adjuvant therapy decisions in women with invasive early-stage breast cancer. Pharm. J. 2020, 20, 27–46. [Google Scholar] [CrossRef] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Giménez-Capitán, A.; Bracht, J.; García, J.J.; Jordana-Ariza, N.; García, B.; Garzón, M.; Mayo-De-Las-Casas, C.; Viteri-Ramirez, S.; Martinez-Bueno, A.; Aguilar, A.; et al. Multiplex Detection of Clinically Relevant Mutations in Liquid Biopsies of Cancer Patients Using a Hybridization-Based Platform. Clin. Chem. 2021, 67, 554–563. [Google Scholar] [CrossRef]
- Kamyabi, N.; Abbasgholizadeh, R.; Maitra, A.; Ardekani, A.; Biswal, S.L.; Grande-Allen, K.J. Isolation and mutational assessment of pancreatic cancer extracellular vesicles using a microfluidic platform. Biomed. Microdevices 2020, 22, 1–11. [Google Scholar] [CrossRef]
- Porras, T.B.; Kaur, P.; Ring, A.; Schechter, N.; Lang, J.E. Challenges in using liquid biopsies for gene expression profiling. Oncotarget 2018, 9, 7036–7053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, T.N.; Boumber, Y.A.; Aggarwal, C.; Pei, J.; Thrash-Bingham, C.; Fittipaldi, P.; Vlasenkova, R.; Rao, C.; Borghaei, H.; Cristofanilli, M.; et al. Circulating tumor cell and cell-free RNA capture and expression analysis identify platelet-associated genes in metastatic lung cancer. BMC Cancer 2019, 19, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossenkov, A.V.; Qureshi, R.; Dawany, N.B.; Wickramasinghe, J.; Liu, Q.; Sonali Majumdar, R.; Chang, C.; Widura, S.; Kumar, T.; Horng, W.H.; et al. A gene expression classifier from whole blood distinguishes benign from malignant lung nodules detected by low-dose CT. Cancer Res. 2019, 79, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracht, J.W.P.; Gimenez-Capitan, A.; Huang, C.Y.; Potie, N.; Pedraz-Valdunciel, C.; Warren, S.; Rosell, R.; Molina-Vila, M.A. Analysis of extracellular vesicle mRNA derived from plasma using the nCounter platform. Sci. Rep. 2021, 11, 3712. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Contreras, M.; Shah, S.H.; Tamayo, A.; Robbins, P.D.; Golberg, R.B.; Mendez, A.J.; Ricordi, C. Plasma-derived exosome characterization reveals a distinct microRNA signature in long duration Type 1 diabetes. Sci. Rep. 2017, 7, 5998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- nCounter NanoString Human Immunology V2 Panel, Omics_Ena_Project, V. 1970. Available online: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA751396 (accessed on 1 August 2021).
- Pedraz-Valdunciel, C.; Giannoukakos, S.; Potie, N.; Giménez-Capitán, A.; Huang, C.Y.; Hackenberg, M.; Fernandez-Hilario, A.; Bracht, J.; Filipska, M.; Aldeguer, E.; et al. Digital multiplexed analysis of circular RNAs in FFPE and fresh non-small cell lung cancer specimens. Mol. Oncol. 2022, 16, 2367–2383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhao, J.; Zou, H.; Cai, K. CircRNA PTPRM Promotes Non-Small Cell Lung Cancer Progression by Modulating the miR-139-5p/SETD5 Axis. Technol. Cancer Res. Treat. 2022, 21, 1–13. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Q.; Xia, N.; Yang, H.; Hu, C. Decreased expression of microRNA-375 in nonsmall cell lung cancer and its clinical significance. J. Int. Med. Res. 2012, 40, 1662–1669. [Google Scholar] [CrossRef]
- Liang, Y.; Li, S.; Tang, L. Microrna 320, an anti-oncogene target mirna for cancer therapy. Biomedicines 2021, 9, 591. [Google Scholar] [CrossRef]
- Hou, L.; Luo, P.; Ma, Y.; Jia, C.; Yu, F.; Lv, Z.; Wu, C.; Fu, D. MicroRNA-125a-3p downregulation correlates with tumorigenesis and poor prognosis in patients with non-small cell lung cancer. Oncol. Lett. 2017, 14, 4441–4448. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Lin, L.; Zhang, Q.; Wu, W.; Su, A. Bioinformatics analysis of the circRNA–miRNA–mRNA network for non-small cell lung cancer. J. Int. Med. Res. 2020, 48, 0300060520929167. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Liu, J.; Jiao, W. Hsa_Circ_0000729, a Potential Prognostic Biomarker in Lung Adenocarcinoma. Thorac. Cancer 2018, 9, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wei, J.; Li, M.; Zhao, Y. A circular RNAs dataset landscape reveals potential signatures for the detection and prognosis of early-stage lung adenocarcinoma. BMC Cancer 2021, 21, 781. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lu, Q.; Lu, Q.; Shen, X.; Yu, L. Matrine regulates proliferation, apoptosis, cell cycle, migration, and invasion of non-small cell lung cancer cells through the circFUT8/miR-944/YES1 axis. Cancer Manag. Res. 2021, 13, 3429–3442. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Ju, J.-G. Circ_0001998 Regulates the Proliferation, Invasion, and Apoptosis of Lung Adenocarcinoma via Sponging miR-145. Evid.-Based Complement. Altern. Med. 2022, 2022, 6446150. [Google Scholar] [CrossRef]
- Sabrkhany, S.; Kuijpers, M.J.E.; Verheul, H.M.W.; Egbrink, M.G.A.O.; Griffioen, A.W. Optimal human blood sampling for platelet research. Curr. Angiogenes. 2013, 2, 157–161. [Google Scholar] [CrossRef]
- Sabrkhany, S.; Kuijpers, M.J.E.; van Kuijk, S.M.J.; Sanders, L.; Pineda, S.; Olde Damink, S.W.M.; Dingemans, A.M.C.; Griffioen, A.W.; oude Egbrink, M.G.A. A combination of platelet features allows detection of early-stage cancer. Eur. J. Cancer 2017, 80, 5–13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Cancer | |
|---|---|---|
| No. of samples | 30 | 30 |
| Age (average; min–max) | 63.1 (42–79) | 63.3 (51–79) |
| Female | 16 | 15 |
| Male | 14 | 15 |
| Lung nodules | 3 | - |
| Early-stage (stage I to IIIa) | - | 20 |
| Late-stage (stage IV) | - | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ambrosi, S.; Giannoukakos, S.; Antunes-Ferreira, M.; Pedraz-Valdunciel, C.; Bracht, J.W.P.; Potie, N.; Gimenez-Capitan, A.; Hackenberg, M.; Fernandez Hilario, A.; Molina-Vila, M.A.; et al. Combinatorial Blood Platelets-Derived circRNA and mRNA Signature for Early-Stage Lung Cancer Detection. Int. J. Mol. Sci. 2023, 24, 4881. https://doi.org/10.3390/ijms24054881
D’Ambrosi S, Giannoukakos S, Antunes-Ferreira M, Pedraz-Valdunciel C, Bracht JWP, Potie N, Gimenez-Capitan A, Hackenberg M, Fernandez Hilario A, Molina-Vila MA, et al. Combinatorial Blood Platelets-Derived circRNA and mRNA Signature for Early-Stage Lung Cancer Detection. International Journal of Molecular Sciences. 2023; 24(5):4881. https://doi.org/10.3390/ijms24054881
Chicago/Turabian StyleD’Ambrosi, Silvia, Stavros Giannoukakos, Mafalda Antunes-Ferreira, Carlos Pedraz-Valdunciel, Jillian W. P. Bracht, Nicolas Potie, Ana Gimenez-Capitan, Michael Hackenberg, Alberto Fernandez Hilario, Miguel A. Molina-Vila, and et al. 2023. "Combinatorial Blood Platelets-Derived circRNA and mRNA Signature for Early-Stage Lung Cancer Detection" International Journal of Molecular Sciences 24, no. 5: 4881. https://doi.org/10.3390/ijms24054881