
Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Links between miR and Gene Regulation
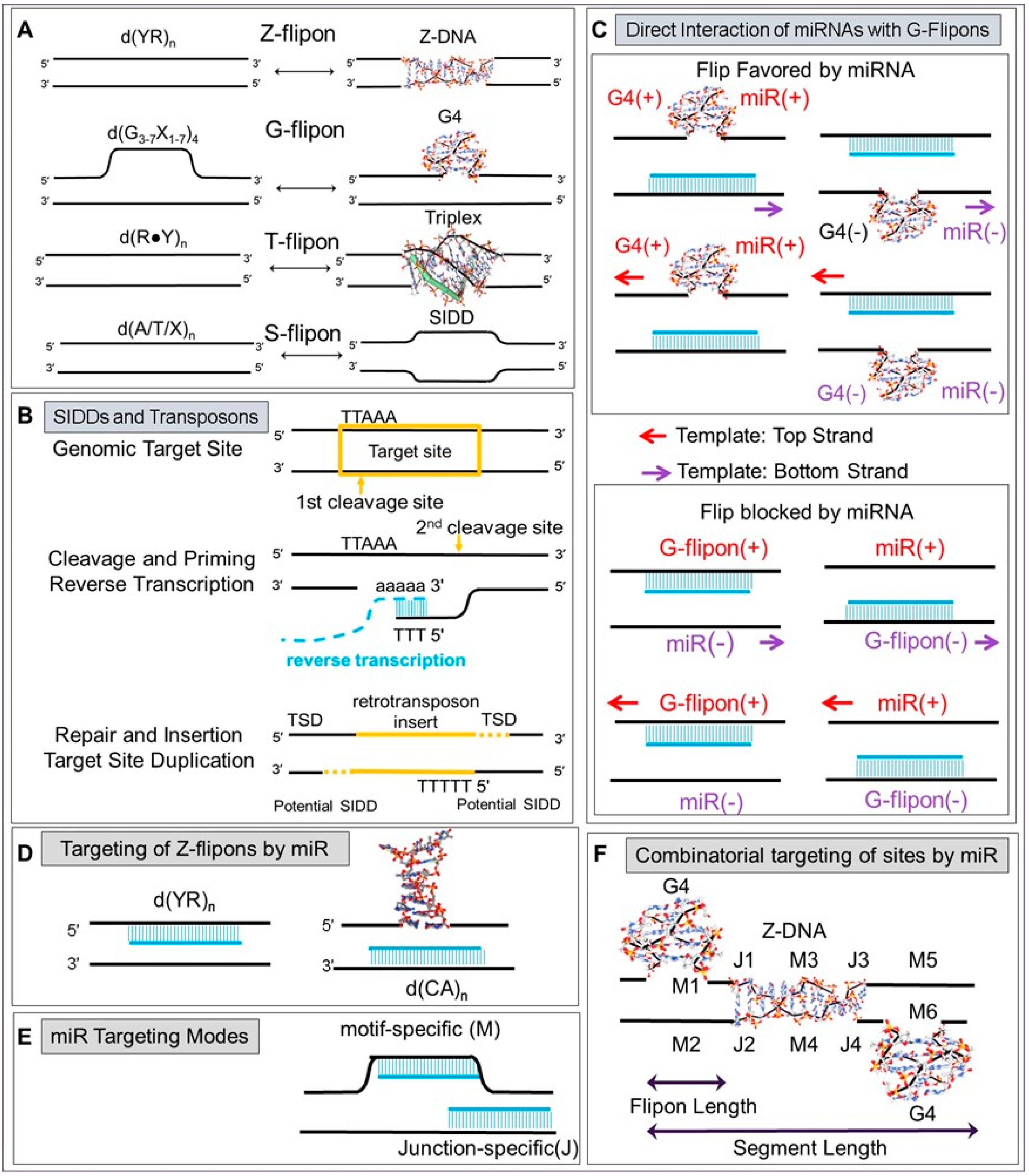
3. Flipons
4. Flipons and miR
5. Results
5.1. SIDDs and miR
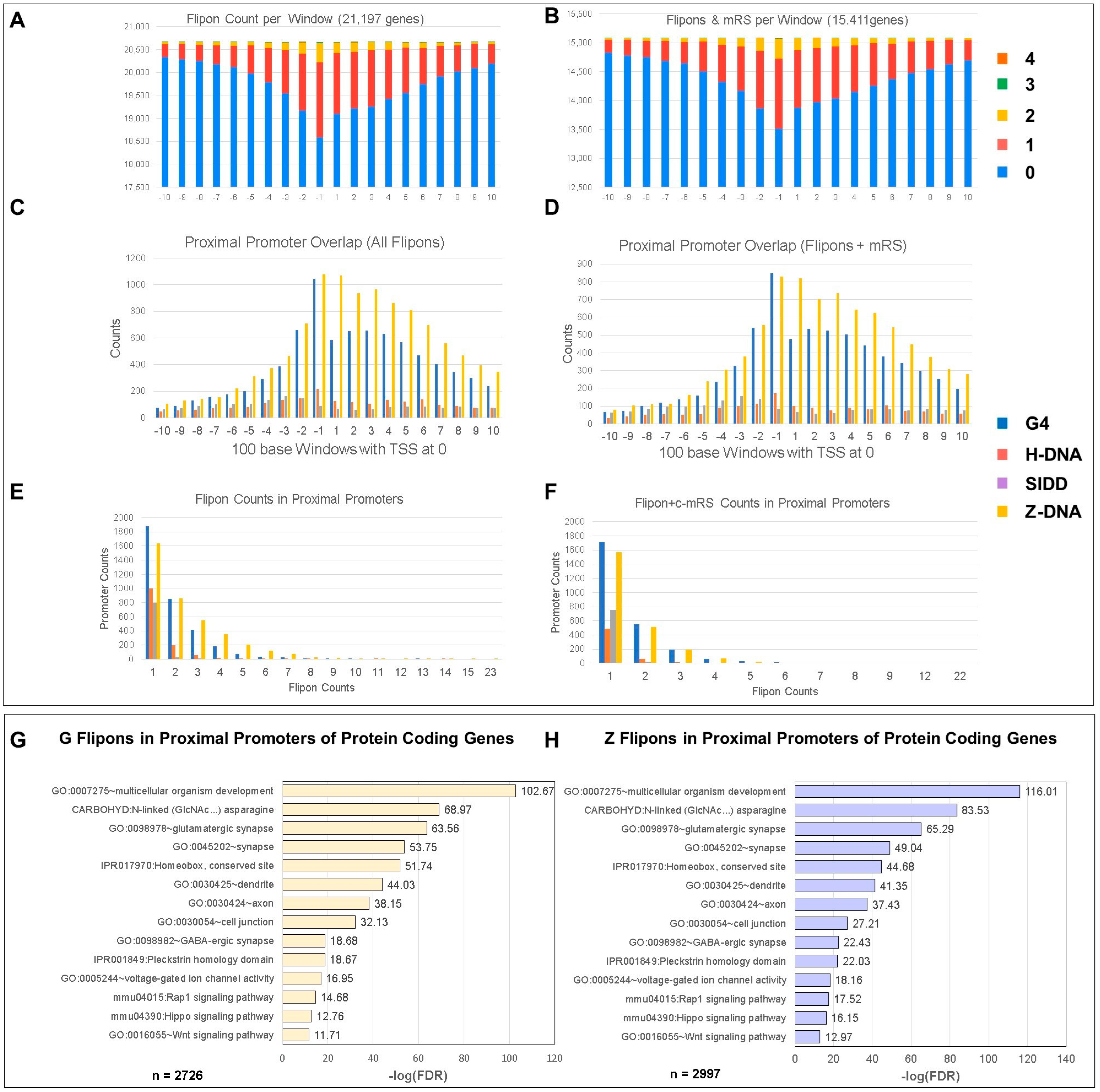
5.2. Promoter Flipons and miR
5.3. Strand Preference for c-mRS Matches

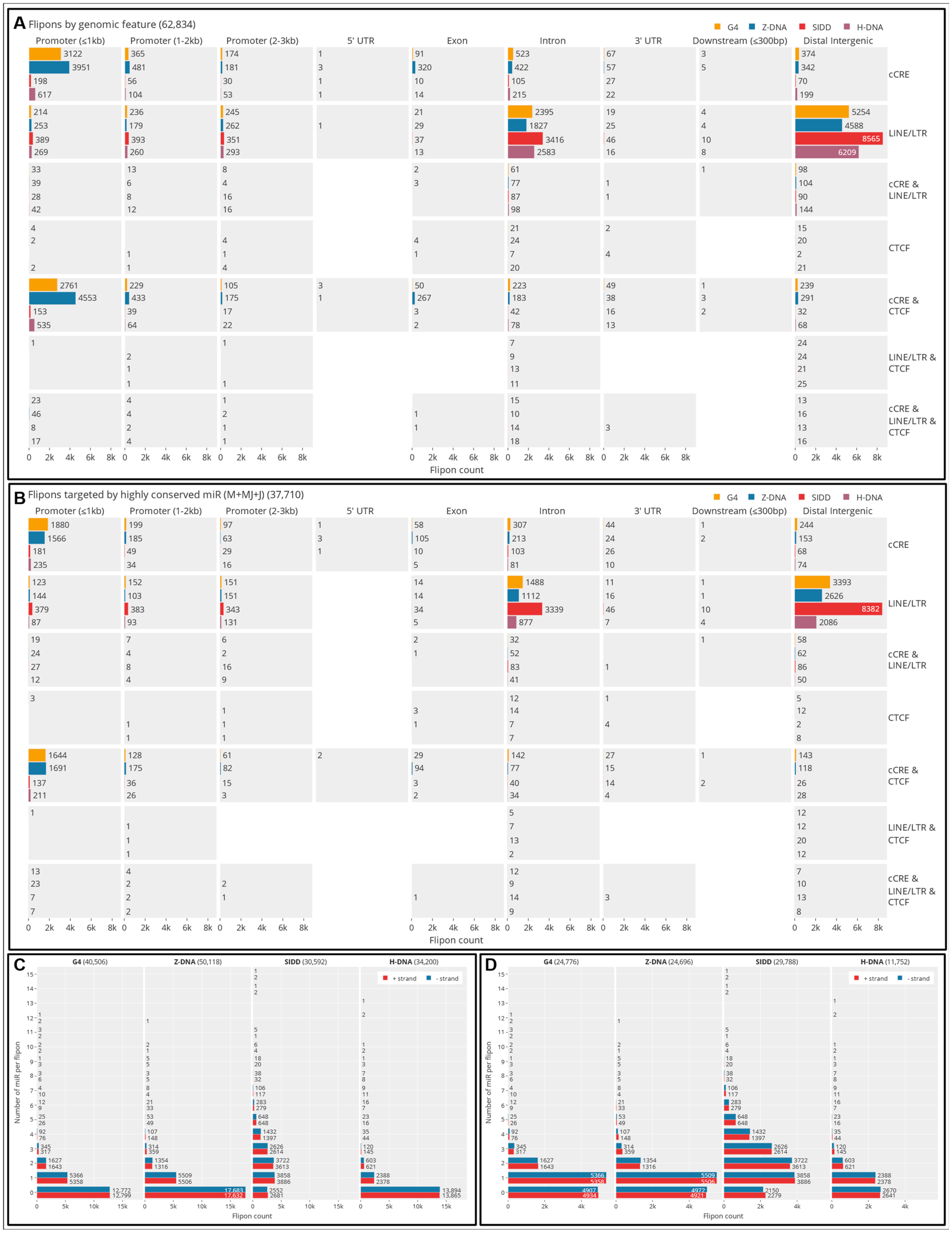
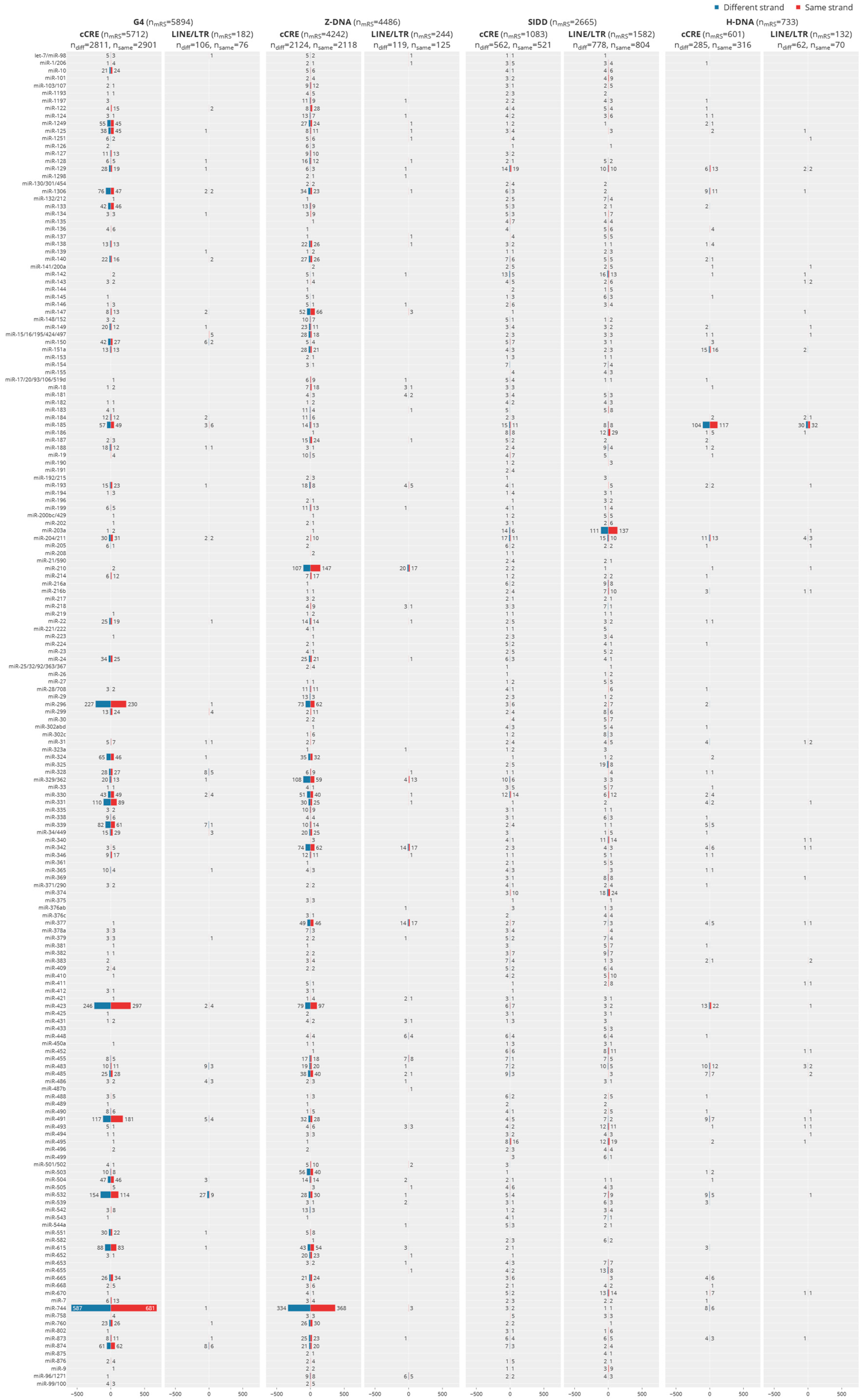
5.4. Interaction of Individual c-miR by Flipon Type, Genomic Region and DNA Strand
5.5. Colocalization of G and Z Flipons in Promoters
5.6. Gene Ontology (GO) of Promoters
5.7. Site Selection of c-mRS in Promoters by Flipon Type
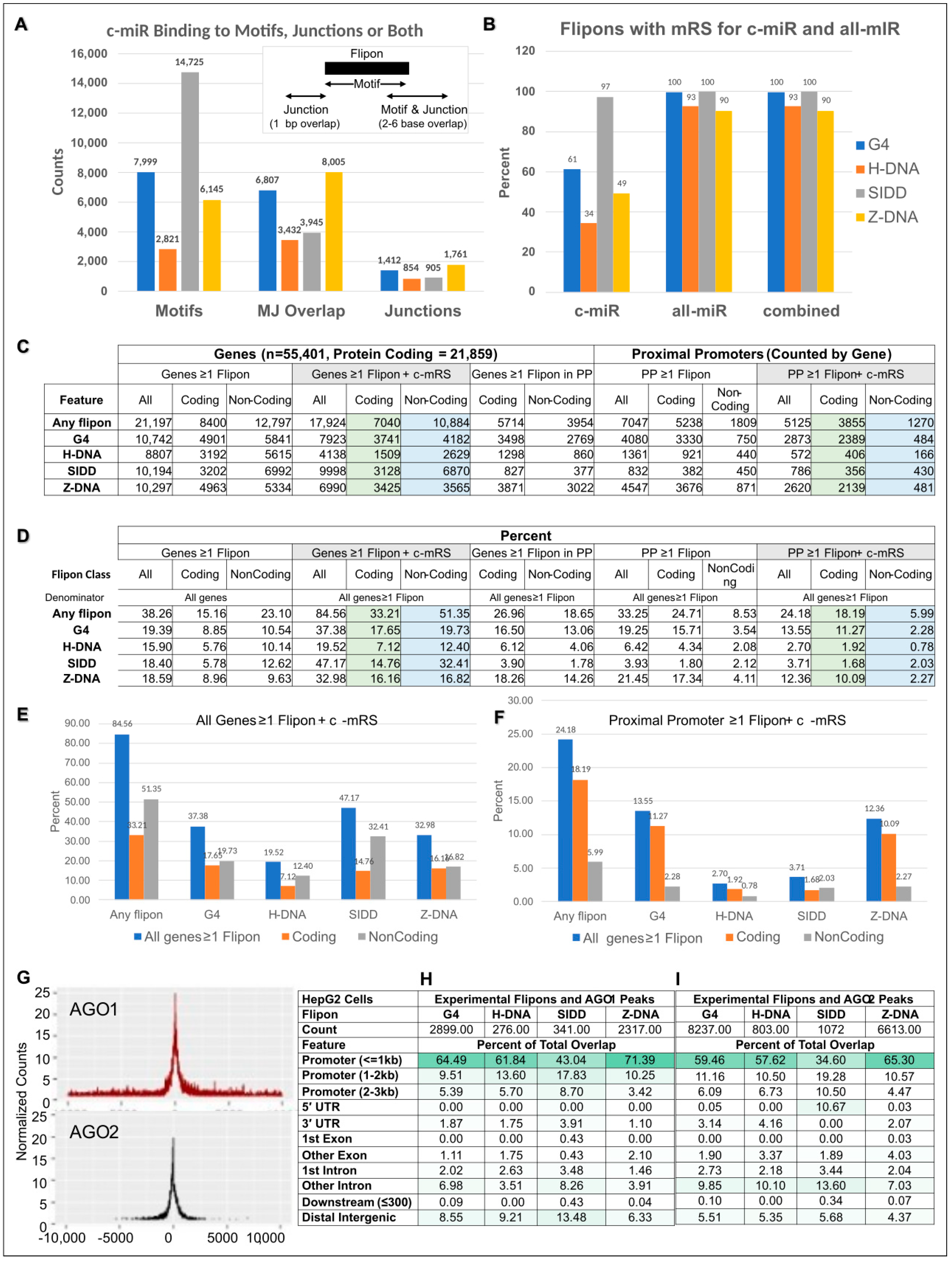
5.8. Enrichment of Flipons in Proximal Promoters of Coding Genes
5.9. Colocalization of Argonaute Protein to Promoter Flipons
5.10. GO analysis of Promoter-Specific miR
5.11. Modeling miR Targeting of Promoter Flipons
5.12. Architecture of Flipon and miR Interactions in Promoters
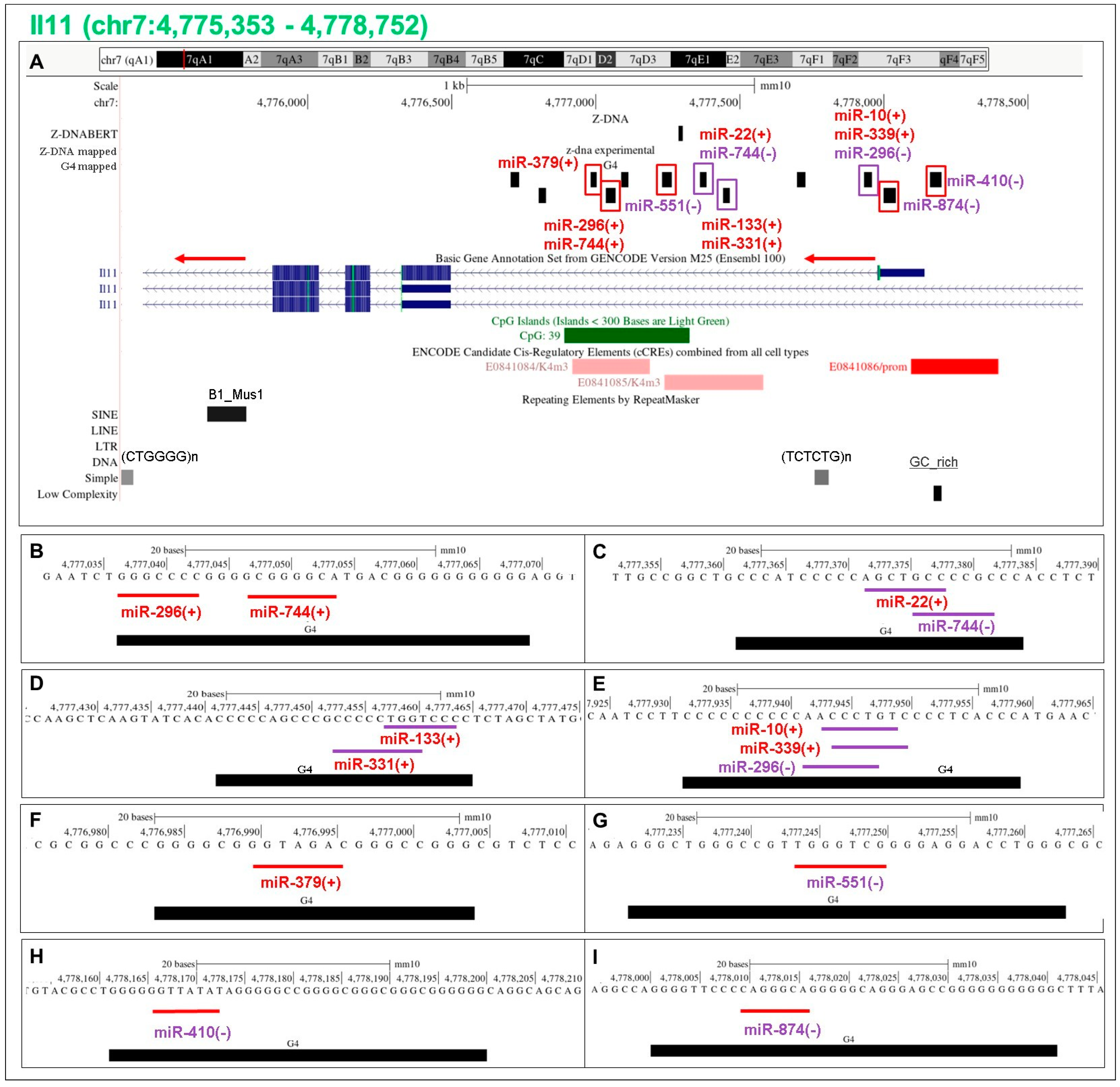
5.13. Flipons in the Il11 Promoter
5.14. Flipons in the Jag2 Promoter
5.15. Flipons in the Acvr2b Promoter
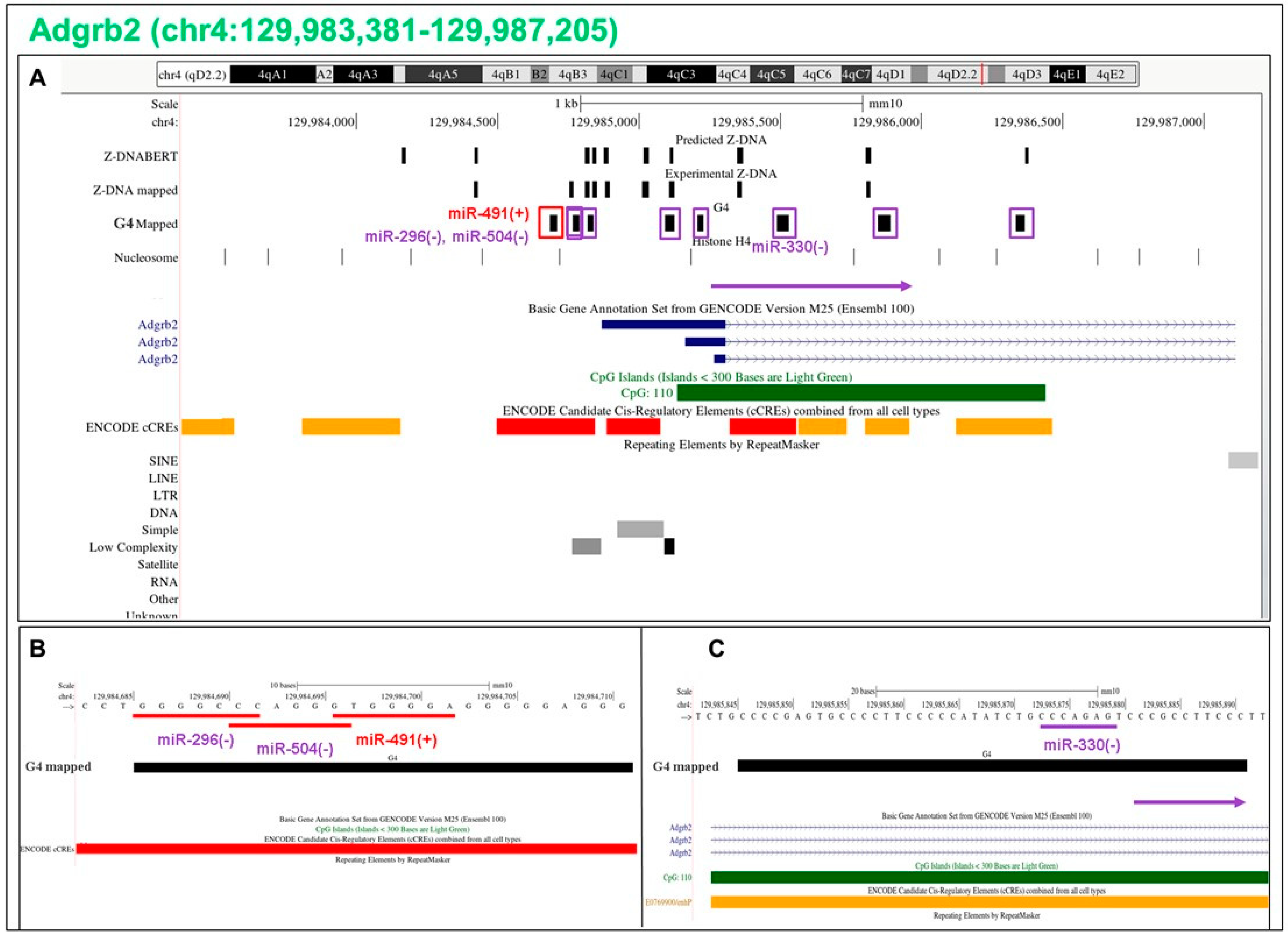
5.16. Flipons in the Adgrb2 Promoter
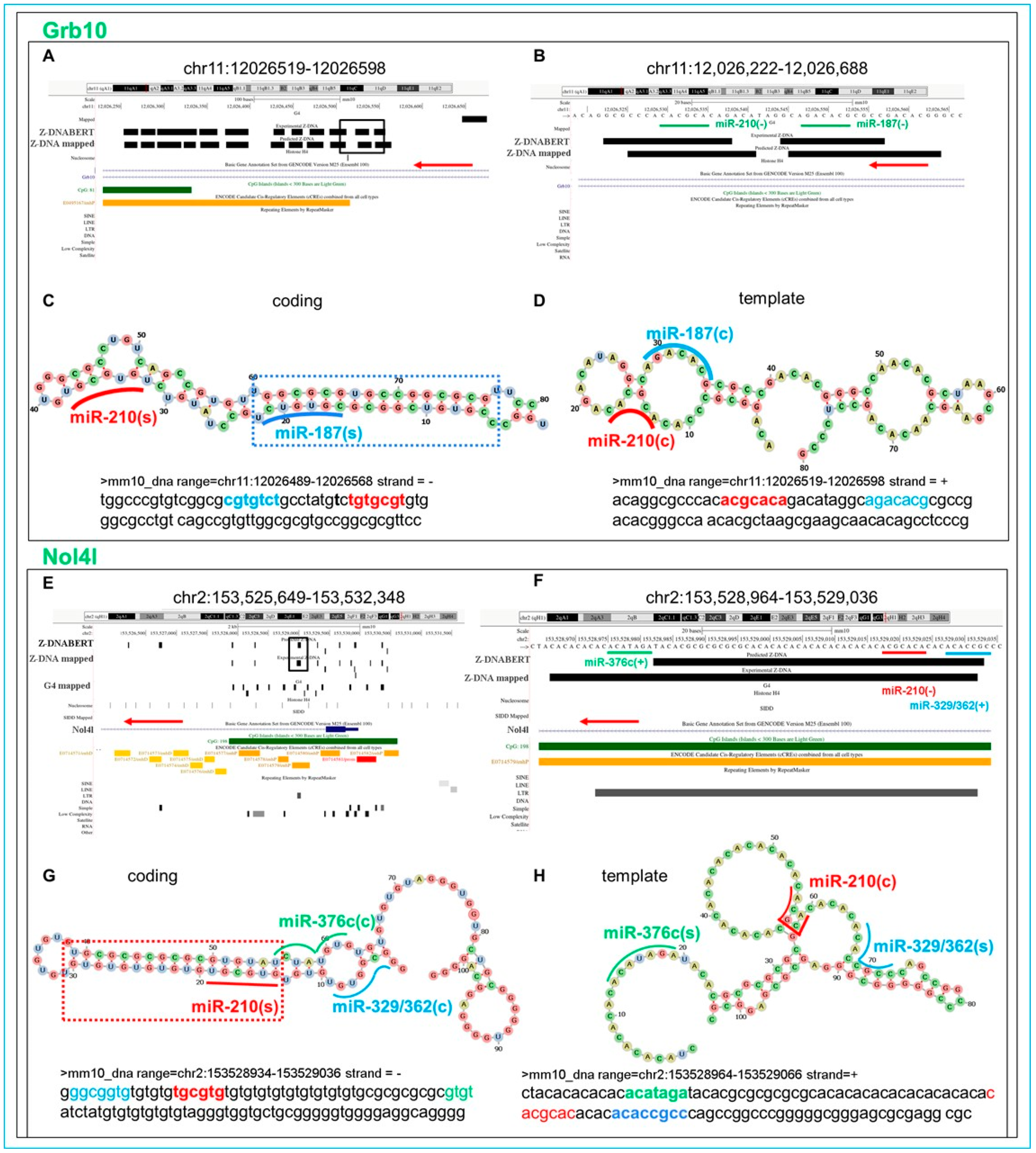
5.17. Flipons in the Grb10 Promoter
6. Discussion
7. Methods
7.1. Data Sources
7.2. Methods
7.2.1. Data Cleaning and Visualization
7.2.2. Gene Enrichment Analysis
7.2.3. Z-DNA Prediction
7.2.4. Flipon Matches with c-mRS
7.2.5. Flipon Matches with Other Features in mm10 and HG38 Genomic annotations
7.2.6. Sites Bound by Each c-miR by DNA Strand
7.2.7. Gene Promoter Analysis
7.2.8. Distances from AGO Peaks to TSS
7.2.9. ChIP-seq Analysis:
7.2.10. AGO Overlap Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Britten, R.J.; Davidson, E.H. Gene Regulation for Higher Cells: A Theory. Science 1969, 165, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, T.; Chen, Q. Exploring the expanding universe of small RNAs. Nat. Cell Biol. 2022, 24, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. A Genetic Instruction Code Based on DNA Conformation. Trends. Genet. 2019, 35, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Horvitz, H.R. Worms, Life, and Death (Nobel Lecture). ChemBioChem 2003, 4, 697–711. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lei, C.; He, Q.; Pan, Z.; Xiao, D.; Tao, Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol. Cancer 2018, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [Green Version]
- Natarelli, L.; Weber, C. A Non-Canonical Link between Non-Coding RNAs and Cardiovascular Diseases. Biomedicines 2022, 10, 445. [Google Scholar] [CrossRef]
- Allo, M.; Agirre, E.; Bessonov, S.; Bertucci, P.; Gomez Acuna, L.; Buggiano, V.; Bellora, N.; Singh, B.; Petrillo, E.; Blaustein, M.; et al. Argonaute-1 binds transcriptional enhancers and controls constitutive and alternative splicing in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15622–15629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agirre, E.; Bellora, N.; Allo, M.; Pages, A.; Bertucci, P.; Kornblihtt, A.R.; Eyras, E. A chromatin code for alternative splicing involving a putative association between CTCF and HP1alpha proteins. BMC Biol. 2015, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Yokota, S.; Liu, J.; Kilikevicius, A.; Johnson, K.C.; Corey, D.R. Argonaute binding within human nuclear RNA and its impact on alternative splicing. Rna 2021, 27, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.; Choi, Y.J.; Han, S.G.; Song, H.; Park, C.; Hong, K.; Kim, J.H. Roles of microRNAs in mammalian reproduction: From the commitment of germ cells to peri-implantation embryos. Biol. Rev. Camb. Philos. Soc. 2019, 94, 415–438. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Totoki, Y.; Toyoda, A.; Watanabe, T.; Yamamoto, Y.; Tokunaga, K.; Sakaki, Y.; Sasaki, H.; Hohjoh, H. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res. 2010, 38, 5141–5151. [Google Scholar] [CrossRef]
- Wang, X.; Ramat, A.; Simonelig, M.; Liu, M.F. Emerging roles and functional mechanisms of PIWI-interacting RNAs. Nat. Rev. Mol. Cell Biol. 2022, 24, 123–141. [Google Scholar] [CrossRef]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Younger, S.T.; Hardy, D.B.; Ram, R.; Huffman, K.E.; Corey, D.R. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 2007, 3, 166–173. [Google Scholar] [CrossRef]
- Portnoy, V.; Lin, S.H.; Li, K.H.; Burlingame, A.; Hu, Z.H.; Li, H.; Li, L.C. saRNA-guided Ago2 targets the RITA complex to promoters to stimulate transcription. Cell Res. 2016, 26, 320–335. [Google Scholar] [CrossRef] [Green Version]
- Voutila, J.; Reebye, V.; Roberts, T.C.; Protopapa, P.; Andrikakou, P.; Blakey, D.C.; Habib, R.; Huber, H.; Saetrom, P.; Rossi, J.J.; et al. Development and Mechanism of Small Activating RNA Targeting CEBPA, a Novel Therapeutic in Clinical Trials for Liver Cancer. Mol. Ther. 2017, 25, 2705–2714. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A. The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules 2021, 26, 4881. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, A. ALU non-B-DNA conformations, flipons, binary codes and evolution. R. Soc. Open Sci. 2020, 7, 200222. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.S.; Frederick, C.A.; Quigley, G.J.; van der Marel, G.A.; van Boom, J.H.; Wang, A.H.; Rich, A.G. T wobble base-pairing in Z-DNA at 1.0 A atomic resolution: The crystal structure of d(CGCGTG). Embo. J. 1985, 4, 3617–3623. [Google Scholar] [CrossRef]
- Escaja, N.; Mir, B.; Garavís, M.; González, C. Non-G Base Tetrads. Molecules 2022, 27, 5287. [Google Scholar] [CrossRef]
- Roschdi, S.; Yan, J.; Nomura, Y.; Escobar, C.A.; Petersen, R.J.; Bingman, C.A.; Tonelli, M.; Vivek, R.; Montemayor, E.J.; Wickens, M.; et al. An atypical RNA quadruplex marks RNAs as vectors for gene silencing. Nat. Struct Mol. Biol. 2022, 29, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Wojtowicz, D.; Yamane, A.; Resch, W.; Kieffer-Kwon, K.R.; Bandle, R.; Nelson, S.; Nakahashi, H.; Awasthi, P.; Feigenbaum, L.; et al. Global regulation of promoter melting in naive lymphocytes. Cell 2013, 153, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Teves, S.S.; Henikoff, S. Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol. 2013, 21, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Baranello, L.; Wojtowicz, D.; Cui, K.; Devaiah, B.N.; Chung, H.J.; Chan-Salis, K.Y.; Guha, R.; Wilson, K.; Zhang, X.; Zhang, H.; et al. RNA Polymerase II Regulates Topoisomerase 1 Activity to Favor Efficient Transcription. Cell 2016, 165, 357–371. [Google Scholar] [CrossRef] [Green Version]
- Saldana-Meyer, R.; Rodriguez-Hernaez, J.; Escobar, T.; Nishana, M.; Jacome-Lopez, K.; Nora, E.P.; Bruneau, B.G.; Tsirigos, A.; Furlan-Magaril, M.; Skok, J.; et al. RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 2019, 76, 412–422.e5. [Google Scholar] [CrossRef]
- Herbert, A. Nucleosomes and flipons exchange energy to alter chromatin conformation, the readout of genomic information, and cell fate. Bioessays 2022, 44, e2200166. [Google Scholar] [CrossRef] [PubMed]
- Schroth, G.P.; Chou, P.J.; Ho, P.S. Mapping Z-DNA in the human genome. Computer-aided mapping reveals a nonrandom distribution of potential Z-DNA-forming sequences in human genes. J. Biol. Chem. 1992, 267, 11846–11855. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.I.; Ham, S.; Park, J.; Seo, S.H.; Lim, C.H.; Jeon, H.; Huh, J.; Roh, T.Y. Z-DNA-forming sites identified by ChIP-Seq are associated with actively transcribed regions in the human genome. DNA Res. 2016, 23, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Chambers, V.S.; Sahakyan, A.B.; McCauley, P.; Boutell, J.M.; Antonio, M.D.; Balasubramanian, S. Whole genome experimental maps of DNA G-quadruplexes in multiple species. Nucleic Acids Res. 2019, 47, 3862–3874. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, R.; Schwartz, U.; Silberhorn, E.; Langst, G. Nucleosomes Stabilize ssRNA-dsDNA Triple Helices in Human Cells. Mol. Cell 2019, 73, 1243–1254.e6. [Google Scholar] [CrossRef] [Green Version]
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K.R.; Benham, C.J.; Casellas, R.; Przytycka, T.M.; et al. Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst. 2017, 4, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Noordewier, M.; Benham, C.J. Stress-Induced DNA Duplex Destabilization (SIDD) in the E. coli Genome: SIDD Sites Are Closely Associated With Promoters. Genome Res. 2004, 14, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Chedin, F.; Benham, C.J. Emerging roles for R-loop structures in the management of topological stress. J. Biol. Chem. 2020, 295, 4684–4695. [Google Scholar] [CrossRef] [Green Version]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb. Perspect. Med. 2013, 3, a014233. [Google Scholar] [CrossRef] [Green Version]
- Valverde, R.; Edwards, L.; Regan, L. Structure and function of KH domains. Febs. J. 2008, 275, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Zhou, Y.; Zhou, D.; Zhang, Y.; Shang, D.; Qi, J. The latest progress on miR-374 and its functional implications in physiological and pathological processes. J. Cell Mol. Med. 2019, 23, 3063–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Tian, Q.; Guan, L.; Niu, S.S. The Dual Role of miR-186 in Cancers: Oncomir Battling With Tumor Suppressor miRNA. Front. Oncol. 2020, 10, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, A.; Alfken, J.; Kim, Y.G.; Mian, I.S.; Nishikura, K.; Rich, A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. USA 1997, 94, 8421–8426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traczyk, A.; Liew, C.W.; Gill, D.J.; Rhodes, D. Structural basis of G-quadruplex DNA recognition by the yeast telomeric protein Rap1. Nucleic Acids Res. 2020, 48, 4562–4571. [Google Scholar] [CrossRef]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferre-D’Amare, A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–469. [Google Scholar] [CrossRef]
- Qiao, Q.; Wang, L.; Meng, F.L.; Hwang, J.K.; Alt, F.W.; Wu, H. AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 2017, 67, 361–373.e4. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225. [Google Scholar] [CrossRef]
- Voong, L.N.; Xi, L.; Sebeson, A.C.; Xiong, B.; Wang, J.P.; Wang, X. Insights into Nucleosome Organization in Mouse Embryonic Stem Cells through Chemical Mapping. Cell 2016, 167, 1555–1570.e15. [Google Scholar] [CrossRef] [Green Version]
- Ng, B.; Widjaja, A.A.; Viswanathan, S.; Dong, J.; Chothani, S.P.; Lim, S.; Shekeran, S.G.; Tan, J.; McGregor, N.E.; Walker, E.C.; et al. Similarities and differences between IL11 and IL11RA1 knockout mice for lung fibro-inflammation, fertility and craniosynostosis. Sci. Rep. 2021, 11, 14088. [Google Scholar] [CrossRef]
- Dillenburg, A.; Ireland, G.; Holloway, R.K.; Davies, C.L.; Evans, F.L.; Swire, M.; Bechler, M.E.; Soong, D.; Yuen, T.J.; Su, G.H.; et al. Activin receptors regulate the oligodendrocyte lineage in health and disease. Acta Neuropathol. 2018, 135, 887–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodberg, A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor. Rev. 2021, 60, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.H.; Toro, C.; Gahl, W.A.; Hall, R.A. A disease-associated mutation in the adhesion GPCR BAI2 (ADGRB2) increases receptor signaling activity. Hum. Mutat. 2017, 38, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.M.; Meador-Woodruff, J.H. Post-translational protein modifications in schizophrenia. NPJ Schizophr. 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habermann, F.A.; Kaltner, H.; Higuero, A.M.; Caballero, G.G.; Ludwig, A.-K.; Manning, J.C.; Abad-Rodríguez, J.; Gabius, H.-J. What Cyto- and Histochemistry Can Do to Crack the Sugar Code. Acta Histochem. Cytochem. 2021, 54, 31–48. [Google Scholar] [CrossRef] [PubMed]
- Levin, M. Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 2021, 184, 1971–1989. [Google Scholar] [CrossRef]
- Raghu, P.; Joseph, A.; Krishnan, H.; Singh, P.; Saha, S. Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 2019, 12, 208. [Google Scholar] [CrossRef] [Green Version]
- Percharde, M.; Bulut-Karslioglu, A.; Ramalho-Santos, M. Hypertranscription in Development, Stem Cells, and Regeneration. Dev. Cell 2017, 40, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Sela, N.; Mersch, B.; Gal-Mark, N.; Lev-Maor, G.; Hotz-Wagenblatt, A.; Ast, G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol. 2007, 8, R127. [Google Scholar] [CrossRef]
- Scruggs, B.S.; Gilchrist, D.A.; Nechaev, S.; Muse, G.W.; Burkholder, A.; Fargo, D.C.; Adelman, K. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 2015, 58, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gapp, K.; Bohacek, J. Epigenetic germline inheritance in mammals: Looking to the past to understand the future. Genes Brain Behav. 2018, 17, e12407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngondo, R.P.; Cirera-Salinas, D.; Yu, J.; Wischnewski, H.; Bodak, M.; Vandormael-Pournin, S.; Geiselmann, A.; Wettstein, R.; Luitz, J.; Cohen-Tannoudji, M.; et al. Argonaute 2 Is Required for Extra-embryonic Endoderm Differentiation of Mouse Embryonic Stem Cells. Stem. Cell Rep. 2018, 10, 461–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissels, U.; Wild, S.; Tomiuk, S.; Holste, A.; Hafner, M.; Tuschl, T.; Bosio, A. Absolute quantification of microRNAs by using a universal reference. Rna 2009, 15, 2375–2384. [Google Scholar] [CrossRef] [Green Version]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA Hypothesis with Quantitative Measurements of miRNA and Target Abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Katahira, J.; Yoneda, Y. Nucleocytoplasmic transport of microRNAs and related small RNAs. Traffic 2011, 12, 1468–1474. [Google Scholar] [CrossRef]
- Wei, Y.; Li, L.; Wang, D.; Zhang, C.Y.; Zen, K. Importin 8 regulates the transport of mature microRNAs into the cell nucleus. J. Biol. Chem. 2014, 289, 10270–10275. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A. The four Rs of RNA-directed evolution. Nat. Genet. 2004, 36, 19–25. [Google Scholar] [CrossRef]
- Beckedorff, F.; Blumenthal, E.; daSilva, L.F.; Aoi, Y.; Cingaram, P.R.; Yue, J.; Zhang, A.; Dokaneheifard, S.; Valencia, M.G.; Gaidosh, G.; et al. The Human Integrator Complex Facilitates Transcriptional Elongation by Endonucleolytic Cleavage of Nascent Transcripts. Cell Rep. 2020, 32, 107917. [Google Scholar] [CrossRef]
- Dharap, A.; Pokrzywa, C.; Murali, S.; Pandi, G.; Vemuganti, R. MicroRNA miR-324-3p induces promoter-mediated expression of RelA gene. PLoS ONE 2013, 8, e79467. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhu, X.; Wang, J.; Li, N.; Li, D.; Sakib, N.; Sha, Z.; Song, W. MiR-744 functions as a proto-oncogene in nasopharyngeal carcinoma progression and metastasis via transcriptional control of ARHGAP5. Oncotarget 2015, 6, 13164–13175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem. Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elements. 2011, 1, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemondy, K.; Wang, X.J.; Torchia, E.C.; Roop, D.R.; Yi, R. MicroRNA-203 represses selection and expansion of oncogenic Hras transformed tumor initiating cells. Elife 2015, 4, e07004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Almeida, M.V.; Vernaz, G.; Putman, A.L.K.; Miska, E.A. Taming transposable elements in vertebrates: From epigenetic silencing to domestication. Trends Genet. 2022, 38, 529–553. [Google Scholar] [CrossRef]
- Carducci, F.; Biscotti, M.A.; Barucca, M.; Canapa, A. Transposable elements in vertebrates: Species evolution and environmental adaptation. Eur. Zool. J. 2019, 86, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Peaston, A.E.; Evsikov, A.V.; Graber, J.H.; de Vries, W.N.; Holbrook, A.E.; Solter, D.; Knowles, B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell 2004, 7, 597–606. [Google Scholar] [CrossRef]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Goncalves, A.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Nazer, E.; Gomez Acuna, L.; Kornblihtt, A.R. Seeking the truth behind the myth: Argonaute tales from “nuclearland”. Mol. Cell 2022, 82, 503–513. [Google Scholar] [CrossRef]
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 2017, 170, 61–71.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Lyu, R.; You, Q.; He, C. Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ. Nat. Methods 2020, 17, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Chetta, M.; Di Pietro, L.; Bukvic, N.; Lattanzi, W. Rising Roles of Small Noncoding RNAs in Cotranscriptional Regulation: In Silico Study of miRNA and piRNA Regulatory Network in Humans. Genes 2020, 11, 482. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbert, A.; Pavlov, F.; Konovalov, D.; Poptsova, M. Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations. Int. J. Mol. Sci. 2023, 24, 4884. https://doi.org/10.3390/ijms24054884
Herbert A, Pavlov F, Konovalov D, Poptsova M. Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations. International Journal of Molecular Sciences. 2023; 24(5):4884. https://doi.org/10.3390/ijms24054884
Chicago/Turabian StyleHerbert, Alan, Fedor Pavlov, Dmitrii Konovalov, and Maria Poptsova. 2023. "Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations" International Journal of Molecular Sciences 24, no. 5: 4884. https://doi.org/10.3390/ijms24054884