

SPRED2: A Novel Regulator of Epithelial-Mesenchymal Transition and Stemness in Hepatocellular Carcinoma Cells


 ,
,  , and
, and
Abstract
1. Introduction
2. Results
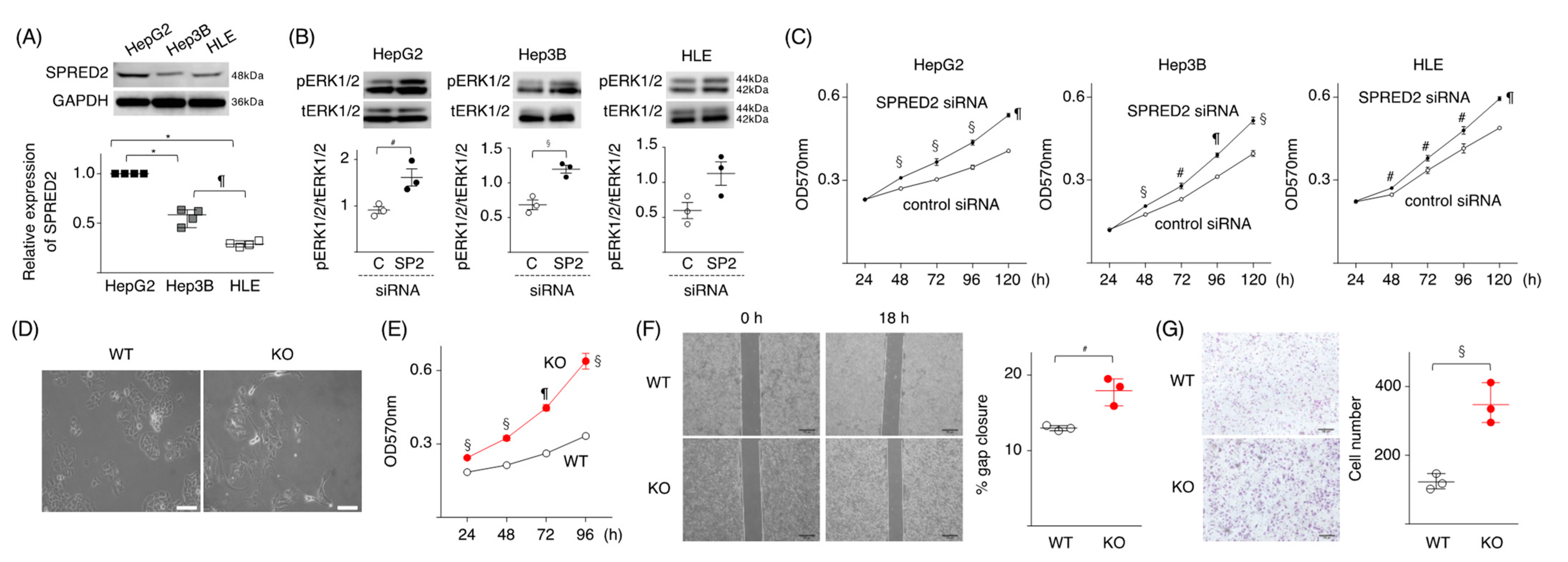
2.1. Loss of SPRED2 Augments HCC Cell Proliferation and Invasiveness
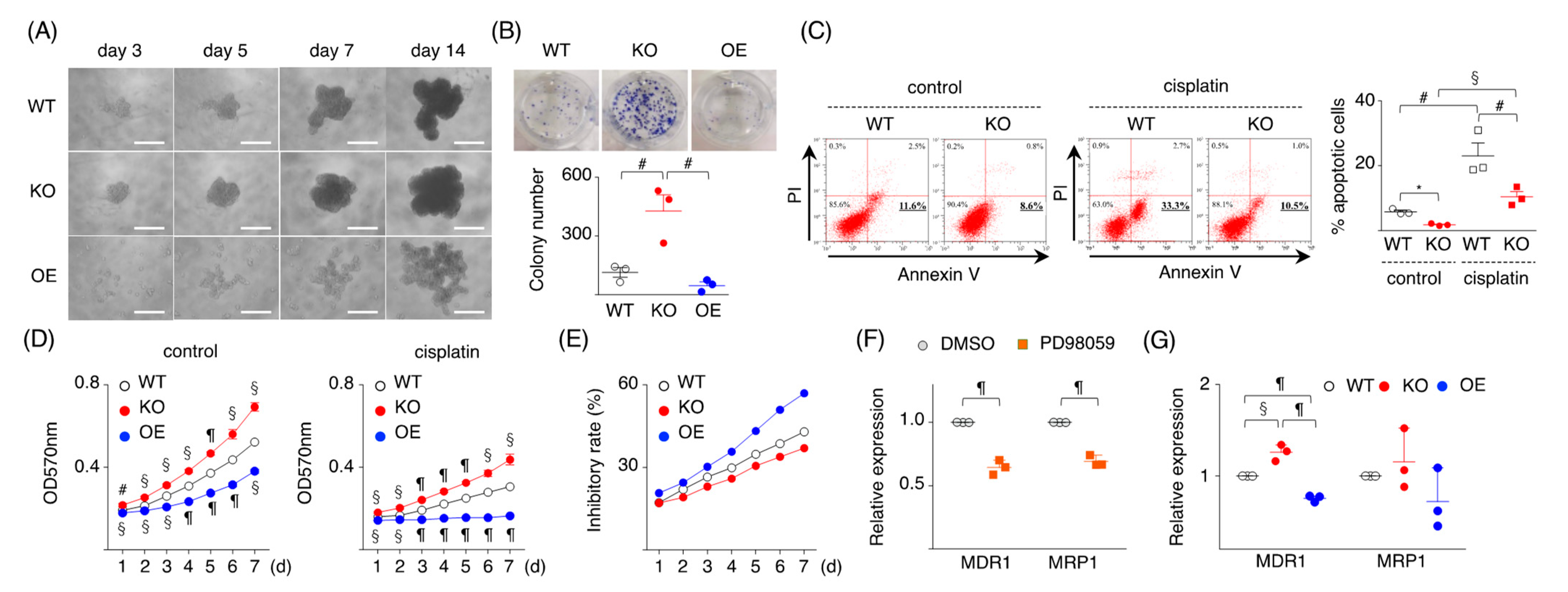
2.2. Loss of SPRED2 Alters the Expression of Molecules Involved in Cancer Cell Growth and Progression
2.3. Loss of SPRED2 Increases Cancer Cell Stemness
2.4. Loss of SPRED2 Upregulates the Expression of Pluripotency and Stemness Markers
2.5. SPRED2 Level Is Associated with the Stemness of HepG2 Cells
2.6. SPRED2 Expression Is Downregulated in HCC Tissues
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection
4.3. Generation of SPRED2 Knockout (SPRED2-KO) Cells
4.4. Real-Time Quantitative PCR (RT-qPCR)
4.5. Western Blotting
4.6. Cell Proliferation and Cytotoxicity Assay
4.7. Scratch Assay
4.8. Transwell Invasion Assay
4.9. Sphere Formation and Colony Formation Assay
4.10. Fluorescence Immunostaining
4.11. Flow Cytometry
4.12. Cell Isolation
4.13. Immunohistochemistry (IHC)
4.14. Human Tissue Samples
4.15. Data Collection
4.16. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Ichida, T.; Sugitani, S.; Genda, T.; Inayoshi, J.; Takamura, M.; Matsuda, Y.; Nomoto, M.; Aoyagi, Y. Overexpression of extracellular signal-regulated protein kinase and its correlation with proliferation in human hepatocellular carcinoma. Liver Int. 2004, 24, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zhao, Y.; Xu, F.; Jia, C.; Xu, Y.; Dai, C. An updated meta-analysis of randomized controlled trials assessing the effect of sorafenib in advanced hepatocellular carcinoma. PLoS ONE 2014, 9, e112530. [Google Scholar] [CrossRef] [PubMed]
- Wakioka, T.; Sasaki, A.; Kato, R.; Shouda, T.; Matsumoto, A.; Miyoshi, K.; Tsuneoka, M.; Komiya, S.; Baron, R.; Yoshimura, A. Spred is a Sprouty-related suppressor of Ras signalling. Nature 2001, 412, 647–651. [Google Scholar] [CrossRef]
- Yoshida, T.; Hisamoto, T.; Akiba, J.; Koga, H.; Nakamura, K.; Tokunaga, Y.; Hanada, S.; Kumemura, H.; Maeyama, M.; Harada, M.; et al. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 2006, 25, 6056–6066. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Yamane, M.; Sugimoto, S.; Hirano, Y.; Kurosaki, T.; Otani, S.; Miyoshi, K.; Ohara, T.; Okazaki, M.; Yoshimura, T.; et al. Negative impact of recipient SPRED2 deficiency on transplanted lung in a mouse model. Transpl. Immunol. 2019, 57, 101242. [Google Scholar] [CrossRef] [PubMed]
- Itakura, J.; Sato, M.; Ito, T.; Mino, M.; Fushimi, S.; Takahashi, S.; Yoshimura, T.; Matsukawa, A. Spred2-deficiecy Protects Mice from Polymicrobial Septic Peritonitis by Enhancing Inflammation and Bacterial Clearance. Sci. Rep. 2017, 7, 12833. [Google Scholar] [CrossRef]
- Ito, T.; Itakura, J.; Takahashi, S.; Sato, M.; Mino, M.; Fushimi, S.; Yamada, M.; Morishima, T.; Kunkel, S.L.; Matsukawa, A. Sprouty-Related Ena/Vasodilator-Stimulated Phosphoprotein Homology 1-Domain-Containing Protein-2 Critically Regulates Influenza A Virus-Induced Pneumonia. Crit. Care Med. 2016, 44, e530–e543. [Google Scholar] [CrossRef]
- Ohkura, T.; Yoshimura, T.; Fujisawa, M.; Ohara, T.; Marutani, R.; Usami, K.; Matsukawa, A. Spred2 Regulates High Fat Diet-Induced Adipose Tissue Inflammation, and Metabolic Abnormalities in Mice. Front. Immunol. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Yamane, M.; Yamamoto, S.; Otani, S.; Miyoshi, K.; Sugimoto, S.; Matsukawa, A.; Toyooka, S.; Oto, T.; Miyoshi, S. SPRED2 deficiency may lead to lung ischemia-reperfusion injury via ERK1/2 signaling pathway activation. Surg. Today 2018, 48, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fujisawa, M.; Ohara, T.; Liu, Q.; Cao, C.; Yang, X.; Yoshimura, T.; Kunkel, S.L.; Matsukawa, A. Spred2 controls the severity of Concanavalin A-induced liver damage by limiting interferon-gamma production by CD4+ and CD8+ T cells. J. Adv. Res. 2022, 35, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yoshimura, T.; Ohkura, T.; Fujisawa, M.; Fushimi, S.; Ito, T.; Itakura, J.; Hiraoka, S.; Okada, H.; Yamamoto, K.; et al. A Novel Role of Spred2 in the Colonic Epithelial Cell Homeostasis and Inflammation. Sci. Rep. 2016, 6, 37531. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, H.; Ito, T.; Fushimi, S.; Nakashima, Y.; Itakura, J.; Qiuying, L.; Win, M.M.; Cuiming, S.; Chen, C.; Sato, M.; et al. Spred-2 deficiency exacerbates acetaminophen-induced hepatotoxicity in mice. Clin. Immunol. 2012, 144, 272–282. [Google Scholar] [CrossRef]
- Xu, Y.; Ito, T.; Fushimi, S.; Takahashi, S.; Itakura, J.; Kimura, R.; Sato, M.; Mino, M.; Yoshimura, A.; Matsukawa, A. Spred-2 deficiency exacerbates lipopolysaccharide-induced acute lung inflammation in mice. PLoS ONE 2014, 9, e108914. [Google Scholar] [CrossRef]
- Yang, X.; Fujisawa, M.; Yoshimura, T.; Ohara, T.; Sato, M.; Mino, M.; San, T.H.; Gao, T.; Kunkel, S.L.; Matsukawa, A. Spred2 Deficiency Exacerbates D-Galactosamine/Lipopolysaccharide -induced Acute Liver Injury in Mice via Increased Production of TNFα. Sci. Rep. 2018, 8, 188. [Google Scholar] [CrossRef]
- Kawara, A.; Mizuta, R.; Fujisawa, M.; Ito, T.; Li, C.; Nakamura, K.; Sun, C.; Kuwabara, M.; Kitabatake, M.; Yoshimura, T.; et al. Spred2-deficiency enhances the proliferation of lung epithelial cells and alleviates pulmonary fibrosis induced by bleomycin. Sci. Rep. 2020, 10, 16490. [Google Scholar] [CrossRef]
- Alysandratos, K.D.; Herriges, M.J.; Kotton, D.N. Epithelial Stem and Progenitor Cells in Lung Repair and Regeneration. Annu. Rev. Physiol. 2021, 83, 529–550. [Google Scholar] [CrossRef]
- Villar, V.; Kocić, J.; Santibanez, J.F. Spred2 inhibits TGF-beta1-induced urokinase type plasminogen activator expression, cell motility and epithelial mesenchymal transition. Int. J. Cancer 2010, 127, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.N.; Liu, X.Y.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Yan, J.; Zhang, Q.W.; Wang, L.S.; Li, X.Y.; Wang, H. Regulation of human hepatocellular carcinoma cells by Spred2 and correlative studies on its mechanism. Biochem. Biophys. Res. Commun. 2011, 410, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Kong, F.; Xiao, F.; Li, Y.; Wang, H.; Zhang, S.; Huang, D.; Wang, L.; Yang, Y. Spred2 inhibits epithelial-mesenchymal transition of colorectal cancer cells by impairing ERK signaling. Oncol. Rep. 2020, 44, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fan, D. The epithelial-mesenchymal transition and cancer stem cells: Functional and mechanistic links. Curr. Pharm. Des. 2015, 21, 1279–1291. [Google Scholar] [CrossRef]
- Lambert, A.W.; Weinberg, R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Xie, D.; Pei, Q.; Wan, X.; Xing, H.R.; Ye, T. Characteristics of the PI3K/AKT and MAPK/ERK pathways involved in the maintenance of self-renewal in lung cancer stem-like cells. Int. J. Biol. Sci. 2021, 17, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun. Signal. 2021, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Vander Borght, S.; Komuta, M.; Libbrecht, L.; Katoonizadeh, A.; Aerts, R.; Dymarkowski, S.; Verslype, C.; Nevens, F.; Roskams, T. Expression of multidrug resistance-associated protein 1 in hepatocellular carcinoma is associated with a more aggressive tumour phenotype and may reflect a progenitor cell origin. Liver Int. 2008, 28, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Wang, X.W. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin Oncol. 2012, 39, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef]
- Mehta, K.; Moravcikova, E.; McFall, D.; Luketich, J.D.; Pennathur, A.; Donnenberg, A.D.; Donnenberg, V.S. The Mesenchymal State Predicts Poor Disease-Free Survival in Resectable Non-Small Cell Lung Cancer. Ann. Thorac. Surg. 2017, 104, 321–328. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Zuñiga-Eulogio, M.D.; Mendoza-Catalán, M.A.; Rodríguez-Ruiz, H.A.; Castañeda-Saucedo, E.; Ortuño-Pineda, C.; Padilla-Benavides, T.; Navarro-Tito, N. Extracellular-Signal Regulated Kinase: A Central Molecule Driving Epithelial-Mesenchymal Transition in Cancer. Int. J. Mol. Sci. 2019, 20, 2885. [Google Scholar] [CrossRef]
- Saitoh, M.; Endo, K.; Furuya, S.; Minami, M.; Fukasawa, A.; Imamura, T.; Miyazawa, K. STAT3 integrates cooperative Ras and TGF-β signals that induce Snail expression. Oncogene 2016, 35, 1049–1057. [Google Scholar] [CrossRef]
- Wang, Y.; van Boxel-Dezaire, A.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, J.; Wang, L.; Tian, Z. Growth inhibition of human hepatocellular carcinoma cells by blocking STAT3 activation with decoy-ODN. Cancer Lett. 2008, 262, 201–213. [Google Scholar] [CrossRef]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef] [PubMed]
- Ellero, A.A.; van den Bout, I.; Vlok, M.; Cromarty, A.D.; Hurrell, T. Continual proteomic divergence of HepG2 cells as a consequence of long-term spheroid culture. Sci. Rep. 2021, 11, 10917. [Google Scholar] [CrossRef]
- Engelhardt, C.M.; Bundschu, K.; Messerschmitt, M.; Renné, T.; Walter, U.; Reinhard, M.; Schuh, K. Expression and subcellular localization of Spred proteins in mouse and human tissues. Histochem. Cell Biol. 2004, 122, 527–538. [Google Scholar] [CrossRef]
- Chiu, C.F.; Ho, M.Y.; Peng, J.M.; Hung, S.W.; Lee, W.H.; Liang, C.M.; Liang, S.M. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene 2013, 32, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [PubMed]
- Nio, K.; Yamashita, T.; Kaneko, S. The evolving concept of liver cancer stem cells. Mol. Cancer 2017, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Tsui, Y.M.; Chan, L.K.; Ng, I.O. Cancer stemness in hepatocellular carcinoma: Mechanisms and translational potential. Br. J. Cancer 2020, 122, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, N.; Valencia, T.; Warren, A.Y.; Gnanapragasam, V.J. Evidence for downregulation of the negative regulator SPRED2 in clinical prostate cancer. Br. J. Cancer 2013, 108, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Fujisawa, M.; Chunning, L.; Ito, T.; Yamaguchi, T.; Yoshimura, T.; Matsukawa, A. Expression of Spred2 in the urothelial tumorigenesis of the urinary bladder. PLoS ONE 2021, 16, e0254289. [Google Scholar] [CrossRef]
- Qin, L.F.; Ng, I.O. Induction of apoptosis by cisplatin and its effect on cell cycle-related proteins and cell cycle changes in hepatoma cells. Cancer Lett. 2002, 175, 27–38. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age | Sex | Stage | Growth Pattern | Tumor Grade | Cirrhosis |
|---|---|---|---|---|---|---|
| 1 | 65 | M | 1b | mixed | moderate | − |
| 2 | 71 | M | 1a | trabecular | well | − |
| 3 | 75 | F | 1b | pseudoglandular | moderate | + |
| 4 | 68 | F | 1b | mixed | poor | − |
| 5 | 73 | M | 1b | trabecular | well | − |
| 6 | 69 | F | 1b | trabecular | moderate | + |
| 7 | 56 | M | 1a | trabecular | moderate | − |
| 8 | 79 | M | 1b | mixed | moderate | + |
| 9 | 61 | M | 2 | trabecular | poor | − |
| 10 | 74 | M | 1a | solid | moderate | + |
| 11 | 79 | M | 2 | mixed | moderate | − |
| 12 | 55 | M | 1a | trabecular | well | + |
| 13 | 82 | F | 2 | trabecular | moderate | − |
| 14 | 52 | M | 1a | trabecular | moderate | + |
| 15 | 71 | M | 1b | mixed | moderate | − |
| 16 | 66 | M | 2 | mixed | well | + |
| 17 | 62 | M | 1a | trabecular | moderate | + |
| 18 | 62 | F | 2 | trabecular | moderate | − |
| 19 | 64 | F | 2 | trabecular | well | + |
| 20 | 68 | M | 2 | mixed | moderate | − |
| 21 | 59 | M | 1a | pseudoglandular | moderate | − |
| 22 | 73 | M | 2 | trabecular | moderate | − |
| 23 | 61 | F | 1a | mixed | well | + |
| 24 | 63 | M | 1a | pseudoglandular | well | − |
| 25 | 73 | F | 1b | mixed | moderate | − |
| 26 | 70 | M | 1b | mixed | moderate | − |
| 27 | 70 | M | 1a | trabecular | moderate | − |
| 28 | 69 | M | 1a | trabecular | moderate | + |
| 29 | 60 | F | 1a | trabecular | well | + |
| 30 | 62 | M | 1b | trabecular | moderate | − |
| 31 | 57 | M | 1b | mixed | moderate | − |
| 32 | 74 | F | 1a | trabecular | well | − |
| 33 | 61 | M | 1b | mixed | moderate | − |
| 34 | 64 | F | 1a | trabecular | well | + |
| 35 | 57 | F | 1b | solid | poor | − |
| 36 | 55 | M | 1b | trabecular | poor | + |
| 37 | 69 | M | 1b | mixed | moderate | − |
| 38 | 50 | M | 2 | trabecular | moderate | + |
| 39 | 77 | F | 2 | mixed | moderate | − |
| 40 | 69 | F | 1a | trabecular | moderate | + |
| Gene | Taqman Gene Expression Assay Kit |
|---|---|
| Nanog | Hs02387400_g1 |
| Myc | Hs00153408_m1 |
| Klf4 | Hs00358836_m1 |
| MDR1 | Hs00184500_m1 |
| MRP1 | Hs02514106_s1 |
| TNFα | Hs00174128_m1 |
| IL-6 | Hs00174131_m1 |
| SPRED2 | Hs00986220_m1 |
| GAPDH | Hs02758991_g1 |
| Antigen | Company (Cat. Number) |
|---|---|
| CyclinD1 | Cell Signaling Technology (92G2) |
| E-cadherin | Cell Signaling Technology (24E10) |
| N-cadherin | Cell Signaling Technology (D4R1H) |
| Snail | Cell Signaling Technology (C15D3) |
| SPRED2 | Proteintech (24091-1-AP) |
| Nanog | Cell Signaling Technology (D73G4) |
| c-Myc | Cell Signaling Technology (D84C12) |
| KLF4 | Cell Signaling Technology (4038) |
| GAPDH | Cell Signaling Technology (5174) |
| p44/42 MAPK (ERK1/2) | Cell Signaling Technology (4695) |
| Phospho-p44/42 MAPK (pERK1/2)(Thr 202/Tyr 204) | Cell Signaling Technology (4370) |
| STAT3 | Cell Signaling Technology (12640) |
| Phospho-STAT3 (Tyr 705) | Cell Signaling Technology (9145) |
| HRP-goat anti-rabbit IgG | Cell Signaling Technology (7074) |
| HRP-anti-mouse IgG | Cell Signaling Technology (7076) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, T.; Yang, X.; Fujisawa, M.; Ohara, T.; Wang, T.; Tomonobu, N.; Sakaguchi, M.; Yoshimura, T.; Matsukawa, A. SPRED2: A Novel Regulator of Epithelial-Mesenchymal Transition and Stemness in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2023, 24, 4996. https://doi.org/10.3390/ijms24054996
Gao T, Yang X, Fujisawa M, Ohara T, Wang T, Tomonobu N, Sakaguchi M, Yoshimura T, Matsukawa A. SPRED2: A Novel Regulator of Epithelial-Mesenchymal Transition and Stemness in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2023; 24(5):4996. https://doi.org/10.3390/ijms24054996
Chicago/Turabian StyleGao, Tong, Xu Yang, Masayoshi Fujisawa, Toshiaki Ohara, Tianyi Wang, Nahoko Tomonobu, Masakiyo Sakaguchi, Teizo Yoshimura, and Akihiro Matsukawa. 2023. "SPRED2: A Novel Regulator of Epithelial-Mesenchymal Transition and Stemness in Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 24, no. 5: 4996. https://doi.org/10.3390/ijms24054996
APA StyleGao, T., Yang, X., Fujisawa, M., Ohara, T., Wang, T., Tomonobu, N., Sakaguchi, M., Yoshimura, T., & Matsukawa, A. (2023). SPRED2: A Novel Regulator of Epithelial-Mesenchymal Transition and Stemness in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 24(5), 4996. https://doi.org/10.3390/ijms24054996