1. Introduction
Introducing conformationally rigid lipophilic moieties into a molecule is a common approach in the medicinal chemistry [
1,
2,
3,
4] that may lead to significant increases in biological activity [
5,
6,
7,
8]. One of the convenient methods for the synthesis of such conformationally rigid polycyclic derivatives is the Diels–Alder reaction of functionalized alkenes containing exocyclic C=C bonds with dienes. Dienesvariation makes it possible to fine-tune the structure of the resulting lipophilic framework [
9,
10,
11], and the presence of a C=C double bond in the [4+2]-cycloaddition adduct opens the way for further modifications of the resulting molecules.
We have recently shown that spiro-2-chalcogenimidazolones, obtained by 1,3-dipolarcycloaddition reactions to 5-methylidene-substituted hydantoins, as well as their 2-thio- and 2-seleno-analogs, possess high cytotoxicity probably due to their ability to inhibit the p53-MDM2 proteins interaction [
12,
13,
14]. However, there were no general methods for the synthesis of 5-methylidene-imidazol-4-onederivatives containing spiro-linked 5- and 6-membered rings described previously, and these compounds have not been previously studied for biological activity.
Diels–Alder reactions of 5-methylene-substituted imidazolones are described in the literature mainly asthe example of highly reactive cyclopentadiene [
15,
16,
17] and as single examples of the reactions with isoprene and 2,3-dimethylbutadiene [
16,
17] (
Scheme 1). In the present article, a systematic study of [4+2]-cycloaddition reactions of cyclopentadiene, cyclohexadiene, 2,3-dimethylbutadiene, and isoprene with 5-methylene-2-chalcogenimidazolone derivatives, both N-unsubstituted and containing substituents of various natures at nitrogen atoms, is carried out. In this work, we first demonstrated the possibility of methylidenethiohydantoins interaction with low active conjugated dienes, which makes it possible to obtain spirocyclic imidazolones containing various lipophilic frameworks. In addition, the possibility of post-synthetic transformations of the formed [4+2]-cycloaddition products using alkylating and acylating agents was demonstrated; these reactions can be an alternative method for the synthesis of those spiro-imidazolones that are formed in the reactions with dienes in low yields or as the inseparable diastereomers mixtures.
Various types of biological activities have been previously described for hydantoins and their spiro derivatives [
18,
19].In this case, if, for example, anticonvulsants require low toxicity, then inhibition of the growth of bacteria and tumor cells is determined by the cytotoxic effect. Therefore, for the initial assessment of the biological effect on cells of various types, a number of compounds synthesized in this work were tested on cytotoxicity and antibacterial activity on several human cell lines of various etiologies and several strains of
E. coli.
2. Results and Discussion
2.1. Synthesis of Dienophiles
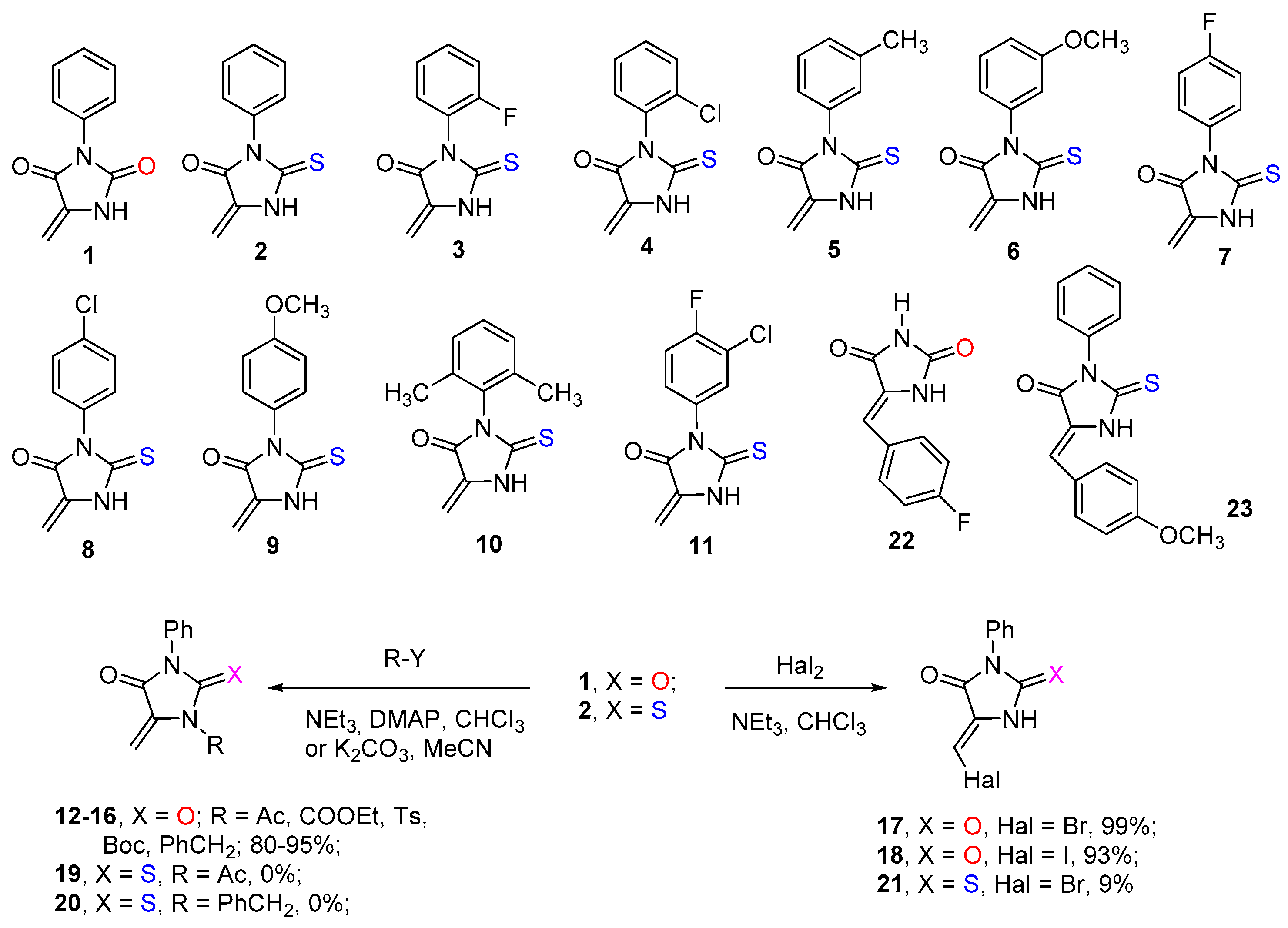
Initial dienophiles
1-
11,
22, and
23 were prepared according to previously described procedures [
20,
21,
22,
23]. Compounds
12-
21, containing both N(1)-substituted and N(1)-unsubstituted 2-chalcogenimidazolone moieties with exocyclic C=CH
2, C=CHHal, and C=CHAr fragments in five positions, were synthesized according to
Scheme 2. The substituents with different electronic and steric effects at the C=C double bond should significantly affect the reactivity of the dienophile, which can allow controlling the rate and selectivity of the Diels–Alder reaction with these substrates. It was found that methylidenehydantoin
1 gives high yields of alkylation and acylation products
12-
16, and its reaction with halogens (Br
2, I
2) in the presence of triethylamine leads to the corresponding halomethylidene-substituted imidazolones
17 and
18 as single isomers (
Scheme 2). The configuration of the C=C double bond in compounds
17 and
18 was confirmed by
1H NMR spectra of the products of their reactions with cyclopentadiene (
Scheme 3 and
Supplementary Information).
Our attempts to introduce methylidenetiohydantoin
2 in the reactions with acetyl chloride and benzyl bromide under the same conditions were unsuccessful. We found that thiohydantoin
2 rapidly degrades in the presence of bases (NEt
3, K
2CO
3), and its alkylation or acylation reactions are not formed even in trace amounts (according to
1H NMR spectra of reaction mixtures). When methylidenethiohydantoin
2 reacted with bromine at 0°C, resinification of the reaction mixture occurred, and the target halogen-substituted methylenethiohydantoin
21 could be isolated from the reaction mixture only in 9% yield (see
Scheme 2 and
Supplementary Information).
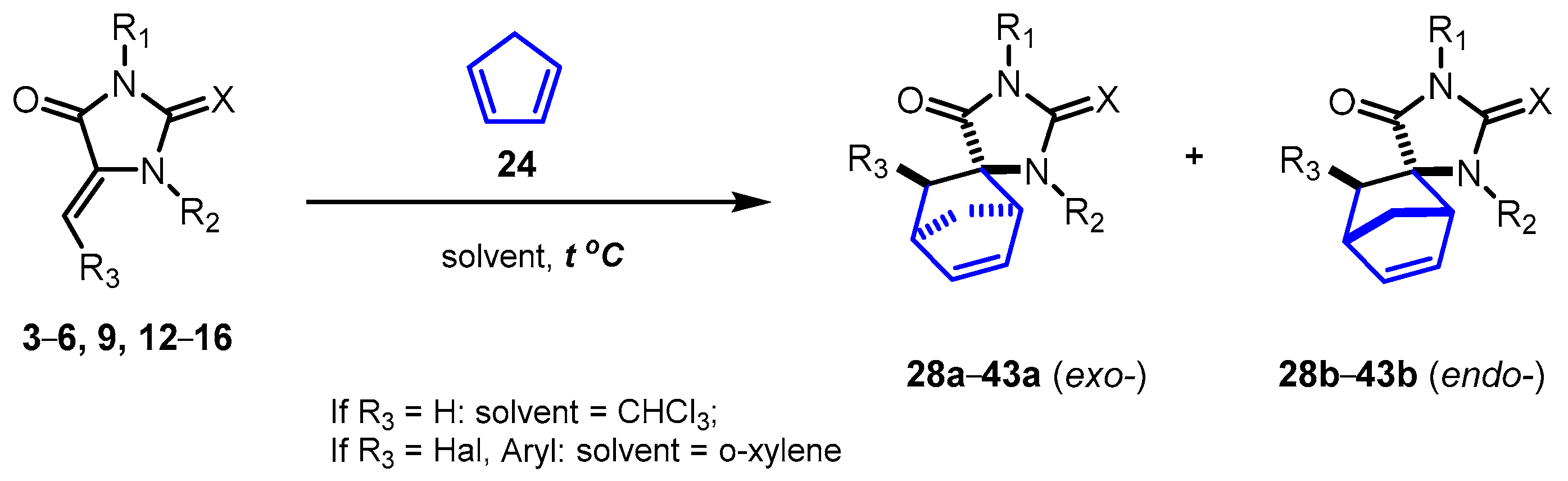
Next, we started studying the Diels–Alder reactions with dienophiles 1-23. It should be noted that for each specific diene (cyclopentadiene 24, cyclohexadiene 25, 2,3-dimethylbutadiene 26, isoprene 27), the optimal reaction conditions turned out to be different and were selected depending on its reactivity.
2.2. Reactions of 5-Methylideneimidazolones 1-6, 9, 12-18, 22, and 23 with Cyclopentadiene
In our previous communication, we demonstrated the possibility of spiro-norbornene derivatives synthesis by the [4+2]-cycloaddition of highly active dienecyclopentadiene with methylideneimidazolones
1 and
2 at an eight-fold excess of the diene in refluxing chloroform [
24]. Hydantoins
3-
6,
9, and
12-
16 were now introduced into the reactions with cyclopentadiene under the same conditions (
Scheme 3,
Table 1). In all cases, cycloaddition reactions proceeded in high yields with the predominant formation of the exo products
28a-
43a.
It should be noted that, unlike exo- and endo-products 29-33a and 29-33b, which do not contain unsubstituted nitrogen atoms, N(1)-unsubstituted compounds 28a, 34a-39a, and 28b, 34b-39b can be separated by column chromatography. Probably, the difference in the chromatographic mobility of the isomers is determined by the ability of the CONH and CSNH fragments of the molecules to form hydrogen bonds with the silica gel surface.
The structures of the resulting spiro-imidazolones
28-
43 were determined using
1H and
13C NMR spectroscopy; for compounds
28 and
34, their spectra correspond to those described in the literature [
24]. According to
1H NMR spectroscopy data, exo- and endo-products
a and
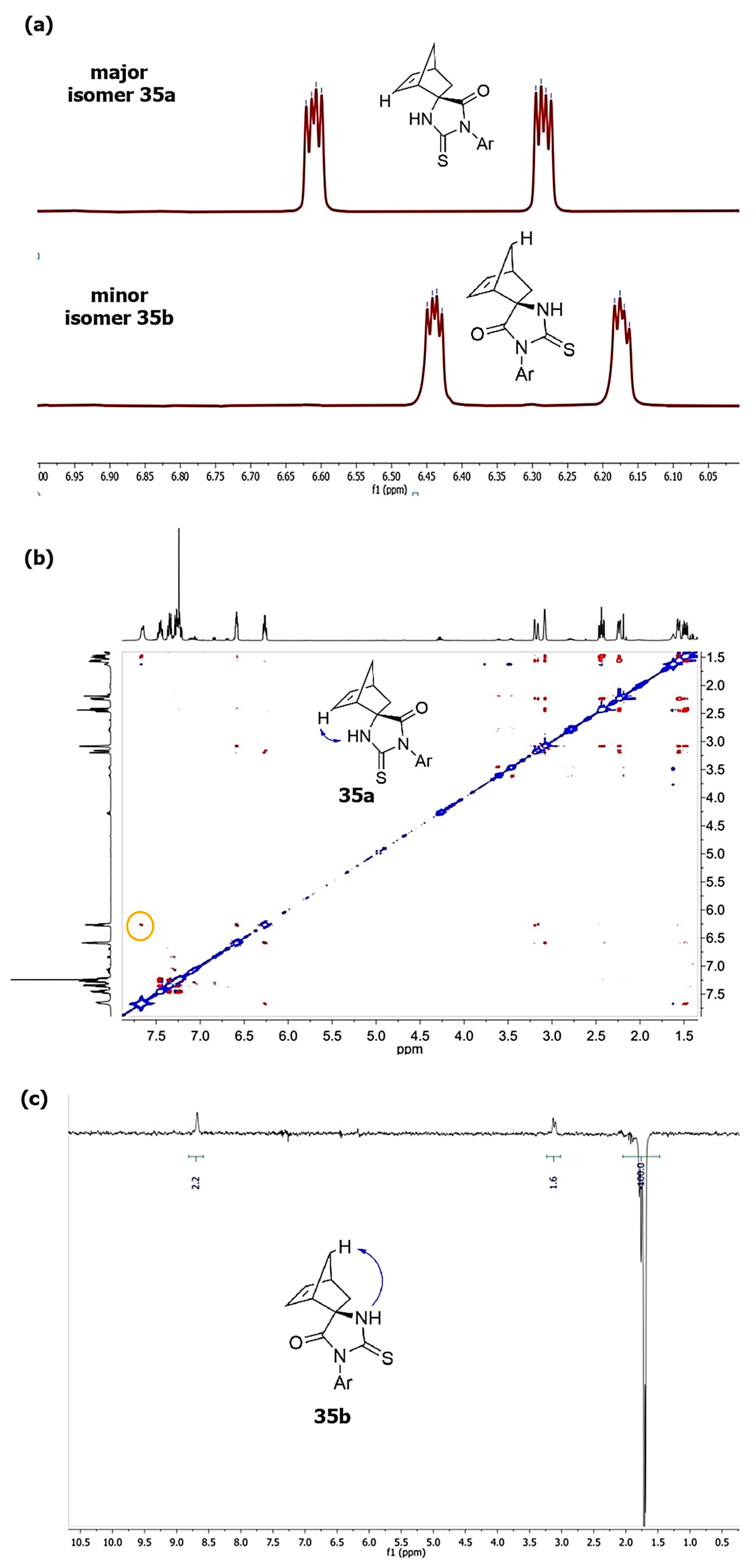
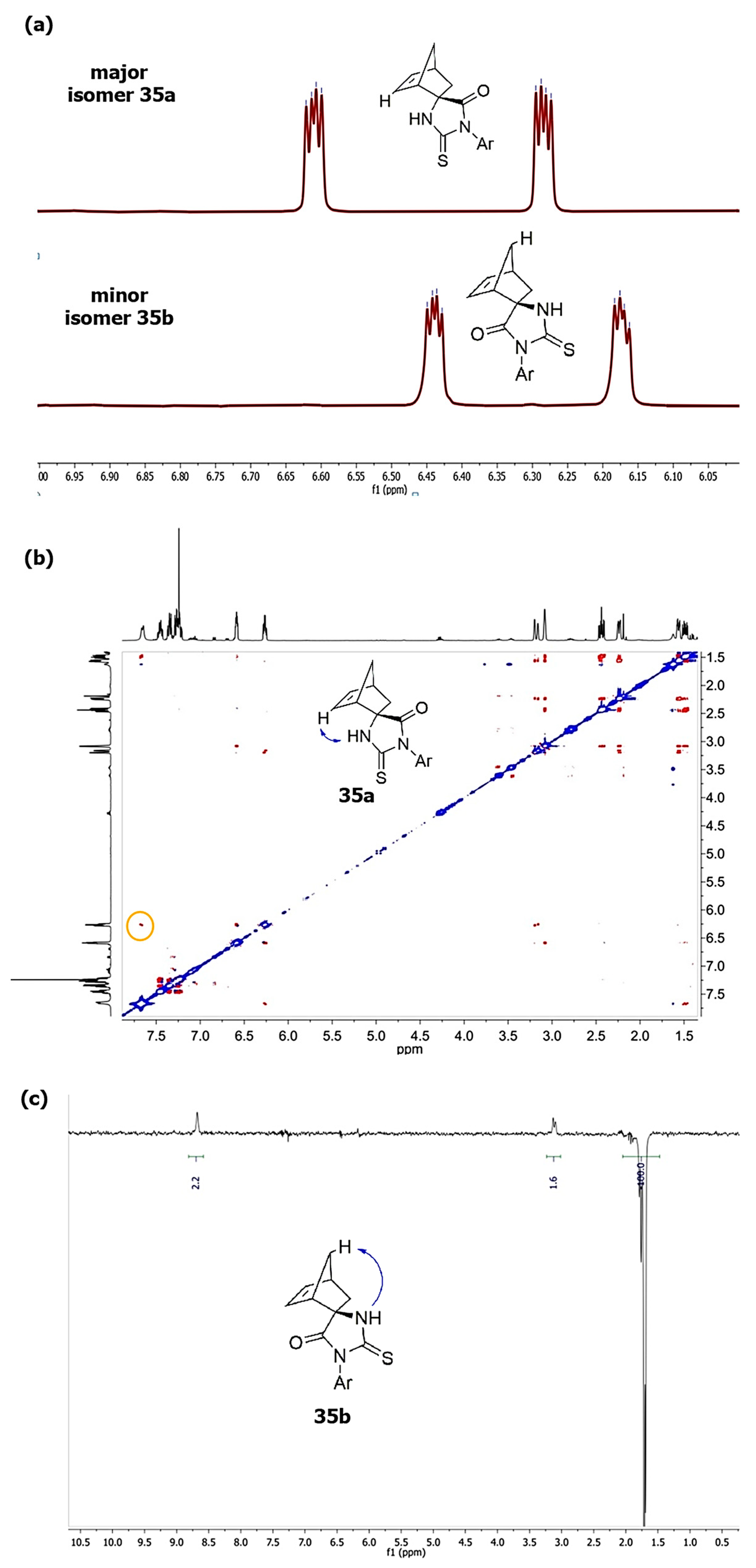
b differ in chemical shifts of HC=CH protons of the norbornene framework: the characteristic doublets of the main products are shifted to a weaker field (at 6.20–6.60 ppm) relative to the signals of minor products (at 6.15–6.45 ppm) (
Figure 1a), which is consistent with the literature data [
15,
17,
24]. In addition, compounds
28,
34-
39 are characterized by a very large difference in the NH protons shifts of exo- and endo-isomers (>1 ppm in CDCl
3), which can be explained by the influence of the anisotropy of the double C=C bond, which is quite close to the amide proton in isomer
a.
The criterion for assigning exo- and endo-stereoisomers of compounds
28,
34-
39, in addition to the chemical shifts of vinyl and NH protons, could be the NOESY spectra shown in the
Supplementary Information for compounds
35a and
35b. In the NOESY spectrum of spiro-derivative
35a, the interaction of the NH proton with the double bond proton, as well as with the proton of the nearest methylene group of the bicycle, is manifested, and there is no interaction with the proton at the C(7) atom of the norbornene framework (see
Figure 1b). On the contrary, for the minor isomer
35b, when the NH proton was irradiated, the Overhauser effect is observed for the proton at the C(7) atom of norbornene and is absent for the proton at the C=C double bond.
The diastereoselectivity of the cyclopentadiene Diels–Alder reaction with methylenehydantoins was found to be sensitive to the nature of the substituent at position 1 of the imidazolone moiety. Despite the increase in steric hindrance near the exocyclic double bond of the dienophile, the interaction of cyclopentadiene with hydantoins
12-
15 containing electron-withdrawing Ac, COOEt, Ts, Boc substituents proceeded less selectively compared to N(1) unsubstituted hydantoin
1. Such results may indicate that the electronic factors of the substituents have a stronger effect on the reactivity of the dienophile than the steric ones. The important role of the substituent in position 1 of the initial heterocycle on the reaction course is also confirmed by the fact that for the imidazolone
16 with a donor benzyl fragment, the yields of the cycloaddition product
33 are low, and only the isomers
33a are formed (see
Table 1).
Variation of the exocyclic chalcogen atom of the starting heterocycle does not affect the stereoselectivity of the Diels–Alder reaction (
Table 1, compare products
28 and
34). The same stereoselectivity of the Diels–Alder reaction is observed for thiohydantoins
2-
6 and
9 with different substituents at N(3) atom. Apparently, the electronic properties of substituents in the C(2) and N(3) positions of the heterocycle have little effect on the reactivity of the dienophile.
An increase in steric hindrance at the C=C bond of methylideneimidazolone should hinder the [4+2]-cycloaddition reaction. Indeed, halomethylidenehydantoins 17, 18 react with cyclopentadiene only when refluxed in ortho-xylene. In the course of these reactions, only exo-isomers 40a and 41a were formed in moderate yields. In the 1H NMR spectra of compounds 40a and 41a, in the region of about 4.7 ppm, there are doublets with J ~3.3 Hz, which confirms the spatial arrangement of the CHBr and CHI groups in the norbornene framework.
Imidazolones 22, 23, containing the bulkier structure and less acceptor compared to halogens aryl substituents at the double C=C bond, did not form the reaction products with cyclopentadiene under similar conditions even in trace amounts (according to 1H NMR spectroscopy of the reaction mixtures), probably for both electronic and steric reasons.
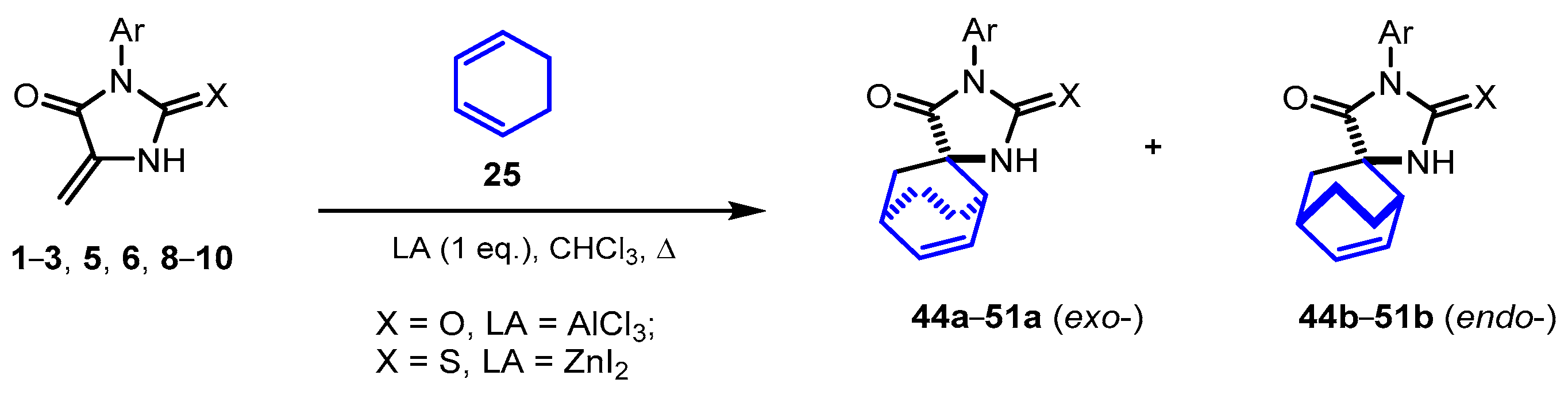
2.3. Reactions of 5-Methylideneimidazolones 1-3, 5, 6, 8-10 with Cyclohexadiene
Reactions of 5-methylideneimidazolones with dienes 25-27, which are less reactive than cyclopentadiene, do not occur even when refluxing in toluene with a 20-fold excess of dienes. Under microwave irradiation, when methylidenehydantoin 1 was heated in benzene to 140°C with an excess of cyclohexadiene 25, the formation of the target products only in trace amounts was detected. The reactions of cyclohexadiene with compounds 2-11 were successfully carried out by catalysis of the reaction with Lewis acids; BF3∙Et2O, AlCl3, and ZnI2 were tested in the target reactions.
It was found that cyclohexadiene 25 rapidly decomposed in the presence of BF3∙Et2O, and no products of its Diels–Alder reaction were formed. In the presence of AlCl3, the decomposition of the diene also occurred, which accelerated upon heating the reaction mixture; however, the target product 44a could be isolated if diene 25 was added slowly into a refluxing solution of dienophile 1 and Lewis acid.
In the presence of 1 equivalent of ZnI
2, cyclohexadiene did not react with 5-methylidenehydantoin
1 even when heated, but methylidenetiohydantoines
2,
3,
5,
6,
8-
10 reacted with this diene under the same conditions to form the mixtures of diastereomeric products
45a-
51a and
45b-
51b in a ratio of ~3:1 (
Scheme 4,
Table 2), which could be separated by column chromatography.
It should be noted that the selectivity of the reaction is practically independent of the substituents in position 3 of the heterocycles 2, 3, 5, 6, 8-10. In the presence of an excess of Lewis acid, the yields of the target products somewhat decreased, presumably due to the acceleration of the decomposition of the diene on the catalyst, and with a submolar amount of ZnI2, the formation of products slowed down.
Different results of the reaction of diene 25 with hydantoin 1 and thiohydantoins 2, 3, 5, 6, 8-10 in the presence of ZnI2 may be due to the fact that ZnI2 as a soft Lewis acid is predominantly coordinated to the sulfur atom of 2-thioimidazolone, and varying the chalcogen (O or S) strongly affects the efficiency of binding the dienophile to the Lewis acid.
The structures of spiro-imidazolones
44-
51 were confirmed by
1H and
13C NMR spectroscopy data; the configuration of compound
46a was determined via two-dimensional NMR techniques COSY and gHSQC (see
Supplementary Information, Figures S67 and S68).
It may be noted that the
1H,
13C (and
19F NMR in cases where the compounds contained fluorine) spectra of all ortho-phenyl-N(3)-substituted spiro-imidazolones (for example, compounds
35,
46, and
51, see
Supplementary Information, Figures S23, S63, and S87 and similar) demonstrate two sets of signals. This can be explained by the hindered rotation around the single bond C(Ar)-N(imidazolone) of the imidazolone fragment and, consequently, the existence of each ortho-phenyl-substituted spiro-compounds as two atropisomers (axially chiral heterocyclic analogs of biaryl derivatives similar to those described for others ortho-substituted 2-thiohydantoins) [
25,
26,
27]. The NMR spectra of these compounds at ambient temperatures do not indicate the presence of intramolecular dynamic processes, apparently due to the high barrier to internal rotation. Thus, weak line broadening in the
19F NMR spectrum of product
35a is observed only at temperatures above 80 °C (
Figure S30); this indicates that the rotational barrier is greater than 20 kcal/mol.
As in the reactions with cyclopentadiene, the isomeric exo- and endo-Diels–Alder products of the reactions of methylidenetiohydatoins 2, 3, 5, 6, 8-10 with cyclohexadiene can be distinguished by the chemical shifts of the protons of the CH=CH group: for the main isomers a, a characteristic doublet of doublets observed in the region of 6.62–6.34 ppm, and for minor products b, such doublet of doublets is present in the region of 6.48–6.30 ppm (in CDCl3).
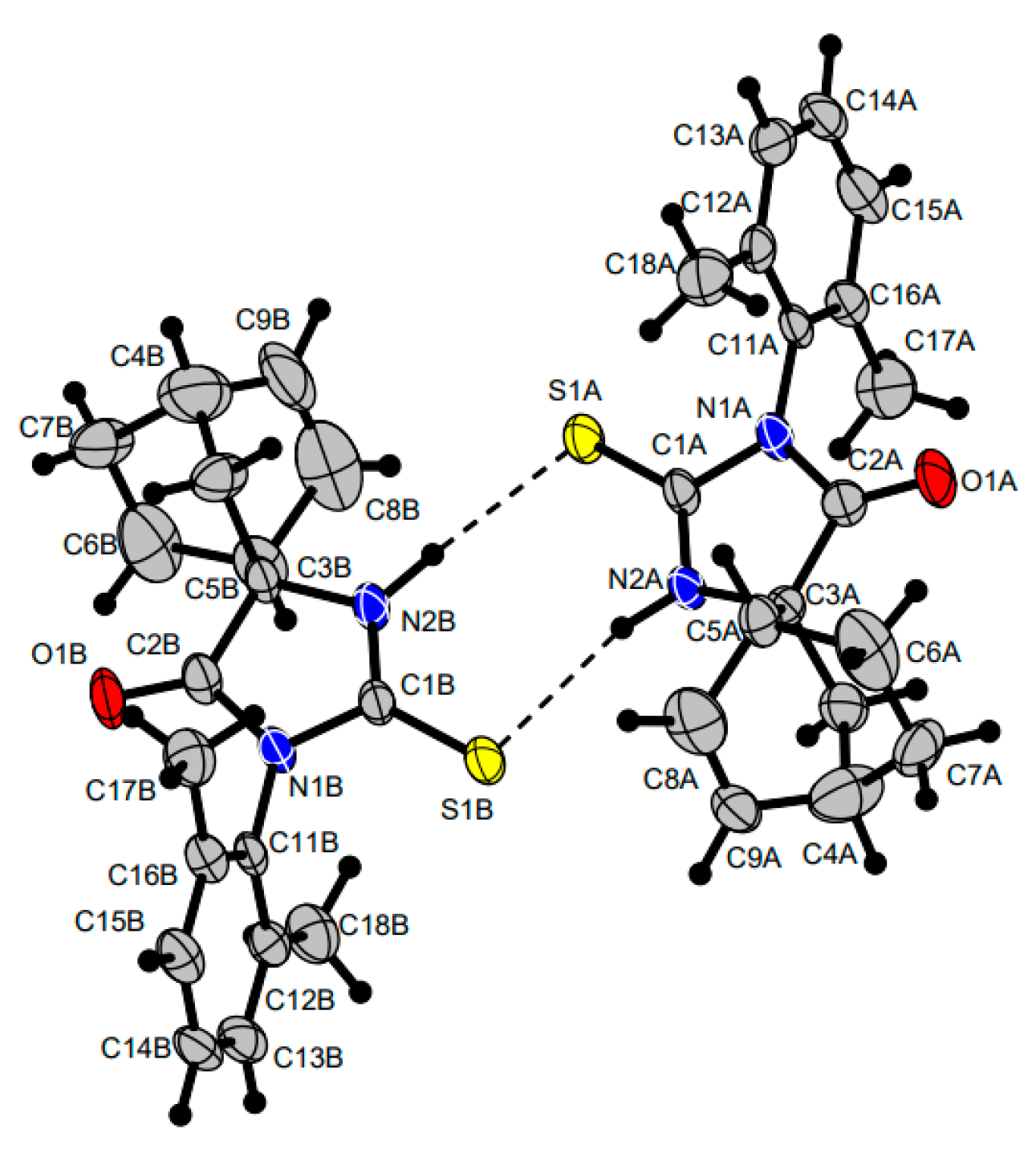
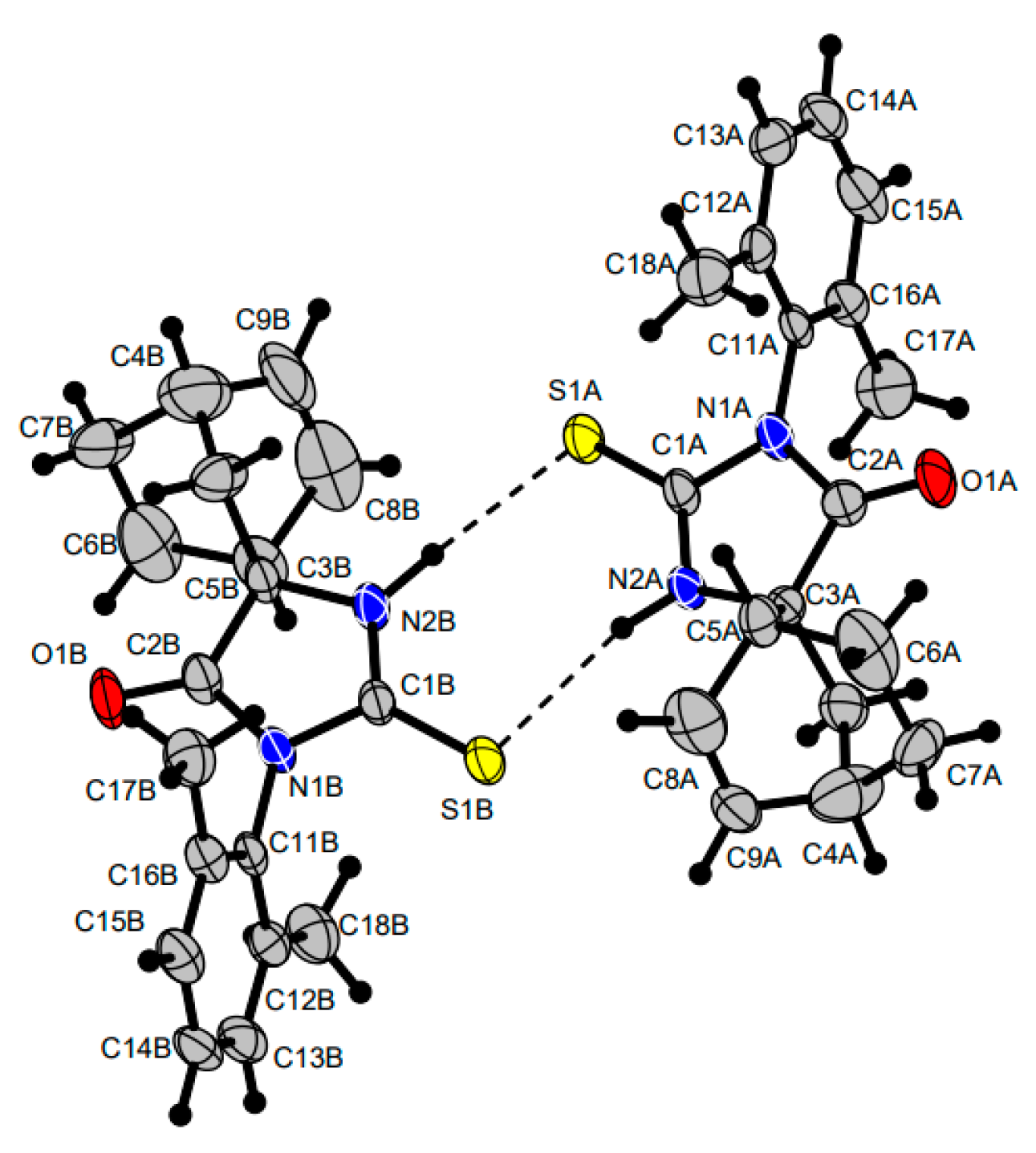
The structure of compound
51a was additionally confirmed by the X-ray diffraction data. The crystal cell shown in
Figure 2 includes two spiro-thiohydantoin molecules linked by NHS hydrogen bonds to form an eight-membered cycle. The dihedral angle between the planes of the cycles at the spiro junction is close to 90°; imidazolone five-membered rings are near the planar.
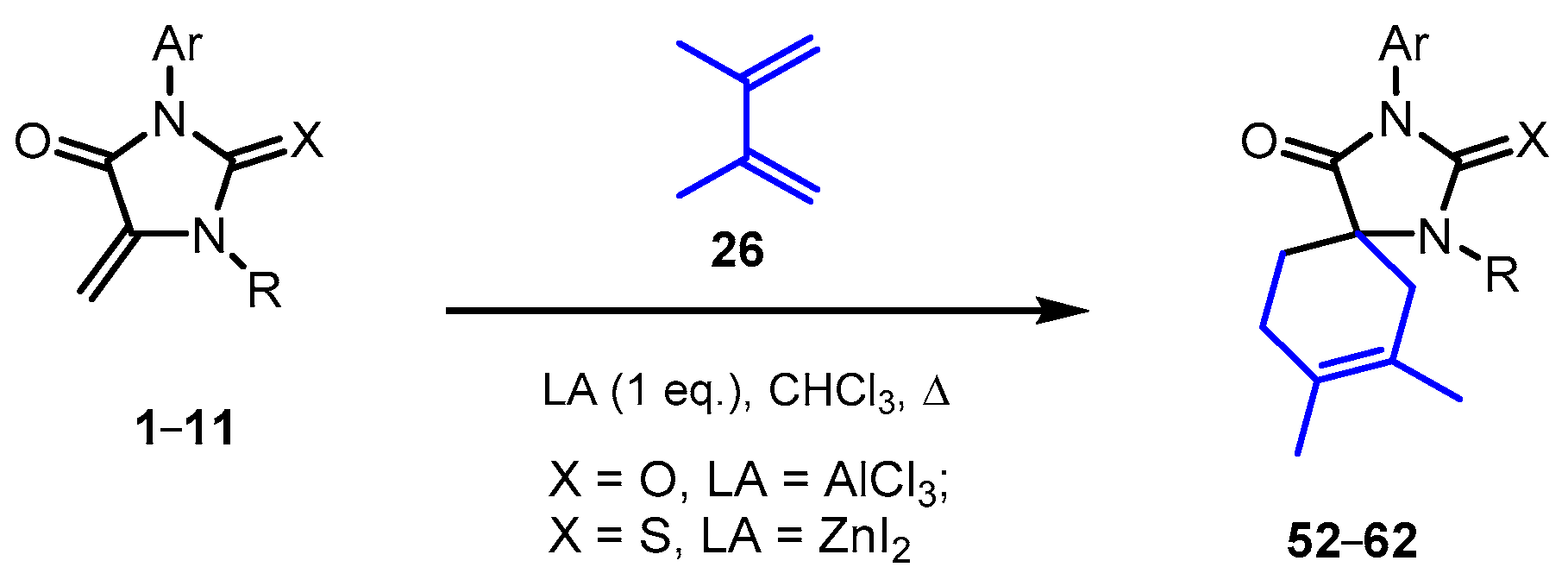
2.4. Reactions of 5-Methylideimidazolones 1-12 with 2,3-Dimethylbutadiene
Reactions of methylideneimidazolones
1-
12 with 2,3-dimethylbutadiene also require Lewis acids as the catalysis but can proceed in the presence of both AlCl
3 and ZnI
2 (
Scheme 5,
Table 3). The reactions of diene
26 with dienophiles
1 and
2 under the action of AlCl
3 proceeded in moderate yields giving the compounds
52 and
53.
As well as in reactions with cyclohexadiene, ZnI2 activates thiohydantoins 2-11 in the reactions with 2,3-dimethylbutadiene with the formation of target compounds 54-62 but does not catalyze the reactions with oxygen analogs, probably due to the selective coordination of the Lewis acid to the C=S bond of the starting heterocycle. A significant increase in the yield of thiohydantoin 53 when AlCl3 was replaced by the softer ZnI2 may be due to a decrease in the rate of the side process of diene polymerization under the action of the Lewis acid.
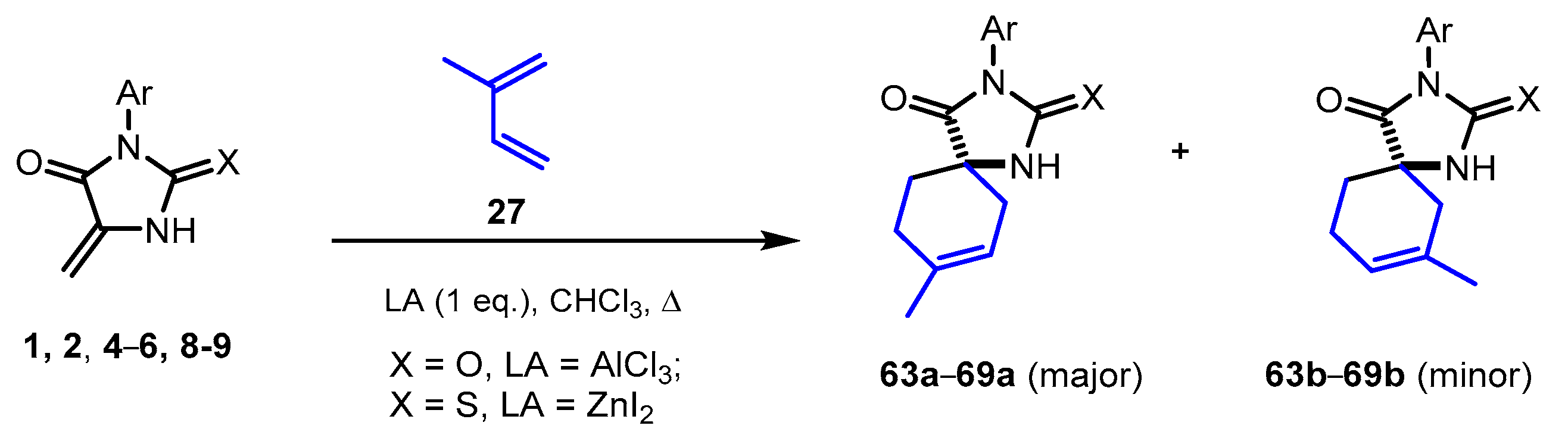
2.5. Reactions of 5-Methylideneimidazolones 1, 2, 4-6, 8, and 9 with Isoprene
Using isoprene
27, the regioselectivity of the Diels–Alder reaction of methylideneimidazolones
1,
2,
4-
6,
8, and
9 were studied. Boiling of hydantoin
1 mixture with a 10-fold excess of this diene in chloroform led to the formation of cycloaddition product
63a in 70% yield and trace amounts of the minor regioisomer
63b. Under similar conditions, thiohydantoins
2,
4-
6,
8, and
9 formed an inseparable mixture of regioisomers
64a-
69a and
64b-
69b in good yields in a ratio of ~87:13 (
Scheme 6,
Table 4 and
Supplementary Information).
The structures of regioisomers were confirmed by
1H NOESY1D,
1H-
13C HSQC, and
1H-
13C HMBC NMR spectroscopy data for the compounds
67a and
67b (see
Supplementary Information, Figures S123–S125). In particular, the position of the methine proton in the six-membered ring of compound
67a was confirmed by the presence of a cross peak in the HMBC spectrum, which is responsible for the vicinal interaction of this proton with the quaternary spiro-carbon.
The results obtained demonstrate that AlCl
3 is an effective catalyst for the reactions of low-activity dienes with hydantoins and ZnI
2 with thiohydantoins. At the same time, AlCl
3 similarly catalyzes reactions with hydantoins and thiohydantoins (see
Table 3). It may be supposed that the harder Lewis acid AlCl
3 is coordinated to the N(1) atom of the starting imidazolone and, thus, catalyzes both reactions with hydantoins and thiohydantoins, while the softer Lewis acid ZnI
2 is coordinated to the S atom and, thus, activates only thiohydantoins; however, exact proof of this assumption requires additional research.
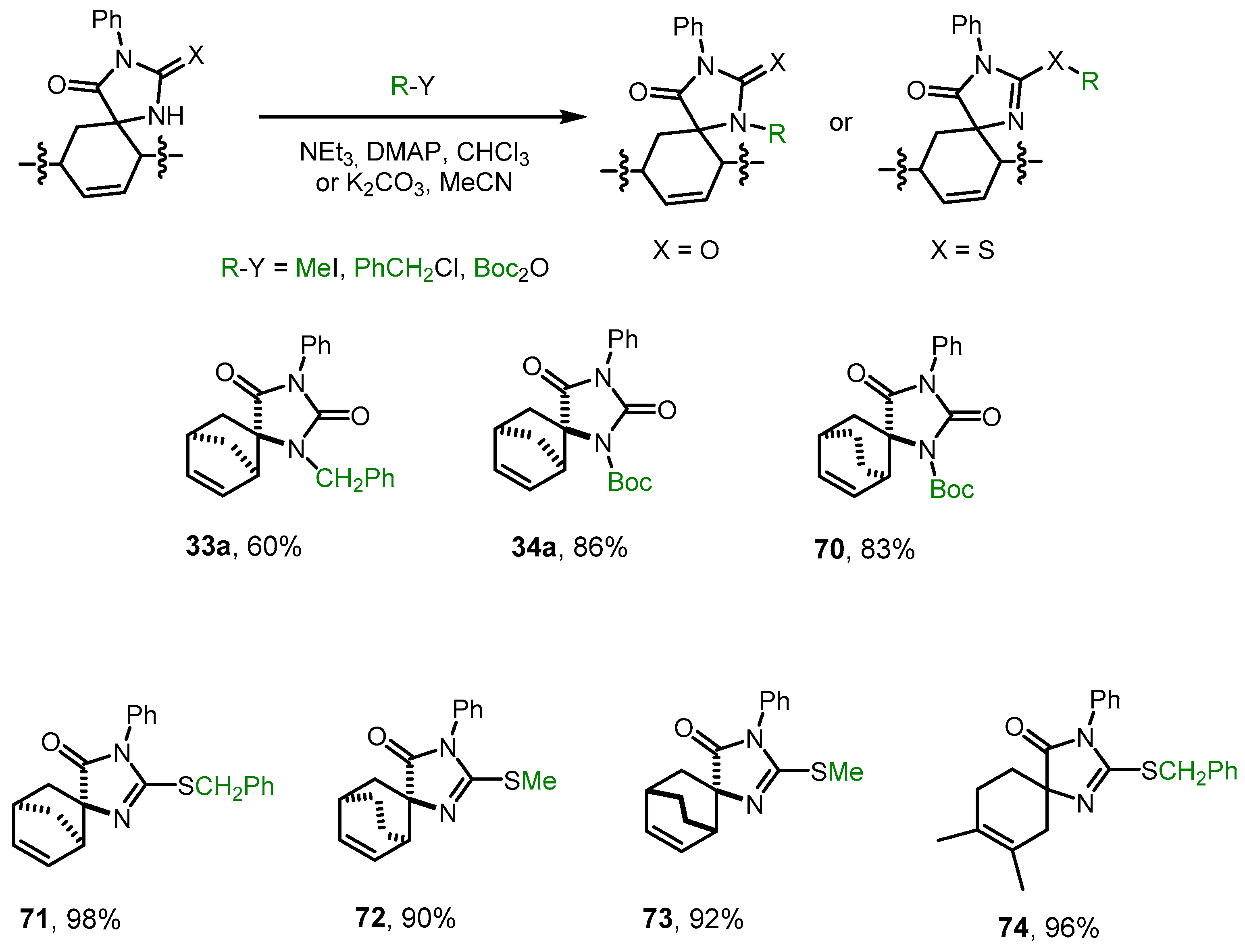
2.6. Alkylation and Acylation of [4+2]-Cycloaddition Reaction Products
Studying the Diels–Alder reactions with methylideneimidazolones
1-
18, it was found that in the case of N(1)-unsubstituted dienophiles
1 and
2, the resulting diastereomeric pair of spiroheterocycles can be successfully separated into individual isomers, while N(1)-substituted derivatives are formed either as an inseparable mixture of diastereomers or in extremely low yields (
Table 1). Therefore, we studied the possibility of post-modification at the nitrogen atom of N(1)-unsubstituted Spiro derivatives
28a,
34a,
44a,
45a,
45b, and
53.
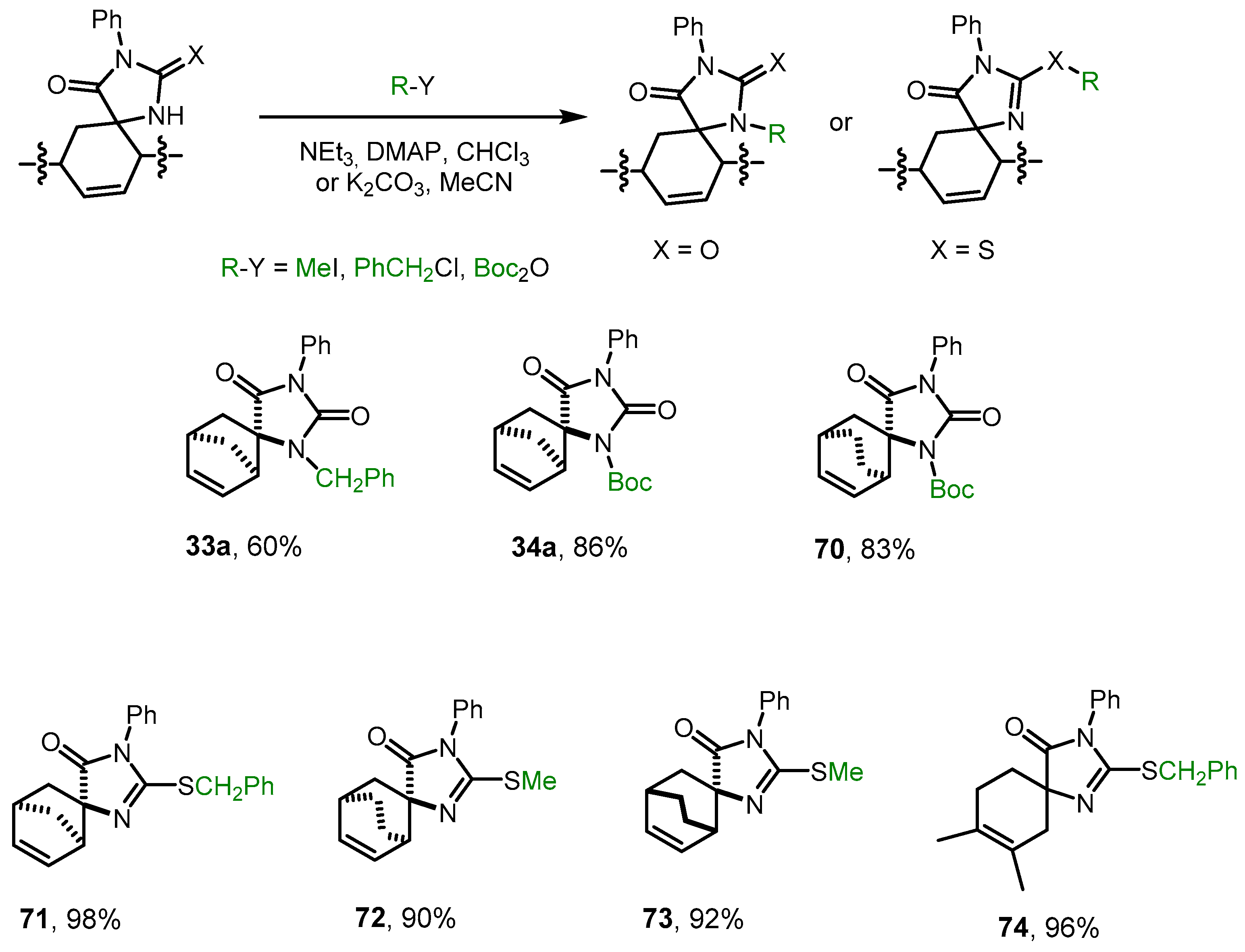
We found that hydantoins
28a and
44a can be alkylated or acylated with PhCH
2Cl or Boc
2O in high yields by refluxing in chloroform or acetonitrile (
Scheme 7). The spectral data of the resulting products
33a and
34a completely coincided with the spectral data of the main products of the Diels–Alder reaction of methylidenehydantoins
15 and
16 with cyclopentadiene (see
Section 2.2).
Thiohydantoins 34a, 45a, 45b, and 54 were alkylated with MeI and PhCH2Cl under milder conditions at room temperature in acetonitrile in the presence of K2CO3, exclusively at the sulfur atom. The formation of S–CH2Ph and S–Me bonds in corresponding imidazolones 72-75 was confirmed by 13C NMR spectroscopy data.
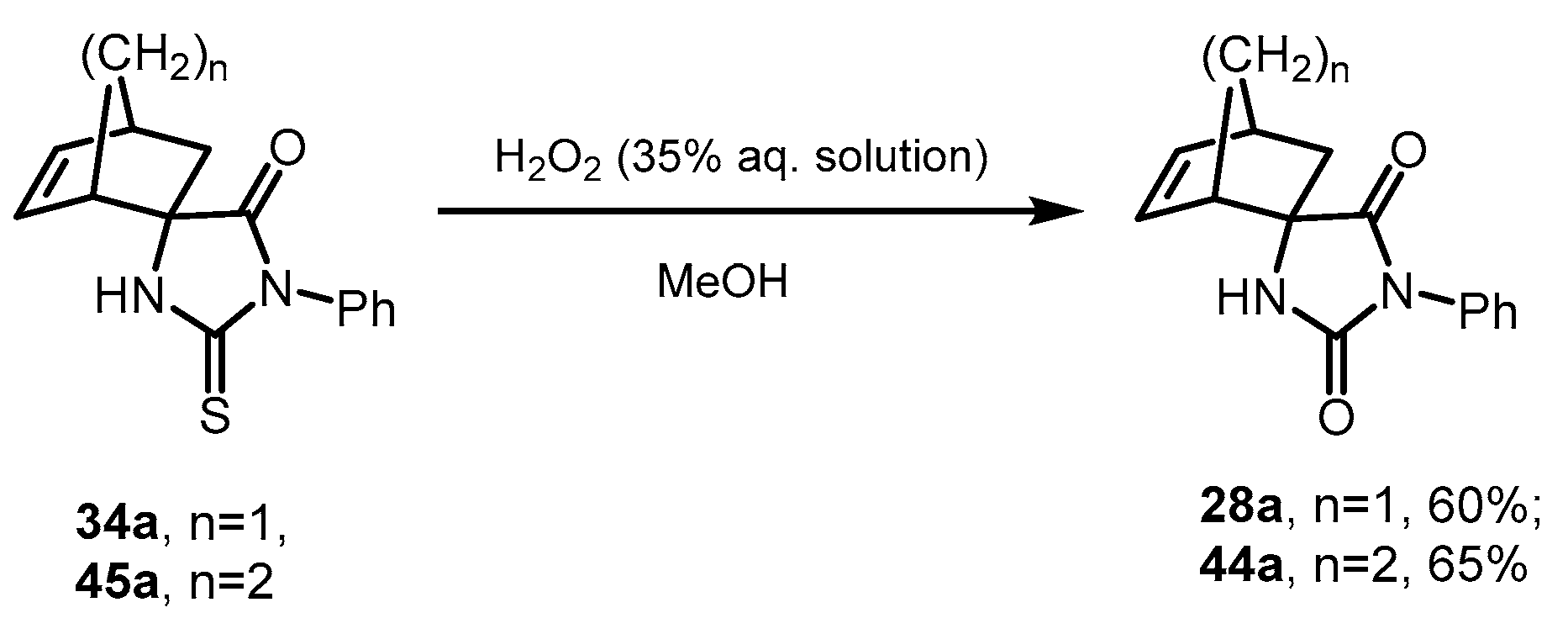
2.7. Desulfurization of Spiro-Thiohydantoins
Since methylidenehydantoin
1 reacted with dienes
25 and
26 with low conversion even in the presence of Lewis acids (
Section 2.3 and
Section 2.4), we proposed an alternative scheme for the synthesis of spirocyclic hydantoins from their thiohydantoin analogs. The procedure described in the literature for the hydrolysis of S-alkylated thiohydantoins under the action of HCl in refluxing ethanol [
13,
28] applied to compound
72 did not lead to the formation of the corresponding hydantoin
28a. In the
1H NMR spectrum of the reaction mixture, there were no characteristic CH=CH signals of the norbornene skeleton protons, which probably indicates the ongoing processes of cationic polymerization under the reaction conditions.
The desired transformation of spirothiohydantoins
34a and
45a into spirohydantoins
28a and
44a was achieved under milder conditions by treating methanolic solutions of the starting compounds with 35% aqueous H
2O
2 at room temperature in good yields (
Scheme 8).
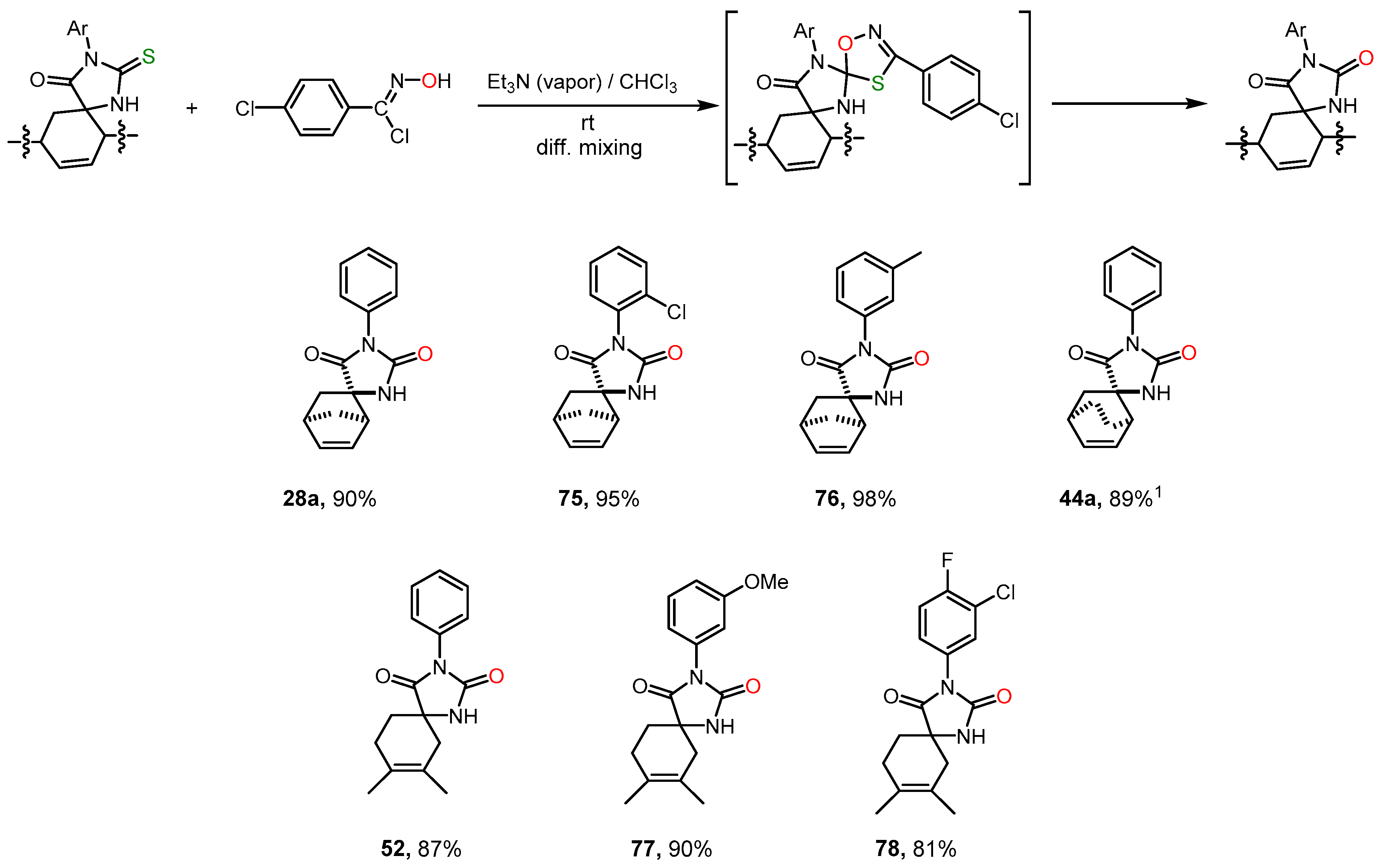
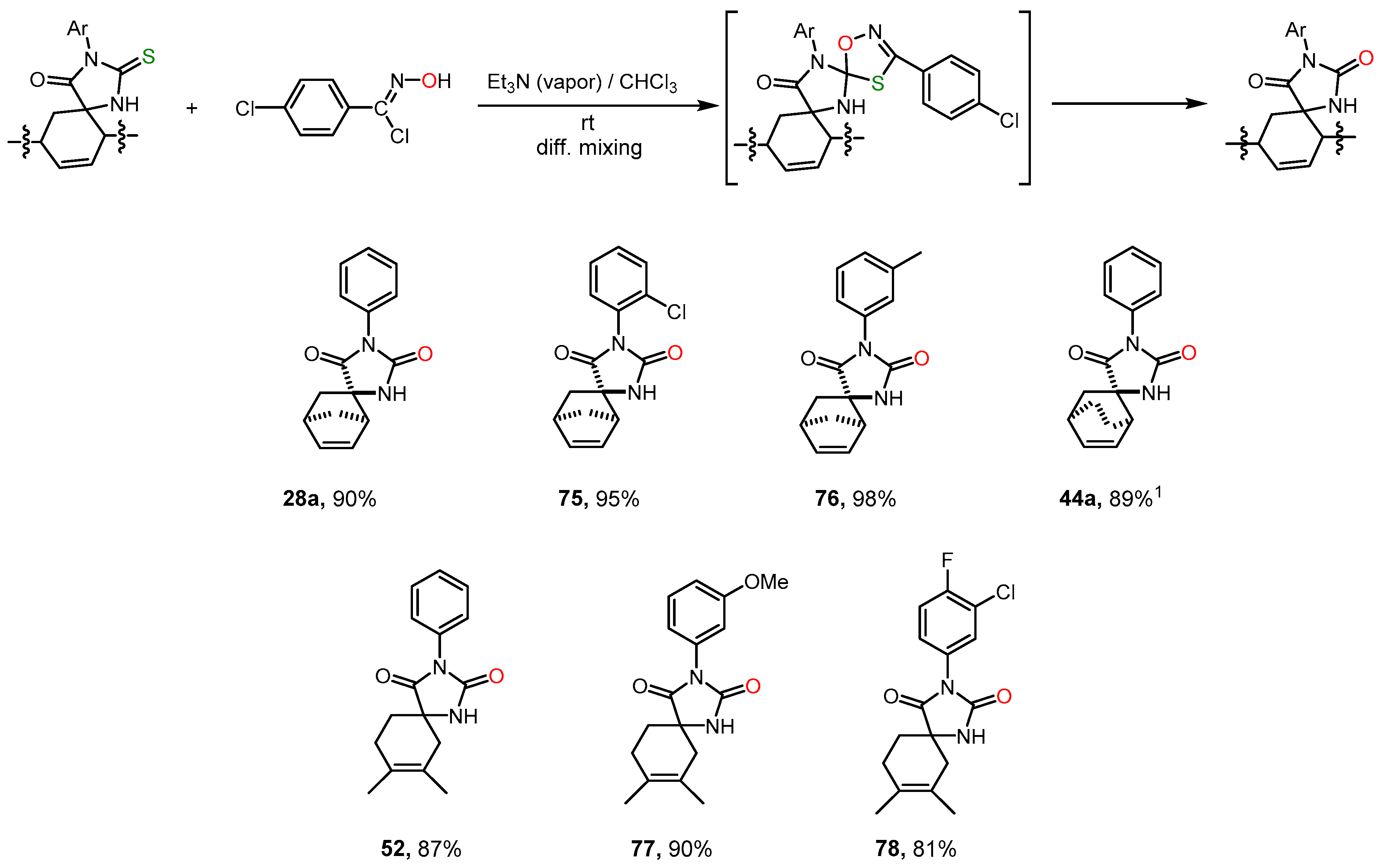
However, an even more effective way of thiohydantoins desulfurizing turned out to be their interaction with nitrile oxides [
29] (
Scheme 9). The reactive nitrile oxide was generated in situ from N-hydroxyimidoyl chloride under the action of a tertiary amine. As a result of subsequent N-hydroxyimidoyl chloride 1,3-dipolar cycloaddition at the C=S bond, an unstable oxatriazole was obtained, which then decomposed to form hydantoin, analogously to that described in [
30]. For this reaction, we used a recently proposed convenient method of diffusion reagents mixing, which made it possible to suppress undesirable dimerization processes of the 1,3-dipole and introduce thiohydantoin and the dipole precursor into the reaction in a strictly equimolar ratio [
31]. Under these conditions, hydantoins
28a,
52,
75-
78 were formed in high yields without the side products of dipole addition to the C=C bond. During the synthesis of compound
44a, a certain amount (no more than 10%) of the addition product of nitrile oxide to the C=C bond was formed.
2.8. Cytotoxicity against Human Cell Lines
Some of the obtained spiro-derivatives were tested for cytotoxicity using the standard MTT assay [
32]. Cytotoxicity was evaluated using the cell lines of various etiologies: breast cancer MCF7, human lung carcinoma A549, non-cancer human embryonic kidney cell line HEK293T, and non-cancer lung fibroblast VA13 cell line. Lung cancers and breast cancers are among the most common causes of tumor lesions and related death in the world [
33], and non-cancerous cells were applied for the specificity of action evaluation. The results are shown in
Table 5; the dose-response dependency graphs are available in
Supplementary Information.
Generally, the tested compounds show rather low or moderate cytotoxicity to all tested cell lines; their IC
50abs values (IC
50abs is the concentration resulting in a two-fold decrease in the number of cells in comparison with untreated cells) against A549 and MCF7 cell lines mostly ranging from 20 to 100 µM. Additionally, the tested compounds show little selectivity to all cell lines tested. Despite the nominal IC
50abs selectivity of the compounds
55 and
61 against A549 compared to VA13, their IC
50 values (IC
50 is the concentration resulting in half of the maximal cytotoxic effect) were quite similar (
Table S1).
It may also be noticed that the introduction into the tested spiro-imidazolones of halogenated phenyl ring instead of non-halogenated one in most cases increased cytotoxicity against all tested cell lines. It can be seen by comparing the data obtained for compound
63, which is the most cytotoxic of all the tested compounds and has a dihalogenated phenyl ring, with the data obtained for compounds
49a,
55,
58, and
59, which have monohalogenated phenyl ring, and the non-halogenated compounds
28,
34. The compounds with OMe- or Me-substitutient in the phenyl rings at N(3) imidazolone atom had no significant cytotoxicity on the most tested cell lines (
Table 5, compounds
37-
39,
48,
50,
56,
60,
66).
2.9. Cytotoxicity against E. coli
All compounds investigated for cytotoxicity effects were also analyzed for antibacterial activity. Tests were carried out on
E. coli BW25113 DTC-pDualrep2 strain that has a normal cell wall and the
E. coli BW25113 LPTD-pDualrep2strain that has damaged cell walls. Some of the tested compounds showed a noticeable antibacterial effect against
E. coli BW25113 DTC-pDualrep2 but were almost inactive against
E. coli BW25113 LPTD-pDualrep2. All the tested compounds were analyzed for the mechanism of antibacterial activity by means of the pDualrep2 [
34] reporter system, which allows for the detection of translational inhibitors and SOS-response inducers, but none of these mechanisms is involved.
Then the minimal inhibitory concentration (MIC) was measured for
E. coli BW25113 DTC and
E. coli K-12 (WT) for the 12 most bacteriotoxic compounds from the plate test. MIC of compound
77 against
E. coli BW25113 DTC is 125 µM, while the other compounds either show MIC ~500 µM, 1 mM, or are inactive in this test (
Table 6).
All the tested compounds were completely inactive against E. coli K-12 at concentrations of 1 mM and below. It indicates that the studied compounds are toxic to E. coli but are effectively removed from the cell by efflux systems and, therefore, do not affect the wild type of E. coli.
3. Materials and Methods
All solvents were purified using standard procedures [
35]. Starting compounds were purchased from commercial sources (Sigma–Aldrich, ABCR, AKSci, Burlington, VT, USA). Reactions were checked by TLC analysis using silica plates
1H, and
13C NMR spectra were recorded on BrukerAvance or Agilent 400-MR spectrometers (400 MHz for
1H, 100 MHz for
13C). Chemical shifts are reported in parts per million relative to TMS.
Electrospray ionization high-resolution mass spectra were recorded on a TripleTOF 5600+ quadrupole time-of-flight mass spectrometer (ABSciex, Concord, Vaughan, ON, Canada) with DuoSpray ion source in positive ion mode. The capillary voltage was 5.5 kV; nebulizing and curtain gas pressure—15 and 25 psi, respectively; ion source temperature ambient; declustering potential 20 V; m/z range 100–1200. Elemental compositions of the detected ions were determined based on accurate masses and isotopic distributions using Formula Finder software (ABSciex, Concord, ON, Canada).
The X-Ray data for compound 51a were collected via STOE diffractometer Pilatus100K detector, Cu Kα (1.54086Å) radiation, rotation method mode. STOE X-AREA software was used for cell refinement and data reduction. Data collection and image processing were performed with X-Area 1.67 (STOE & Cie GmbH, Darmstadt, Germany, 2013). Intensity data were scaled with LANA (part of X-Area) in order to minimize differences in intensities of symmetry-equivalent reflections (multi-scan method).
The structures were solved and refined with SHELX(1) program [
36]. The non-hydrogen atoms were refined by using the anisotropic full matrix least-square procedure. Hydrogen atoms were placed in the calculated positions and allowed to ride on their parent atoms [C-H 0.93–0.98; Uiso 1.2 Ueq(parent atom)]. Hydrogen atoms at nitrogen atoms N2A and N2B (see
Figure 2) were localized from Fourier syntheses and refined freely. Molecular geometry calculations were performed with the SHELX program, and the molecular graphics were prepared by using DIAMOND(2) version 4 software [
37].
Cell cultures. Human embryonic kidney HEK293T cell line was kindly provided by Dr. E. Knyazhanskaya; immortalized human fibroblasts cell line VA13 was kindly provided by Dr. M. Rubtsova; human breast cancer cell line MCF7 and human lung adenocarcinoma cell line A549 were kindly provided by Dr. S. Dmitriev. MCF7, VA13, A549, and HEK293T cell lines were maintained in DMEM/F-12 (Thermo Fisher Scientific, Waltham, Massachusetts, USA) culture medium containing 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and 50 µg/mL penicillin and 0.05 mg/mL streptomycin at 37°C (Thermo Fisher Scientific, Waltham, Massachusetts, USA) in 5% CO2. Cells were maintained at 37°C in a humidified incubator MCO-18AC (Sanyo, Japan) supplied with 5% CO2. Cell cultures were tested for the absence of mycoplasma.
In Vitro Survival Assay (MTT Assay). The cytotoxicity was tested using the MTT (3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl tetrazolium bromide) assay [
32] with some modifications. A total of 2500 cells per well for the MCF7, HEK293T cell lines, 3000 cells for the A549 cell line, or 4000 cells per well for the VA-13 cell line were plated out in 135 µL of DMEM-F12 media (Gibco, USA) in a 96-well plate and then incubated in the 5% CO
2 incubator for first 20 h without treating. After this, 15 µL of media-DMSO solutions of tested substances were added to the cells (final DMSO concentrations in the media were 0,5% or less) and treated for65 h with 45 nM–100 µM (eight dilutions) of our substances (triplicate each) and doxorubicin as a control substance. Then the MTT reagent (Paneco LLC, Moscow, Russia) was added to cells up to the final concentration of 0.5 g/L (10 × stock solution in PBS was used), and cells were incubated for 2 h at 37 °C under an atmosphere of 5% CO
2. Then the MTT solution was discarded, and 140 µL of DMSO (PharmaMed LLC, Russia) was added. The plates were swayed on a shaker (200 rpm) to dissolve the formazan. The absorbance was measured using a microplate reader (VICTOR ×5 Light Plate Reader, PerkinElmer, USA) at a wavelength of 555 nm (in order to measure formazan concentration). The results were used to construct dose-response graphs and to estimate IC
50absvalues (IC
50abs is the concentration resulting in a two-fold decrease in the number of cells in comparison with the untreated cells) and, in some cases, IC
50 values (IC
50 is the concentration resulting in half of the maximal cytotoxic effect) with GraphPadSoftware, Inc., San Diego, CA, USA.
E. coli cytotoxicity assay with a screening of Mechanism of Action. A total of 50 µL of 20 mM solution of each compound in DMSO and DMSO as a control substance was added into wells punched in an agar plate containing a lawn of the
E. coli BW25113 DTC-pDualrep2 or
E. coli BW25113 LPTD-pDualrep2, which are hypersensitive to antibiotics.
E. coli DTC [
38] lacks the
tolC gene, which encodes the outer membrane component of several multidrug transporters [
39].
E. coli LPTD [
40] has the 23-amino-acid deletion in the
lptD gene, an inessential protein functioning in the final stages of the assembly of lipopolysaccharides into the outer membrane [
41]. After overnight incubation at 37 °C, the plate was scanned with the ChemiDoc (Bio-Rad, Hercules, CA, USA) system with two channels, including “Cy3-blot” (553/574 nm, green pseudocolor) for RFP fluorescence and “Cy5-blot” (588/633 nm, red pseudocolor) for Katushka2S fluorescence. Cells without reporter construction could be detected in both channels. The induction of the expression of Katushka2S is triggered by translation inhibitors, while RFP is up-regulated by the induction of DNA damage and SOS response. In addition, 2 µL of levofloxacin (25 µg/mL) and erythromycin (5 mg/mL) were used as positive controls for DNA biosynthesis and ribosome inhibitors, respectively.
Determination of Minimal Inhibitory Concentration (MIC). MIC values were determined by monitoring growth in 96-well plates of E. coli cultures exposed to serial dilutions of compounds. Specifically, overnight E. coli BW25113 DTC and K12 cultures were diluted in 96-well plates to an OD590 of 0.01 in LB medium. The wells were then supplemented with the solution of each tested compound at concentrations of 1 mM, 500 µM, 250 µM, 125 µM, 62.5 µM, 31.25 µM, 15.63 µM, 7.81 µM, 3.91 µM, and 1.95 µM. Then the plates were incubated with shaking (200 r.p.m.) overnight at 37°C, and cell growth was assessed by scanning OD590 each well with a VictorX5 reader.


 ,
, 








{kind=link}
{kind=link}
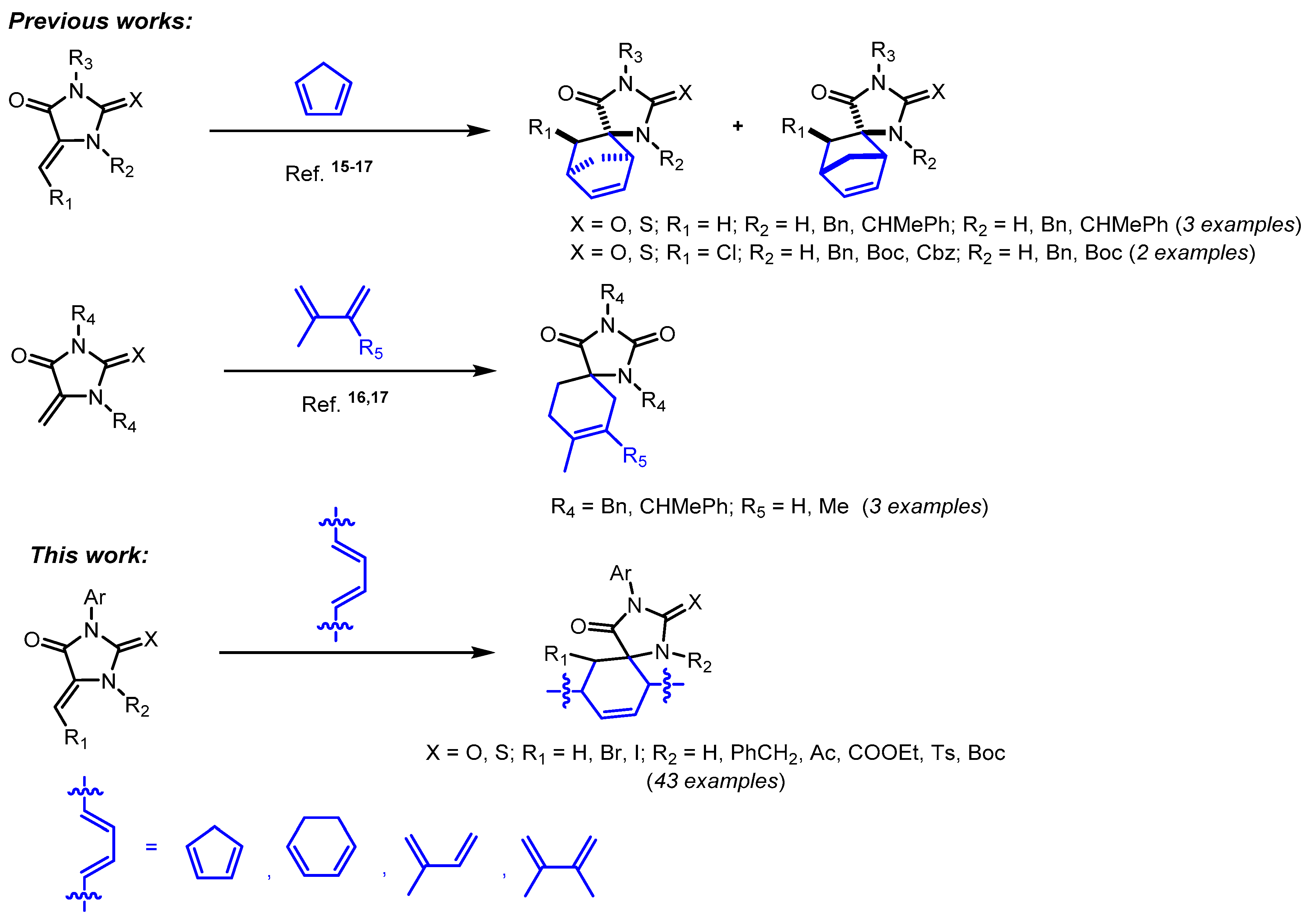{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}