
Regulation of MAPK Signaling Pathways by the Large HERC Ubiquitin Ligases
 , and
, and
Abstract
:1. Introduction
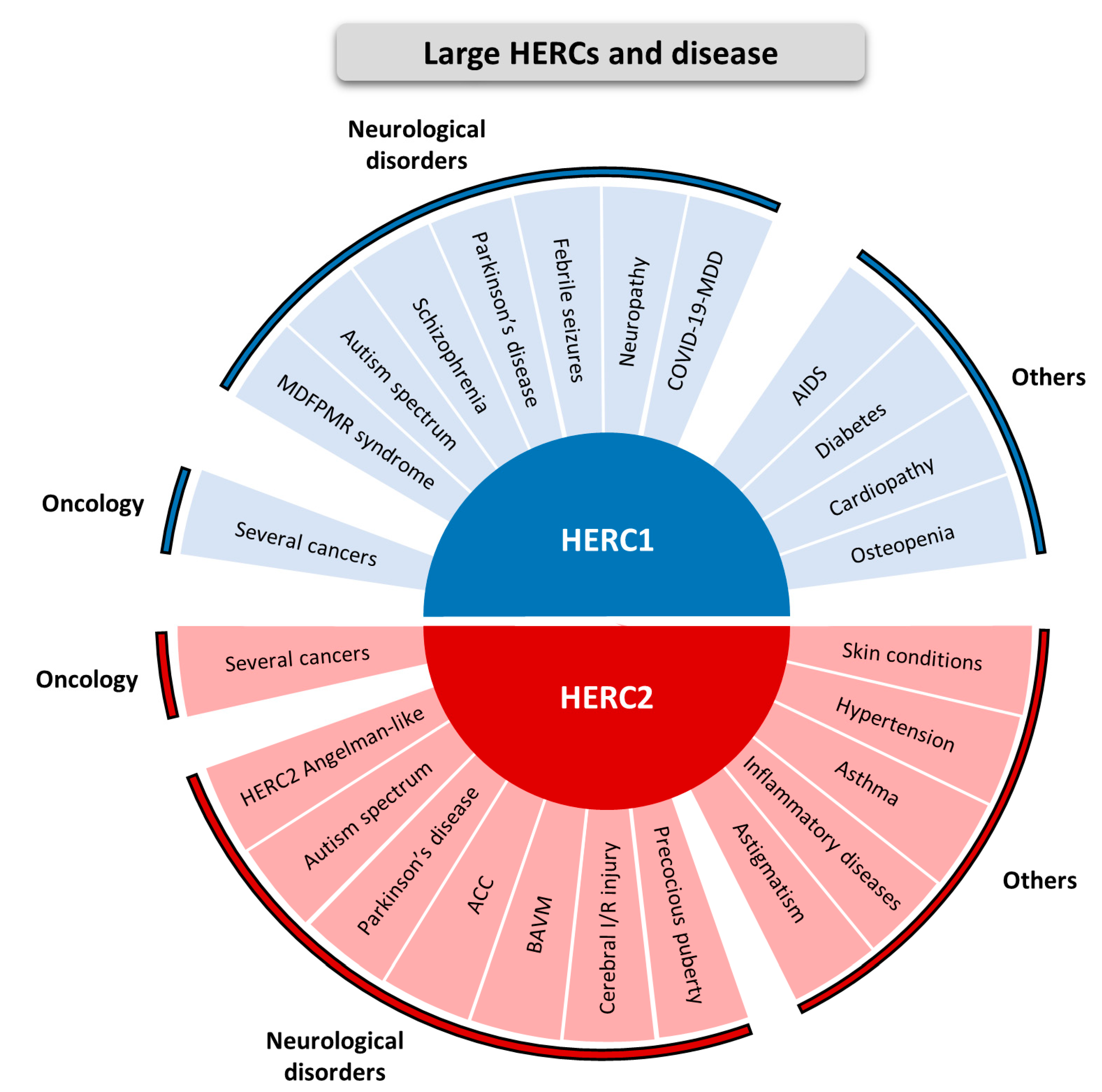
2. Large HERCs and Disease
3. The Role of HECT Ubiquitin Ligases in the Regulation of MAPK Signaling Pathways
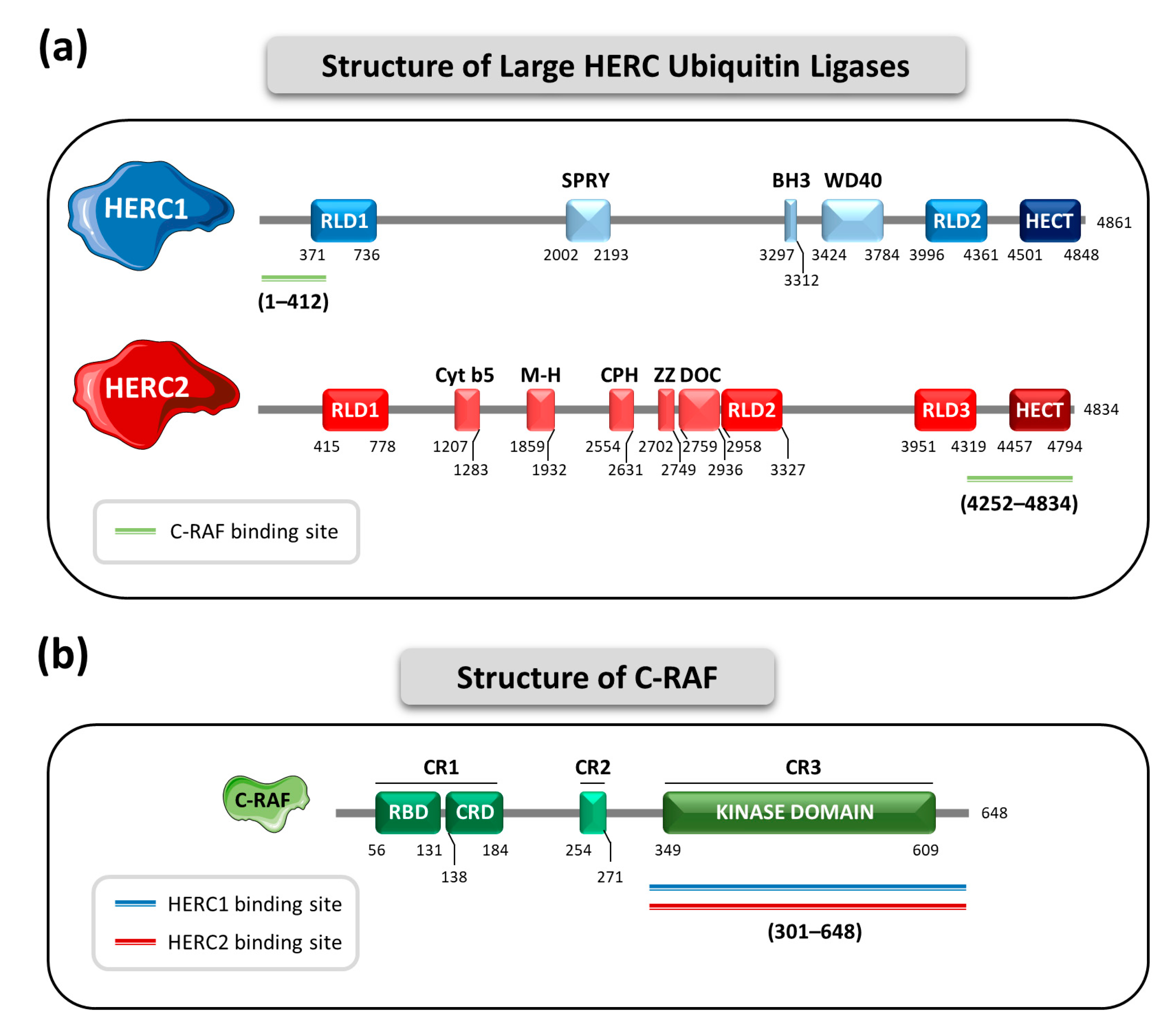
4. The Large HERC Ubiquitin Ligases in MAPK Signaling
5. Future Perspectives and Therapeutic Implications
6. Concluding Remarks
- Large HERCs are involved in several diseases, with a notable implication in neurological diseases and cancer.
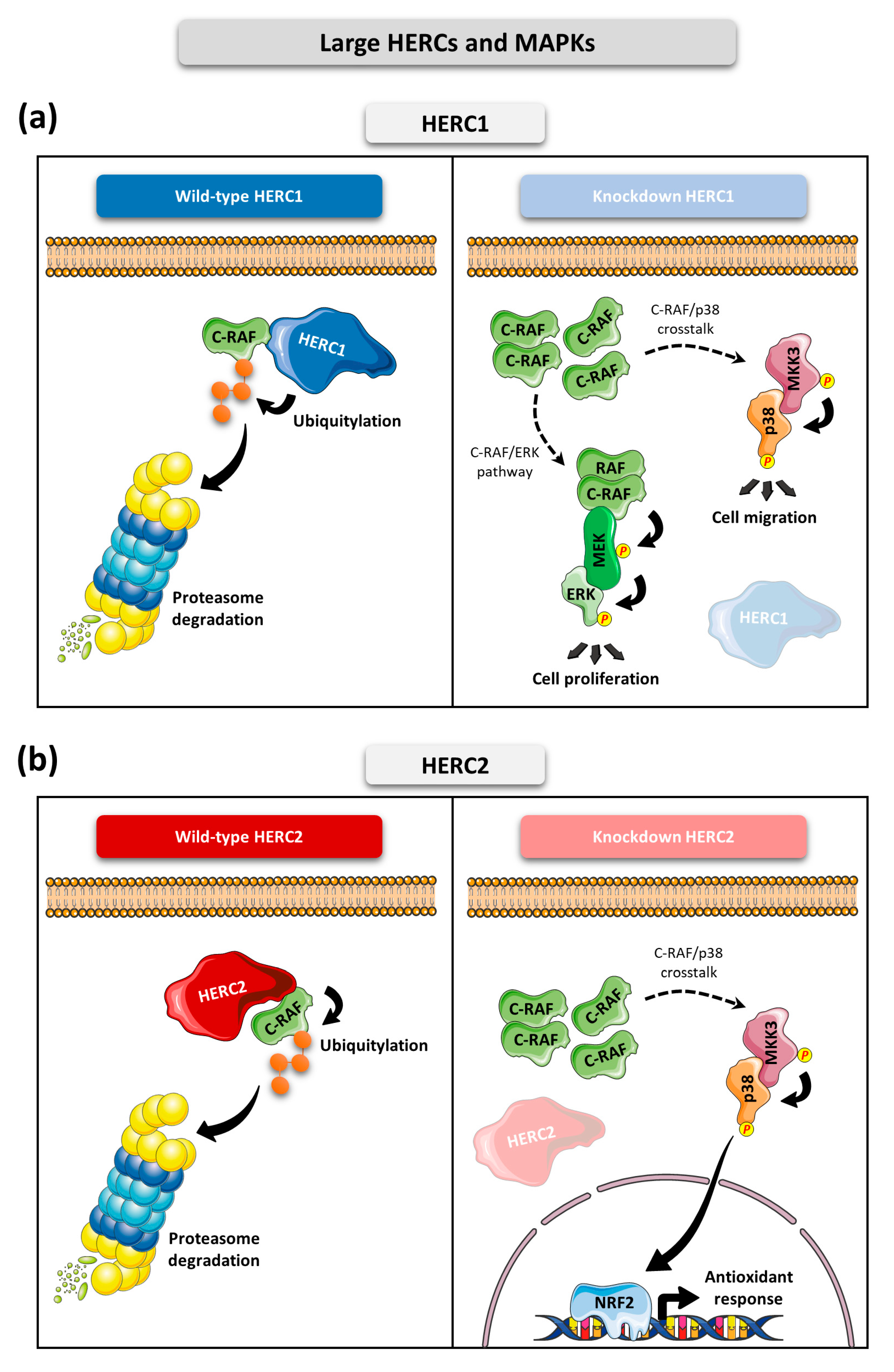
- HERC1 regulates ERK and p38 signaling pathways through controlling C-RAF protein levels.
- HERC2 regulates C-RAF protein levels, affecting the p38 signaling pathway.
- Downregulation of HERC1 or HERC2 causes accumulation of C-RAF protein levels which alters MAPK signaling.
- The use of RAF inhibitors such as sorafenib, or the development of specific PROTACs against C-RAF, may represent a promising therapeutic option to counteract alterations in MAPK signaling caused by HERC1 or HERC2 deficiency.
Author Contributions
Funding
Conflicts of Interest
References
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- McClellan, A.J.; Laugesen, S.H.; Ellgaard, L. Cellular Functions and Molecular Mechanisms of Non-Lysine Ubiquitination. Open Biol. 2019, 9, 190147. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Schulman, B.A. An Expanded Lexicon for the Ubiquitin Code. Nat. Rev. Mol. Cell Biol. 2022, 1–15. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 Ubiquitin Ligases: Styles, Structures and Functions. Mol. Biomed. 2021, 2, 23. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e1. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The U Box Is a Modified RING Finger—A Common Domain in Ubiquitination. Curr. Biol. 2000, 10, R132–R134. [Google Scholar] [CrossRef] [Green Version]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- García-Cano, J.; Martinez-Martinez, A.; Sala-Gaston, J.; Pedrazza, L.; Rosa, J.L. HERCing: Structural and Functional Relevance of the Large HERC Ubiquitin Ligases. Front. Physiol. 2019, 10, 1014. [Google Scholar] [CrossRef] [Green Version]
- Dove, K.K.; Klevit, R.E. RING-Between-RING E3 Ligases: Emerging Themes amid the Variations. J. Mol. Biol. 2017, 429, 3363–3375. [Google Scholar] [CrossRef]
- Marin, I. Animal HECT Ubiquitin Ligases: Evolution and Functional Implications. BMC Evol. Biol. 2010, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Tena, S.; Cubillos-Rojas, M.; Schneider, T.; Rosa, J.L. Functional and Pathological Relevance of HERC Family Proteins: A Decade Later. Cell. Mol. Life Sci. 2016, 73, 1955–1968. [Google Scholar] [CrossRef]
- Damgaard, R.B. The Ubiquitin System: From Cell Signalling to Disease Biology and New Therapeutic Opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK Cascades: Signaling Components, Nuclear Roles and Mechanisms of Nuclear Translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Baek, K. E3 Ligases and Deubiquitinating Enzymes Regulating the MAPK Signaling Pathway in Cancers. Biochim. Biophys. Acta 2022, 1877, 188736. [Google Scholar] [CrossRef]
- Pérez-Villegas, E.M.; Ruiz, R.; Bachiller, S.; Ventura, F.; Armengol, J.A.; Rosa, J.L. The HERC Proteins and the Nervous System. Semin. Cell Dev. Biol. 2022, 132, 5. [Google Scholar] [CrossRef]
- Ortega-Recalde, O.; Beltrán, O.I.; Gálvez, J.M.; Palma-Montero, A.; Restrepo, C.M.; Mateus, H.E.; Laissue, P. Biallelic HERC1 Mutations in a Syndromic Form of Overgrowth and Intellectual Disability. Clin. Genet. 2015, 88, e1–e3. [Google Scholar] [CrossRef]
- Aggarwal, S.; Das Bhowmik, A.; Ramprasad, V.L.; Murugan, S.; Dalal, A. A Splice Site Mutation in HERC1 Leads to Syndromic Intellectual Disability with Macrocephaly and Facial Dysmorphism: Further Delineation of the Phenotypic Spectrum. Am. J. Med. Genet. 2016, 170, 1868–1873. [Google Scholar] [CrossRef]
- Nguyen, L.S.; Schneider, T.; Rio, M.; Moutton, S.; Siquier-pernet, K.; Verny, F.; Boddaert, N.; Desguerre, I.; Munich, A.; Rosa, J.L.; et al. A Nonsense Variant in HERC1 Is Associated with Intellectual Disability, Megalencephaly, Thick Corpus Callosum and Cerebellar Atrophy. Eur. J. Hum. Genet. 2016, 24, 455–458. [Google Scholar] [CrossRef]
- Utine, G.E.; Taşkıran, E.Z.; Koşukcu, C.; Karaosmanoğlu, B.; Güleray, N.; Akgün Doğan, Ö.; Şimşek Kiper, P.Ö.; Boduroğlu, K.; Alikaşifoğlu, M. HERC1 Mutations in Idiopathic Intellectual Disability. Eur. J. Med. Genet. 2017, 60, 279–283. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Pedrazza, L.; Stenzel, W.; Rosa, J.L.; Schuelke, M.; Straussberg, R. A New Homozygous HERC1 Gain-of-Function Variant in MDFPMR Syndrome Leads to MTORC1 Hyperactivation and Reduced Autophagy during Cell Catabolism. Mol. Genet. Metab. 2020, 131, 126–134. [Google Scholar] [CrossRef]
- Hashimoto, R.; Nakazawa, T.; Tsurusaki, Y.; Yasuda, Y.; Nagayasu, K.; Matsumura, K.; Kawashima, H.; Yamamori, H.; Fujimoto, M.; Ohi, K.; et al. Whole-Exome Sequencing and Neurite Outgrowth Analysis in Autism Spectrum Disorder. J. Hum. Genet. 2016, 61, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Afshar, S.; Rajadhyaksha, A.M.; Potash, J.B.; Han, S. A Machine Learning Approach to Predicting Autism Risk Genes: Validation of Known Genes and Discovery of New Candidates. Front. Genet. 2020, 11, 500064. [Google Scholar] [CrossRef]
- Gu, X.; Hou, Y.; Chen, Y.; Ou, R.; Cao, B.; Wei, Q.; Zhang, L.; Song, W.; Zhao, B.; Wu, Y.; et al. Enrichment of Rare Variants in E3 Ubiquitin Ligase Genes in Early Onset Parkinson’s Disease. Neurobiol. Aging 2022, 109, 273–278. [Google Scholar] [CrossRef]
- Guo, A.; Lun, P.; Chen, J.; Li, Q.; Chang, K.; Li, T.; Pan, D.; Zhang, J.; Zhou, J.; Wang, K.; et al. Association Analysis of Risk Genes Identified by SCHEMA with Schizophrenia in the Chinese Han Population. Psychiatr. Genet. 2022, 32, 188–193. [Google Scholar] [CrossRef]
- Skotte, L.; Fadista, J.; Bybjerg-Grauholm, J.; Appadurai, V.; Hildebrand, M.S.; Hansen, T.F.; Banasik, K.; Grove, J.; Albiñana, C.; Geller, F.; et al. Genome-Wide Association Study of Febrile Seizures Implicates Fever Response and Neuronal Excitability Genes. Brain 2022, 145, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Roca-ceballos, M.A.; García-domínguez, I.; Pérez-Villegas, E.M.; Martos-Carmona, D.; Pérez-Castro, M.Á.; Real, L.M.; Rosa, J.L.; Tabares, L.; Venero, J.L.; et al. HERC1 Ubiquitin Ligase Is Required for Normal Axonal Myelination in the Peripheral Nervous System. Mol. Neurobiol. 2018, 55, 8856–8868. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, F.-J.; Zhang, L.-L.; Xian, D.-X.; Li, Y.; Li, J.-J.; Tang, S.-X.; Li, X.-J.; Liu, Y.; Peng, M.; et al. An Approach Combining Bioinformatics and Machine Learning to Identify Eight Autophagy- Related Biomarkers and Construct Molecular Mechanisms Underlying COVID-19 and Major Depressive Disorders. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 8129–8143. [Google Scholar] [CrossRef]
- Duarte, R.R.R.; Pain, O.; Furler, R.L.; Nixon, D.F.; Powell, T.R. Transcriptome-Wide Association Study of HIV-1 Acquisition Identifies HERC1 as a Susceptibility Gene. iScience 2022, 25, 104854. [Google Scholar] [CrossRef]
- Chatenoud, L.; Marquet, C.; Valette, F.; Scott, L.; Quan, J.; Bu, C.H.; Hildebrand, S.; Moresco, E.M.Y.; Bach, J.; Beutler, B. Modulation of Autoimmune Diabetes by N-Ethyl-N-Nitrosourea- Induced Mutations in Non-Obese Diabetic Mice. Dis. Model. Mech. 2022, 15, dmm049484. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.E.; Muoio, D.M.; Stevens, R.; Craig, D.; Bain, J.R.; Grass, E.; Haynes, C.; Kwee, L.; Qin, X.; Slentz, D.H.; et al. Metabolomic Quantitative Trait Loci (MQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis. PLoS Genet. 2015, 11, e1005553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrazza, L.; Martinez-martinez, A.; Sánchez-de-diego, C.; Valer, J.A.; Pimenta-Lopes, C.; Sala-Gaston, J.; Szpak, M.; Tyler-Smith, C.; Ventura, F.; Rosa, J.L. HERC1 Deficiency Causes Osteopenia through Transcriptional Program Dysregulation during Bone Remodeling. Cell Death Dis. 2023, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Puffenberger, E.G.; Jinks, R.N.; Wang, H.; Xin, B.; Fiorentini, C.; Sherman, E.A.; Degrazio, D.; Shaw, C.; Sougnez, C.; Cibulskis, K.; et al. A Homozygous Missense Mutation in HERC2 Associated with Global Developmental Delay and Autism Spectrum Disorder. Hum. Mutat. 2012, 33, 1639–1646. [Google Scholar] [CrossRef]
- Harlalka, G.V.; Baple, E.L.; Cross, H.; Kühnle, S.; Cubillos-Rojas, M.; Matentzoglu, K.; Patton, M.A.; Wagner, K.; Coblentz, R.; Ford, D.L.; et al. Mutation of HERC2 Causes Developmental Delay with Angelman-like Features. J. Med. Genet. 2013, 50, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Morice-Picard, F.; Benard, G.; Rezvani, H.R.; Lasseaux, E.; Simon, D.; Moutton, S.; Rooryck, C.; Lacombe, D.; Baumann, C.; Arveiler, B. Complete Loss of Function of the Ubiquitin Ligase HERC2 Causes a Severe Neurodevelopmental Phenotype. Eur. J. Hum. Genet. 2016, 25, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Abraham, J.R.; Barnard, J.; Wang, H.; Noritz, G.H.; Yeganeh, M.; Buhas, D.; Natowicz, M.R. Proteomic Investigations of Human HERC2 Mutants: Insights into the Pathobiology of a Neurodevelopmental Disorder. Biochem. Biophys. Res. Commun. 2019, 512, 421–427. [Google Scholar] [CrossRef]
- Ueda, K.; Ogawa, S.; Matsuda, K.; Hasegawa, Y.; Nishi, E.; Yanagi, K.; Kaname, T.; Yamamoto, T.; Okamoto, N. Blended Phenotype of Combination of HERC2 and AP3B2 Deficiency and Angelman Syndrome Caused by Paternal Isodisomy of Chromosome 15. Am. J. Med. Genet. Part A 2021, 185, 3092–3098. [Google Scholar] [CrossRef]
- Elpidorou, M.; Best, S.; Poulter, J.A.; Hartill, V.; Hobson, E.; Sheridan, E.; Johnson, C.A. Novel Loss-of-Function Mutation in HERC2 Is Associated with Severe Developmental Delay and Paediatric Lethality. J. Med. Genet. 2021, 58, 334–341. [Google Scholar] [CrossRef]
- Chai, J.; Locke, D.P.; Greally, J.M.; Knoll, J.H.M.; Ohta, T.; Dunai, J.; Yavor, A.; Eichler, E.E.; Nicholls, R.D. Identification of Four Highly Conserved Genes between Breakpoint Hotspots BP1 and BP2 of the Prader-Willi/Angelman Syndromes Deletion Region That Have Undergone Evolutionary Transposition Mediated by Flanking Duplicons. Am. J. Hum. Genet. 2003, 73, 898–925. [Google Scholar] [CrossRef] [Green Version]
- Neubert, G.; Von Au, K.; Drossel, K.; Tzschach, A.; Horn, D.; Nickel, R.; Kaindl, A.M. Angelman Syndrome and Severe Infections in a Patient with de Novo 15q11.2–Q13.1 Deletion and Maternally Inherited 2q21.3 Microdeletion. Gene 2013, 512, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Park, J.; Jang, W.; Chae, H.; Kim, M.; Kim, Y. A Twin Sibling with Prader-Willi Syndrome Caused by Type 2 Microdeletion Following Assisted Reproductive Technology: A Case Report. Biomed. Rep. 2016, 5, 18–22. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Filipek, P.A.; Wu, C.; Bocian, M.; Hakin, S.; Modahl, C.; Spence, M.A. Analysis of a 1-Megabase Deletion in 15q22-Q23 in an Autistic Patient: Identification of Candidate Genes for Autism and of Homologous DNA Segments in 15q22-Q23 and 15q11-Q13. Am. J. Med. Genet. 2000, 96, 765–770. [Google Scholar] [CrossRef]
- Vincent, K.M.; Eaton, A.; Reza Yassaee, V.; Miryounesi, M.; Hashemi-Gorji, F.; Rudichuk, L.; Goez, H.; Leonard, N.; Lazier, J. Delineating the Expanding Phenotype of HERC2-Related Disorders: The Impact of Biallelic Loss of Function versus Missense Variation. Clin. Genet. 2021, 100, 637–640. [Google Scholar] [CrossRef]
- Imai, Y.; Kobayashi, Y.; Inoshita, T.; Meng, H.; Arano, T.; Uemura, K.; Asano, T.; Yoshimi, K.; Zhang, C.L.; Matsumoto, G.; et al. The Parkinson’s Disease-Associated Protein Kinase LRRK2 Modulates Notch Signaling through the Endosomal Pathway. PLoS Genet. 2015, 11, e1005503. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Brunet, V.; Gagnon, P.A.; Lavoie, M.-È.; Bouchard, J.B.; Nadaf, J.; Majewski, J.; Morin, C.; Laprise, C. Exome Sequencing Study of Partial Agenesis of the Corpus Callosum in Men with Developmental Delay, Epilepsy, and Microcephaly. Mol. Genet. Genom. Med. 2020, 8, e992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Ding, X.; Zhang, Q.; Liu, J.; Zhang, Y.; Zhang, Y.; Tian, Z.; Li, W.; Zhu, W.; Kang, H.; et al. Exome Sequencing of 112 Trios Identifies Recessive Genetic Variants in Brain Arteriovenous Malformations. J. Neurointerv. Surg. 2021, 13, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Q.; Zheng, Y.Y.; Zhou, H.J.; Zhang, X.X.; Wu, P.; Zhu, S.M. LncRNA-Fendrr Protects against the Ubiquitination and Degradation of NLRC4 Protein through HERC2 to Regulate the Pyroptosis of Microglia. Mol. Med. 2021, 27, 39. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Jeong, H.R.; Rho, J.G.; Kum, C.D.; Hee, K.; Kim, D.W.; Cheong, J.Y.; Jeong, S.; Hwang, J.S. Identification of Rare Missense Mutations in NOTCH2 and HERC2 Associated with Familial Central Precocious Puberty via Whole-Exome Sequencing. Gynecol. Endocrinol. 2020, 36, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.L.; Guggenheim, J.A. UK BioBank Eye and Vision Consortium Genome-Wide Association Studies for Corneal and Refractive Astigmatism in UK Biobank Demonstrate a Shared Role for Myopia Susceptibility Loci. Hum. Genet. 2018, 137, 881–896. [Google Scholar] [CrossRef] [Green Version]
- Yucesoy, B.; Kaufman, K.M.; Lummus, Z.L.; Weirauch, M.T.; Zhang, G.; Boulet, L.; Sastre, J.; Quirce, S.; Tarlo, S.M.; Cruz, M.; et al. Genome-Wide Association Study Identifies Novel Loci Associated With Diisocyanate-Induced Occupational Asthma. Toxicol. Sci. 2015, 146, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Man, A.W.C.; Yang, K.; Guo, Y.; Xu, C.; Tse, H.-F.; Han, W.; Bloksgaard, M.; De Mey, J.G.R.; Vanhoutte, P.M.; et al. Endothelial SIRT1 Prevents Adverse Arterial Remodeling by Facilitating HERC2-Mediated Degradation of Acetylated LKB1. Oncotarget 2016, 7, 39065–39081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Liu, G.; Liao, Y.-Y.; Chu, C.; Zheng, W.-L.; Wang, Y.; Hu, J.-W.; Ma, Q.; Wang, K.-K.; Yan, Y.; et al. Identification of Candidate Biomarkers for Salt Sensitivity of Blood Pressure by Integrated Bioinformatics Analysis. Front. Genet. 2020, 11, 988. [Google Scholar] [CrossRef]
- Jin, Y.; Birlea, S.A.; Fain, P.R.; Ferrara, T.M.; Ben, S.; Riccardi, S.L.; Cole, J.B.; Gowan, K.; Holland, P.J.; Bennett, D.C.; et al. Genome-Wide Association Analyses Identify 13 New Susceptibility Loci for Generalized Vitiligo. Nat. Genet. 2012, 44, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Aponte, J.L.; Chiano, M.N.; Yerges-armstrong, L.M.; Hinds, D.A.; Tian, C.; Gupta, A.; Guo, C.; Fraser, D.J.; Freudenberg, J.M.; Rajpal, D.K.; et al. Assessment of Rosacea Symptom Severity by Genome-Wide Association Study and Expression Analysis Highlights Immuno-Inflammatory and Skin Pigmentation Genes. Hum. Mol. Genet. 2018, 27, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yin, J.; Huang, H.; Jorgenson, E.; Choquet, H.; Asgari, M.M. Genome-Wide Association Study of Actinic Keratosis Identifies New Susceptibility Loci Implicated in Pigmentation and Immune Regulation Pathways. Commun. Biol. 2022, 5, 386. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; May, S.; Lu, T.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; Schreiber, S. Replication of Signals from Recent Studies of Crohn’s Disease Identifies Previously Unknown Disease Loci for Ulcerative Colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef]
- Wang, K.; Baldassano, R.; Zhang, H.; Qu, H.; Imielinski, M.; Kugathasan, S.; Annese, V.; Dubinsky, M.; Rotter, J.I.; Russell, R.K.; et al. Comparative Genetic Analysis of Inflammatory Bowel Disease and Type 1 Diabetes Implicates Multiple Loci with Opposite Effects. Hum. Mol. Genet. 2010, 19, 2059–2067. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Nothnagel, M.; Franke, A.; Jacobs, G.; Saadati, H.R.; Gaede, K.I.; Rosenstiel, P.; Schu, M.; Mu, J.; Schreiber, S.; et al. Association of Inflammatory Bowel Disease Risk Loci with Sarcoidosis, and Its Acute and Chronic Subphenotypes. Eur. Respir. J. 2011, 37, 610–616. [Google Scholar] [CrossRef] [Green Version]
- García-heredia, J.M.; Carnero, A. The Cargo Protein MAP17 (PDZK1IP1) Regulates the Immune Microenvironment. Oncotarget 2017, 8, 98580–98597. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Q.; Deng, F.; Zheng, Z.; Luo, J.; Wang, P.; Zhou, J.; Lu, X.; Zhang, L.; Chen, Z.; et al. HERC2 Promotes Inflammation-driven Cancer Stemness and Immune Evasion in Hepatocellular Carcinoma by Activating STAT3 Pathway. J. Exp. Clin. Cancer Res. 2023, 42, 38. [Google Scholar] [CrossRef] [PubMed]
- Sala-Gaston, J.; Martinez-Martinez, A.; Pedrazza, L.; Lorenzo-Martín, L.F.; Caloto, R.; Bustelo, X.R.; Ventura, F.; Rosa, J.L. Herc Ubiquitin Ligases in Cancer. Cancers 2020, 12, 1653. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.; Meloche, S. Atypical Mitogen-Activated Protein Kinases: Structure, Regulation and Functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Liu, J.; Zhang, C. Evolutionary History of the Vertebrate Mitogen Activated Protein Kinases Family. PLoS ONE 2011, 6, e26999. [Google Scholar] [CrossRef]
- Laine, A.; Ronai, Z. Ubiquitin Chains in the Ladder of MAPK Signaling. Sci. STKE 2005, 2005, re5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, S.; Lee, H.; Han, S.; Song, J.M.; Han, D.; Suh, Y.H. Nedd4 E3 Ligase and Beta-Arrestins Regulate Ubiquitination, Trafficking, and Stability of the MGlu7 Receptor. Elife 2019, 8, e44502. [Google Scholar] [CrossRef]
- Filonova, I.; Trotter, J.H.; Banko, J.L.; Weeber, E.J. Activity-Dependent Changes in MAPK Activation in the Angelman Syndrome Mouse Model. Learn. Mem. 2014, 21, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.R.; Shi, P.; Bryant, J.; Chen, J.; Dukhande, V.; Gentry, M.S.; Jang, H.; Jeoung, M.; Galperin, E. HUWE1 Is a Molecular Link Controlling RAF-1 Activity Supported by the Shoc2 Scaffold. Mol. Cell. Biol. 2014, 34, 3579–3593. [Google Scholar] [CrossRef] [Green Version]
- Sala-Gaston, J.; Pedrazza, L.; Ramirez, J.; Martinez-Martinez, A.; Rawlins, L.E.; Baple, E.L.; Crosby, A.H.; Mayor, U.; Ventura, F.; Rosa, J.L. HERC2 Deficiency Activates C-RAF/MKK3/P38 Signalling Pathway Altering the Cellular Response to Oxidative Stress. Cell. Mol. Life Sci. 2022, 79, 548. [Google Scholar] [CrossRef]
- Wang, K.; Tang, J.; Liu, X.; Wang, Y.; Chen, W.; Zheng, R. UBR5 Regulates Proliferation and Radiosensitivity in Human Laryngeal Carcinoma via the P38/MAPK Signaling Pathway. Oncol. Rep. 2020, 44, 685–697. [Google Scholar] [CrossRef]
- Carpentier, I.; Coornaert, B.; Beyaert, R. Smurf2 Is a TRAF2 Binding Protein That Triggers TNF-R2 Ubiquitination and TNF-R2-Induced JNK Activation. Biochem. Biophys. Res. Commun. 2008, 374, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 Ubiquitin Ligase HERC1 Controls the ERK Signaling Pathway Targeting C-RAF for Degradation. Oncotarget 2018, 9, 31531–31548. [Google Scholar] [CrossRef] [PubMed]
- Pedrazza, L.; Schneider, T.; Bartrons, R.; Ventura, F.; Rosa, J.L. The Ubiquitin Ligase HERC1 Regulates Cell Migration via RAF-Dependent Regulation of MKK3/P38 Signaling. Sci. Rep. 2020, 10, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhu, L.; Sun, J.; Zhang, Y.; Cui, Q.; Wu, L.; Chen, S.; Lu, J. Pan-Cancer Analysis of NEDD4L and Its Tumor Suppressor Effects in Clear Cell Renal Cell Carcinoma. J. Cancer 2021, 12, 6242–6253. [Google Scholar] [CrossRef]
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin Plays an Atypical Role in GPCR-Induced P38 MAP Kinase Activation on Endosomes. J. Cell Biol. 2015, 210, 1117–1131. [Google Scholar] [CrossRef] [Green Version]
- Grimsey, N.J.; Narala, R.; Rada, C.C.; Mehta, S.; Stephens, B.S.; Kufareva, I.; Lapek, J.; Gonzalez, D.J.; Handel, T.M.; Zhang, J.; et al. A Tyrosine Switch on NEDD4-2 E3 Ligase Transmits GPCR Inflammatory Signaling. Cell Rep. 2018, 24, 3312–3323.e5. [Google Scholar] [CrossRef] [Green Version]
- Ge, Q.Y.; Chen, J.; Li, G.X.; Tan, X.L.; Song, J.; Ning, D.; Mo, J.; Du, P.C.; Liu, Q.M.; Liang, H.F.; et al. GRAMD4 Inhibits Tumour Metastasis by Recruiting the E3 Ligase ITCH to Target TAK1 for Degradation in Hepatocellular Carcinoma. Clin. Transl. Med. 2021, 11, e635. [Google Scholar] [CrossRef]
- Yin, Q.; Han, T.; Fang, B.; Zhang, G.; Zhang, C.; Roberts, E.R.; Izumi, V.; Zheng, M.; Jiang, S.; Yin, X.; et al. K27-Linked Ubiquitination of BRAF by ITCH Engages Cytokine Response to Maintain MEK-ERK Signaling. Nat. Commun. 2019, 10, 1870. [Google Scholar] [CrossRef] [Green Version]
- Otaki, Y.; Takahashi, H.; Watanabe, T.; Funayama, A.; Netsu, S.; Honda, Y.; Narumi, T.; Kadowaki, S.; Hasegawa, H.; Honda, S.; et al. HECT-Type Ubiquitin E3 Ligase ITCH Interacts with Thioredoxin-Interacting Protein and Ameliorates Reactive Oxygen Species-Induced Cardiotoxicity. J. Am. Heart Assoc. 2016, 5, e002485. [Google Scholar] [CrossRef] [Green Version]
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.-Z.; Massoumi, R.; Venuprasad, K. The E3 Ubiquitin Ligase Itch Inhibits P38α Signaling and Skin Inflammation through the Ubiquitylation of Tab1. Sci. Signal. 2015, 8, ra22. [Google Scholar] [CrossRef]
- Ahn, Y.-H.; Kurie, J.M. MKK4/SEK1 Is Negatively Regulated through a Feedback Loop Involving the E3 Ubiquitin Ligase Itch. J. Biol. Chem. 2009, 284, 29399–29404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Yang, B.; Qi, R.; Xie, Q.; Li, T.; Yang, J.; Tong, T.; Niu, K.; Li, M.; Pan, W.; et al. Targeting WWP1 Ameliorates Cardiac Ischemic Injury by Suppressing KLF15-Ubiquitination Mediated Myocardial Inflammation. Theranostics 2023, 13, 417–437. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.W.; Xu, W.C.; Luo, J.G.; Guo, X.J.; Sun, T.; Zhao, X.L.; Fu, Z.J. WW Domain Containing E3 Ubiquitin Protein Ligase 1 (WWP1) Negatively Regulates TLR4-Mediated TNF-α and IL-6 Production by Proteasomal Degradation of TNF Receptor Associated Factor 6 (TRAF6). PLoS ONE 2013, 8, e67633. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yue, Y.; Xiong, S. Mycobacterial PPE36 Modulates Host In Fl Ammation by Promoting E3 Ligase Smurf1-Mediated MyD88 Degradation. Front. Immunol. 2022, 13, 690667. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, D.; Li, N.; Gao, P.; Zhang, M.; Zhang, Y. Hippo Kinase NDR2 Inhibits IL-17 Signaling by Promoting Smurf1-Mediated MEKK2 Ubiquitination and Degradation. Mol. Immunol. 2019, 105, 131–136. [Google Scholar] [CrossRef]
- Yamashita, M.; Ying, S.X.; Zhang, G.M.; Li, C.; Cheng, S.Y.; Deng, C.X.; Zhang, Y.E. Ubiquitin Ligase Smurf1 Controls Osteoblast Activity and Bone Homeostasis by Targeting MEKK2 for Degradation. Cell 2005, 121, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Hadjebi, O.; Casas-Terradellas, E.; Garcia-Gonzalo, F.R.; Rosa, J.L. The RCC1 Superfamily: From Genes, to Function, to Disease. Biochim. Biophys. Acta 2008, 1783, 1467–1479. [Google Scholar] [CrossRef] [Green Version]
- Desideri, E.; Cavallo, A.L.; Baccarini, M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 2015, 161, 967–970. [Google Scholar] [CrossRef] [Green Version]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF Proteins Take Centre Stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Noeparast, A.; Giron, P.; Noor, A.; Bahadur Shahi, R.; De Brakeleer, S.; Eggermont, C.; Vandenplas, H.; Boeckx, B.; Lambrechts, D.; De Grève, J.; et al. CRAF Mutations in Lung Cancer Can Be Oncogenic and Predict Sensitivity to Combined Type II RAF and MEK Inhibition. Oncogene 2019, 38, 5933–5941. [Google Scholar] [CrossRef] [Green Version]
- Galligan, T.; Martinez-noe, G.; Arndt, V.; Hayes, S.; Chittenden, T.W.; Harper, J.W.; Howley, P.M. Proteomic Analysis and Identification of Cellular Interactors of the Giant Ubiquitin Ligase HERC2. J. Proteome Res. 2015, 14, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, T.; Martinez-Martinez, A.; Cubillos-Rojas, M.; Bartrons, R.; Ventura, F.; Rosa, J.L. Large HERCs Function as Tumor Suppressors. Front. Oncol. 2019, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Noel, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P.M. Identification and Proteomic Analysis of Distinct UBE3A/E6AP Protein Complexes. Mol. Cell. Biol. 2012, 32, 3095–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Noël, G.; Luck, K.; Kühnle, S.; Desbuleux, A.; Szajner, P.; Galligan, J.T.; Rodriguez, D.; Zheng, L.; Boyland, K.; Leclere, F.; et al. Network Analysis of UBE3A/E6AP-Associated Proteins Provides Connections to Several Distinct Cellular Processes. J. Mol. Biol. 2018, 430, 1024–1050. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 Controls Iron Homeostasis and Ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef]
- Iroegbu, J.D.; Ijomone, O.K.; Femi-akinlosotu, O.M.; Ijomone, O.M. ERK/MAPK Signalling in the Developing Brain: Perturbations and Consequences. Neurosci. Biobehav. Rev. 2021, 131, 792–805. [Google Scholar] [CrossRef]
- Murtaza, N.; Cheng, A.A.; Brown, C.O.; Praveen Meka, D.; Hong, S.; Uy, J.A.; El-Hajjar, J.; Pipko, N.; Unda, B.K.; Schwanke, B.; et al. Neuron-Specific Protein Network Mapping of Autism Risk Genes Identifies Shared Biological Mechanisms and Disease-Relevant Pathologies. Cell Rep. 2022, 41, 111678. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological Roles of MAPK Signaling Pathways in Human Diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kang, L.; Wang, Y.-Z.; Yang, B.-R.; Zhang, C.; Lu, Y.; Kang, L. Microglia in Motor Neuron Disease: Signaling Evidence from Last 10 Years. Dev. Neurobiol. 2022, 82, 625–638. [Google Scholar] [CrossRef]
- U.S Food and Drug Administration (FDA). Nexavar (Sorafenib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021923_s000_nexavartoc.cfm (accessed on 27 January 2023).
- European Medicines Agengy (EMA). Nexavar (Sorafenib). Available online: https://www.ema.europa.eu/en/documents/overview/nexavar-epar-summary-public_en.pdf (accessed on 27 January 2023).
- Lito, P.; Rosen, N.; Solit, D.B. Tumor Adaptation and Resistance to RAF Inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The RAF Inhibitor Paradox Revisited. Cancer Cell 2012, 21, 147–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, C.W.; Dale, I.L.; Thomas, A.P.; Hunt, J.; Chin, J.W. Selective CRAF Inhibition Elicits Transactivation. J. Am. Chem. Soc. 2021, 143, 4600–4606. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.T.; Navas, C.; Martín-Serrano, G.; Graña-Castro, O.; Lechuga, C.G.; Martín-Díaz, L.; Djurec, M.; Li, J.; Morales-Cacho, L.; Esteban-Burgos, L.; et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell 2019, 35, 573–587.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Cao, C.; Ni, Z.; Liu, Y.; Song, P.; Hao, S.; He, Y.; Sun, X.; Rao, Y. PROTACs: Great Opportunities for Academia and Industry (an Update from 2020 to 2021). Signal Transduct. Target. Ther. 2022, 7, 181. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug. Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Burke, M.R.; Smith, A.R.; Zheng, G. Overcoming Cancer Drug Resistance Utilizing PROTAC Technology. Cell Dev. Biol. 2022, 10, 872729. [Google Scholar] [CrossRef]
- Posternak, G.; Tang, X.; Maisonneuve, P.; Jin, T.; Lavoie, H.; Daou, S.; Orlicky, S.; De Rugy, T.G.; Caldwell, L.; Chan, K.; et al. Functional Characterization of a PROTAC Directed against BRAF Mutant V600E. Nat. Chem. Biol. 2020, 16, 1170–1178. [Google Scholar] [CrossRef]
- Alabi, S.; Jaime-figueroa, S.; Yao, Z.; Gao, Y.; Hines, J.; Samarasinghe, K.T.G.; Vogt, L.; Rosen, N.; Crews, C.M. Mutant-Selective Degradation by BRAF-Targeting PROTACs. Nat. Commun. 2021, 12, 920. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| HECT E3s | MAPK Pathway | Mechanism | References |
|---|---|---|---|
| HERC1 | ERK and p38 | HERC1 regulates ubiquitylation-mediated degradation of C-RAF affecting ERK and p38 signaling | [34,66,67] |
| HERC2 | p38 | HERC2 regulates ubiquitylation-mediated degradation of C-RAF affecting p38 signaling | [68] |
| NEDD4 | ERK1/2 | Nedd4 regulates ubiquitylation and degradation of mGlu7, which mediates MAPK signaling | [69] |
| NEDD4L | ERK1/2 | NEDD4L overexpression inhibits ERK1/2 phosphorylation | [70] |
| p38 | NEDD4L mediates K63-linked ubiquitylation of PAR1 inducing TAB1-mediated p38 activation | [71,72] | |
| ITCH | ERK1/2, p38 and JNK | ITCH is recruited by GRAMD4 to target TAK1 ubiquitylation and degradation | [73] |
| ERK1/2 | ITCH ubiquitylates BRAF leading to its activation and subsequent elevation of MEK/ERK signaling | [74] | |
| p38 | ITCH targets TXNIP for ubiquitin-proteasome degradation, decreasing p38 signaling | [75] | |
| p38 | ITCH catalyzes the K48-linked ubiquitylation of TAB1, which modulates p38 signaling | [76] | |
| JNK | ITCH regulates MKK4 ubiquitylation and stability, affecting JNK signaling | [77] | |
| WWP1 | ERK1/2 and p38 | WWP1 regulates ubiquitylation and subsequent degradation of KLF15, which acts inhibiting MAPK signaling | [78] |
| ERK1/2, p38 and JNK | WWP1 modulates stability and LPS-induced TRAF6 ubiquitylation, affecting ERK, JNK, and p38 phosphorylation | [79] | |
| SMURF1 | ERK1/2, p38 and JNK | SMURF1 interacts and regulates ubiquitylation-mediated proteasomal degradation of MyD88, a MAPK signaling adapter | [80] |
| ERK1/2, p38 and JNK | SMURF1 promotes MEKK2 ubiquitylation and degradation | [81,82] | |
| SMURF2 | JNK | SMURF2 induces TNF-R2 ubiquitylation and relocalization, which enhances JNK signaling | [83] |
| UBE3A | ERK1/2 | UBE3A deficiency impairs ERK1/2 activation | [84] |
| HUWE1 | ERK1/2 | HUWE1 mediates ubiquitylation of C-RAF and regulates its stability, affecting ERK1/2 signaling | [85] |
| UBR5 | p38 | UBR5 silencing activates the p38 signaling pathway | [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala-Gaston, J.; Costa-Sastre, L.; Pedrazza, L.; Martinez-Martinez, A.; Ventura, F.; Rosa, J.L. Regulation of MAPK Signaling Pathways by the Large HERC Ubiquitin Ligases. Int. J. Mol. Sci. 2023, 24, 4906. https://doi.org/10.3390/ijms24054906
Sala-Gaston J, Costa-Sastre L, Pedrazza L, Martinez-Martinez A, Ventura F, Rosa JL. Regulation of MAPK Signaling Pathways by the Large HERC Ubiquitin Ligases. International Journal of Molecular Sciences. 2023; 24(5):4906. https://doi.org/10.3390/ijms24054906
Chicago/Turabian StyleSala-Gaston, Joan, Laura Costa-Sastre, Leonardo Pedrazza, Arturo Martinez-Martinez, Francesc Ventura, and Jose Luis Rosa. 2023. "Regulation of MAPK Signaling Pathways by the Large HERC Ubiquitin Ligases" International Journal of Molecular Sciences 24, no. 5: 4906. https://doi.org/10.3390/ijms24054906