
New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy


Abstract
1. Introduction
2. Classification of Cardiorenal Syndrome
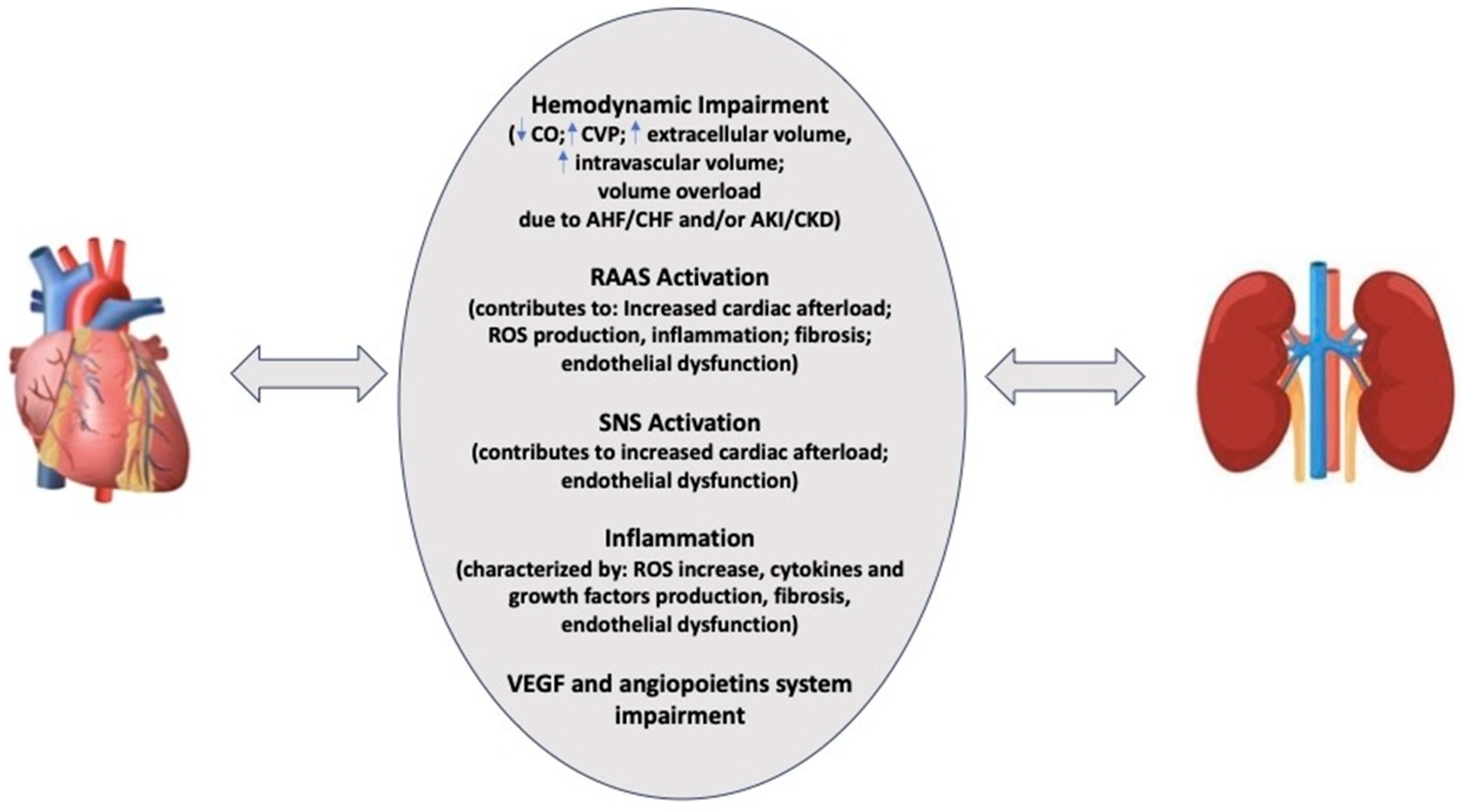
3. Pathophysiology of Cardiorenal Syndrome
3.1. Hemodynamic Factors
3.2. Endothelial Dysfunction, Oxidative Stress and Inflammation
3.3. Micro RNAs
4. Biomarkers in Cardiorenal Syndrome
4.1. Cardiac Biomarkers
4.2. Renal Biomarkers
5. Therapeutic Strategies in CRS
5.1. Diuretic and Ultrafiltration Therapy
5.2. Inotropic Agents and Beta Blockers
5.3. Renin Angiotensin System Inhibitors
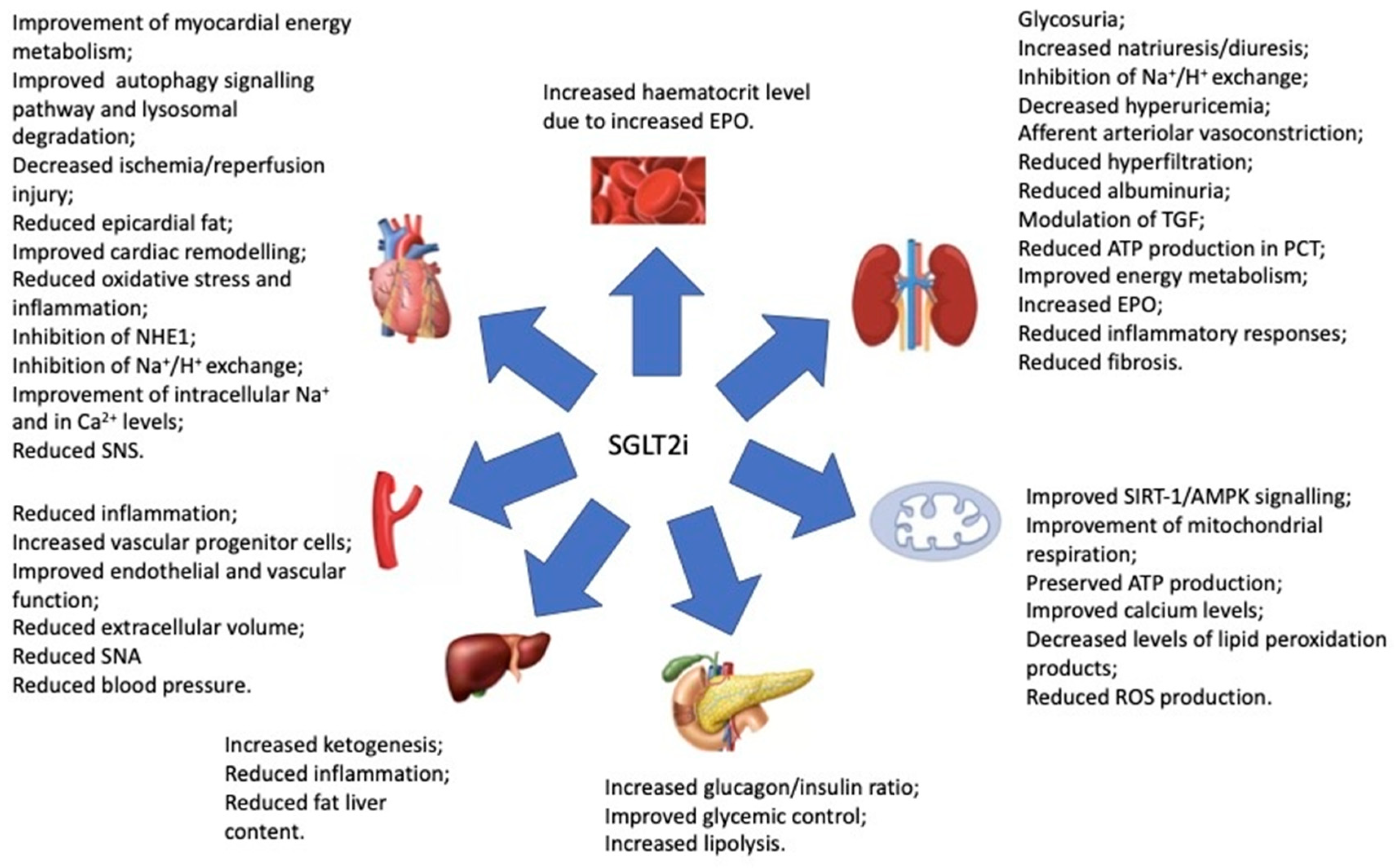
5.4. SGLT2 Inhibitors: An Emerging Therapeutic Tool in CRS
5.5. Novel Therapeutic Strategies
5.6. Non-Pharmacological Approaches
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.E.; Butler, J.; Wang, Y.; Abraham, W.T.; O’Connor, C.M.; Gottlieb, S.S.; Loh, E.; Massie, B.M.; Rich, M.W.; Stevenson, L.W.; et al. Incidence, predictors at admission, and impact of worsening renal function among patients hospitalized with heart failure. J. Am. Coll. Cardiol. 2004, 43, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Santoro, D.; Gembillo, G.; Andò, G. Glomerular Filtration Rate as a Predictor of Outcome in Acute Coronary Syndrome Complicated by Atrial Fibrillation. J. Clin. Med. 2020, 9, 1466. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Müller, C.; Damman, K.; Murray, P.T.; Kellum, J.A.; Ronco, C.; McCullough, P.A. Pathogenesis of cardiorenal syndrome type 1 in acute decompensated heart failure: Workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 99–116. [Google Scholar] [PubMed]
- Lorin, J.; Guilland, J.-C.; Stamboul, K.; Guenancia, C.; Cottin, Y.; Rochette, L.; Vergely, C.; Zeller, M. Increased Symmetric Dimethylarginine Level Is Associated with Worse Hospital Outcomes through Altered Left Ventricular Ejection Fraction in Patients with Acute Myocardial Infarction. PLoS ONE 2017, 12, e0169979. [Google Scholar] [CrossRef]
- Tharaux, P.L. Histamine provides an original vista on cardiorenal syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 5550–5552. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L.; Acute Kidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Xue, J.L.; Daniels, F.; Star, R.A.; Kimmel, P.L.; Eggers, P.W.; Molitoris, B.A.; Himmelfarb, J.; Collins, A.J. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J. Am. Soc. Nephrol. JASN 2006, 17, 1135–1142. [Google Scholar] [CrossRef]
- Cowie, M.R.; Komajda, M.; Murray-Thomas, T.; Underwood, J.; Ticho, B.; POSH Investigators. Prevalence and impact of worsening renal function in patients hospitalized with decompensated heart failure: Results of the prospective outcomes study in heart failure (POSH). Eur. Heart J. 2006, 27, 1216–1222. [Google Scholar] [CrossRef]
- Pimienta González, R.; Couto Comba, P.; Rodríguez Esteban, M.; Alemán Sánchez, J.J.; Hernández Afonso, J.; Rodríguez Pérez, M.D.; Marcelino Rodríguez, I.; Brito Díaz, B.; Elosua, R.; Cabrera de León, A. Incidence, Mortality and Positive Predictive Value of Type 1 Cardiorenal Syndrome in Acute Coronary Syndrome. PLoS ONE 2016, 11, e0167166. [Google Scholar] [CrossRef]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef]
- Damman, K.; Testani, J.M. The kidney in heart failure: An update. Eur. Heart J. 2015, 36, 1437–1444. [Google Scholar] [CrossRef]
- Ceravolo, G.; Macchia, T.; Cuppari, C.; Dipasquale, V.; Gambadauro, A.; Casto, C.; Ceravolo, M.D.; Cutrupi, M.; Calabrò, M.P.; Borgia, P.; et al. Update on the Classification and Pathophysiological Mechanisms of Pediatric Cardiorenal Syndromes. Children 2021, 8, 528. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Cozzi, M.; Bush, E.L.; Rabb, H. Distant Organ Dysfunction in Acute Kidney Injury: A Review. Am. J. Kidney Dis. 2018, 72, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Firoz, A.; Garlapati, S. Emerging Treatments of Cardiorenal Syndrome: An Update on Pathophysiology and Management. Cureus 2021, 13, e17240. [Google Scholar] [CrossRef]
- Nangaku, M.; Fujita, T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2008, 31, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Malvin, R.L. Stimulation of renal sodium reabsorption by angiotensin II. Am. J. Physiol. 1977, 232, F298–F306. [Google Scholar] [CrossRef]
- Ghionzoli, N.; Sciaccaluga, C.; Mandoli, G.E.; Vergaro, G.; Gentile, F.; D’Ascenzi, F.; Mondillo, S.; Emdin, M.; Valente, S.; Cameli, M. Cardiogenic shock and acute kidney injury: The rule rather than the exception. Heart Fail. Rev. 2021, 26, 487–496. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Ruocco, G.; Pellicori, P.; Incampo, E.; Di Tommaso, C.; Favilli, R.; Evangelista, I.; Nuti, R.; Testani, J.M. The prognostic role of different renal function phenotypes in patients with acute heart failure. Int. J. Cardiol. 2019, 276, 198–203. [Google Scholar] [CrossRef]
- Ljungman, S.; Laragh, J.H.; Cody, R.J. Role of the kidney in congestive heart failure. Relationship of cardiac index to kidney function. Drugs 1990, 39 (Suppl. 4), 10–24. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Navis, G.; Smilde, T.D.; Voors, A.A.; van der Bij, W.; van Veldhuisen, D.J.; Hillege, H.L. Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur. J. Heart Fail. 2008, 9, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Raine, A.E.; Ledingham, J.G. Raised venous pressure: A direct cause of renal sodium retention in oedema? Lancet 1988, 1, 1033–1035. [Google Scholar] [CrossRef]
- Winton, F.R. The influence of venous pressure on the isolated mammalian kidney. J. Physiol. 1931, 72, 49–61. [Google Scholar] [CrossRef]
- Mullens, W.; Abrahams, Z.; Skouri, H.N.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Paganini, E.; Tang, W.H. Elevated intra-abdominal pressure in acute decompensated heart failure: A potential contributor to worsening renal function? J. Am. Coll. Cardiol. 2008, 51, 300–306. [Google Scholar] [CrossRef]
- Nohria, A.; Hasselblad, V.; Stebbins, A.; Pauly, D.F.; Fonarow, G.C.; Shah, M.; Yancy, C.W.; Califf, R.M.; Stevenson, L.W.; Hill, J.A. Cardiorenal interactions: Insights from the ESCAPE trial. J. Am. Coll. Cardiol. 2008, 51, 1268–1274. [Google Scholar] [CrossRef]
- Kanjanahattakij, N.; Sirinvaravong, N.; Aguilar, F.; Agrawal, A.; Krishnamoorthy, P.; Gupta, S. High Right Ventricular Stroke Work Index Is Associated with Worse Kidney Function in Patients with Heart Failure with Preserved Ejection Fraction. Cardiorenal Med. 2018, 8, 123–129. [Google Scholar] [CrossRef]
- Redant, S.; Honoré, P.M.; De Bels, D. Fifty shades of central venous pressure in the cardiorenal syndrome. J. Transl. Int. Med. 2020, 8, 1–2. [Google Scholar] [CrossRef]
- Clementi, A.; Virzì, G.M.; Battaglia, G.G.; Ronco, C. Neurohormonal, Endocrine, and Immune Dysregulation and Inflammation in Cardiorenal Syndrome. Cardiorenal Med. 2019, 9, 265–273. [Google Scholar] [CrossRef]
- Prastaro, M.; Nardi, E.; Paolillo, S.; Santoro, C.; Parlati, A.L.M.; Gargiulo, P.; Basile, C.; Buonocore, D.; Esposito, G.; Filardi, P.P. Cardiorenal syndrome: Pathophysiology as a key to the therapeutic approach in an under-diagnosed disease. J. Clin. Ultrasound JCU 2022, 50, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Jentzer, J.C.; Chawla, L.S. A Clinical Approach to the Acute Cardiorenal Syndrome. Crit. Care Clin. 2015, 31, 685–703. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Zhang, J.; Nalesso, F.; Ronco, C.; McCullough, P.A. The Role of Dendritic and Endothelial Cells in Cardiorenal Syndrome. Cardiorenal Med. 2018, 8, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bottiglieri, T.; McCullough, P.A. The Central Role of Endothelial Dysfunction in Cardiorenal Syndrome. Cardiorenal Med. 2017, 7, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Castrop, H.; Schweda, F.; Mizel, D. Permissive role of nitric oxide in macula densa control of renin secretion. Am. J. Physiol. 2004, 286, F848–F857. [Google Scholar] [CrossRef]
- Neves, K.B.; Rios, F.J.; van der Mey, L.; Alves-Lopes, R.; Cameron, A.C.; Volpe, M.; Montezano, A.C.; Savoia, C.; Touyz, R.M. VEGFR (Vascular Endothelial Growth Factor Receptor) Inhibition Induces Cardiovascular Damage via Redox-Sensitive Processes. Hypertension 2018, 71, 638–647. [Google Scholar] [CrossRef]
- Gallo, G.; Volpe, M.; Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front. Med. 2022, 8, 798958. [Google Scholar] [CrossRef]
- Peesapati, V.; Sadik, M.; Verma, S. Panoramic Dominance of the Immune System in Cardiorenal Syndrome Type I. Cureus 2020, 12, e986. [Google Scholar] [CrossRef]
- Buliga-Finis, O.N.; Ouatu, A.; Badescu, M.C.; Dima, N.; Tanase, D.M.; Richter, P.; Rezus, C. Beyond the Cardiorenal Syndrome: Pathophysiological Approaches and Biomarkers for Renal and Cardiac Crosstalk. Diagnostics 2022, 12, 773. [Google Scholar] [CrossRef]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chu, M. Differential roles of VEGF: Relevance to tissue fibrosis. J. Cell. Biochem. 2019, 120, 10945–10951. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Allen, B.; Korhonen, E.A.; Nitschké, M.; Yang, H.W.; Baluk, P.; Saharinen, P.; Alitalo, K.; Daly, C.; Thurston, G.; et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Investig. 2016, 126, 3511–3525. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Lee, C.S.; Chiu, Y.W.; Lee, J.J.; Lee, S.C.; Hsu, Y.L.; Kuo, M.C. Angiopoietin-2, Renal Deterioration, Major Adverse Cardiovascular Events and All-Cause Mortality in Patients with Diabetic Nephropathy. Kidney Blood Press. Res. 2018, 43, 545–554. [Google Scholar] [CrossRef]
- Ricciardi, C.A.; Gnudi, L. Vascular growth factors as potential new treatment in cardiorenal syndrome in diabetes. Eur. J. Clin. Investig. 2021, 51, e13579. [Google Scholar] [CrossRef]
- Breitkreuz, M.; Hamdani, N. A change of heart: Oxidative stress in governing muscle function? Biophys. Rev. 2015, 7, 321–341. [Google Scholar] [CrossRef]
- Rababa’h, A.M.; Guillory, A.N.; Mustafa, R.; Hijjawi, T. Oxidative Stress and Cardiac Remodeling: An Updated Edge. Curr. Cardiol. Rev. 2018, 14, 53–59. [Google Scholar] [CrossRef]
- Hatamizadeh, P. Cardiorenal Syndrome An Important Subject in Nephrocardiology. Cardiol. Clin. 2021, 39, 455–464. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Wu, L.; Mei, Y.; Cui, S.; Feng, Z.; Chen, X. New insights into the pathophysiological mechanisms underlying cardiorenal syndrome. Aging 2020, 12, 12422–12431. [Google Scholar] [CrossRef] [PubMed]
- Sumida, M.; Doi, K.; Ogasawara, E.; Yamashita, T.; Hamasaki, Y.; Kariya, T.; Takimoto, E.; Yahagi, N.; Nangaku, M.; Noiri, E. Regulation of Mitochondrial Dynamics by Dynamin-Related Protein-1 in Acute Cardiorenal Syndrome. J. Am. Soc. Nephrol. JASN 2015, 26, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. JASN 2013, 24, 1512–1518. [Google Scholar] [CrossRef]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef]
- Callera, G.; Tostes, R.; Savoia, C.; Muscara, M.N.; Touyz, R.M. Vasoactive peptides in cardiovascular (patho)physiology. Expert. Rev. Cardiovasc Ther. 2007, 5, 531–552. [Google Scholar] [CrossRef]
- Vianello, A.; Caponi, L.; Galetta, F.; Franzoni, F.; Taddei, M.; Rossi, M.; Pietrini, P.; Santoro, G. β2-Microglobulin and TIMP1 Are Linked Together in Cardiorenal Remodeling and Failure. Cardiorenal Med. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Liu, X.; Chen, J.; Zhang, K.; Huang, F.; Wang, J.F.; Tang, W.; Huang, H. Apocynin Attenuates Cardiac Injury in Type 4 Cardiorenal Syndrome via Suppressing Cardiac Fibroblast Growth Factor-2 with Oxidative Stress Inhibition. J. Am. Heart Assoc. 2015, 4, e001598. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef]
- Virzì, G.M.; Breglia, A.; Brocca, A.; de Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Levels of Proinflammatory Cytokines, Oxidative Stress, and Tissue Damage Markers in Patients with Acute Heart Failure with and without Cardiorenal Syndrome Type 1. Cardiorenal Med. 2018, 8, 321–331. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Kanemitsu, Y.; Saigusa, D.; Mukawa, C.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Tachikawa, T.; Tsukimi, T.; et al. Canagliflozin reduces plasma uremic toxins and alters the intestinal microbiota composition in a chronic kidney disease mouse model. Am. J. Physiol. Physiol. 2018, 315, F824–F833. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Ishrat, R.; Tazyeen, S.; Alam, A.; Farooqui, A.; Ali, R.; Imam, N.; Tamkeen, N.; Ali, S.; Malik, M.D.; et al. In Silico Integrative Approach Revealed Key MicroRNAs and Associated Target Genes in Cardiorenal Syndrome. Bioinform. Biol. Insights 2021, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Clementi, A.; Brocca, A.; Cal, M.; Ronco, C. Molecular and Genetic Mechanisms Involved in the Pathogenesis of Cardiorenal Cross Talk. Pathobiology 2016, 83, 201–207. [Google Scholar] [CrossRef]
- Xu, X.; Kriegel, A.J.; Liu, Y.; Usa, K.; Mladinov, D.; Liu, H.; Fang, Y.; Ding, X.; Liang, M. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012, 82, 1167–1175. [Google Scholar] [CrossRef]
- Di, J.; Yang, M.; Zhou, H.; Li, M.; Zhao, J. MicroRNA-21-containing microvesicles from tubular epithelial cells promote cardiomyocyte hypertrophy. Ren. Fail. 2021, 43, 391–400. [Google Scholar] [CrossRef]
- Gembillo, G.; Visconti, L.; Giusti, M.A.; Siligato, R.; Gallo, A.; Santoro, D.; Mattina, A. Cardiorenal Syndrome: New Pathways and Novel Biomarkers. Biomolecules 2021, 11, 1581. [Google Scholar] [CrossRef]
- Huang, C.-K.; Bar, C.; Thum, T. miR-21, Mediator, and Potential Therapeutic Target in the Cardiorenal Syndrome. Front. Pharmacol. 2020, 11, 726. [Google Scholar] [CrossRef]
- Ronco, C.; House, A.A.; Haapio, M. Cardiorenal syndrome: Refining the definition of a complex symbiosis gone wrong. Intensive Care Med. 2008, 34, 957–962. [Google Scholar] [CrossRef]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef]
- Wang, J.; Tan, G.J.; Han, L.N.; Bai, Y.Y.; He, M.; Liu, H.B. Novel biomarkers for cardiovascular risk prediction. J. Geriatr. Cardiol. JGC 2017, 14, 135–150. [Google Scholar] [PubMed]
- Van Kimmenade, R.R.; Januzzi, J.L.; Baggish, A.L., Jr.; Lainchbury, J.G.; Bayes-Genis, A.; Richards, A.M.; Pinto, Y.M. Amino-terminal pro-brain natriuretic Peptide, renal function, and outcomes in acute heart failure: Redefining the cardiorenal interaction? J. Am. Coll. Cardiol. 2006, 48, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Zhao, S.; Ye, P.; Luo, L. Biomarkers in Cardiorenal Syndromes. BioMed Res. Int. 2018, 2018, 9617363. [Google Scholar] [CrossRef] [PubMed]
- Pickering, J.W.; Than, M.P.; Cullen, L.; Aldous, S.; Ter Avest, E.; Body, R.; Carlton, E.W.; Collinson, P.; Dupuy, A.M.; Ekelund, U.; et al. Rapid Rule-out of Acute Myocardial Infarction With a Single High-Sensitivity Cardiac Troponin T Measurement Below the Limit of Detection: A Collaborative Meta-analysis. Ann. Intern. Med. 2017, 166, 715–724. [Google Scholar] [CrossRef]
- Núñez, J.; de la Espriella, R.; Rossignol, P.; Voors, A.A.; Mullens, W.; Metra, M.; Chioncel, O.; Januzzi, J.L.; Mueller, C.; Richards, A.M.; et al. Congestion in heart failure: A circulating biomarker-based perspective. A review from the Biomarkers Working Group of the Heart Failure Association, European Society of Cardiology. Eur. J. Heart Fail. 2022, 24, 1751–1766. [Google Scholar] [CrossRef]
- Seliger, S.L.; Hong, S.N.; Christenson, R.H.; Kronmal, R.; Daniels, L.B.; Lima, J.A.; De Lemos, J.A.; Bertoni, A.; Defilippi, C.R. High-Sensitive Cardiac Troponin T as an Early Biochemical Signature for Clinical and Subclinical Heart Failure: MESA (Multi-Ethnic Study of Atherosclerosis). Circulation 2017, 135, 1494–1505. [Google Scholar] [CrossRef]
- Morawiec, B.; Fournier, S.; Tapponnieretal, M. Performance of highly sensitive cardiac troponin T assay to detect ischaemia at PET-CT in low-risk patients with acute coronary syndrome: A prospective observational study. BMJ Open 2017, 7, e014655. [Google Scholar] [CrossRef]
- Savoia, C.; Volpe, M.; Alonzo, A.; Rossi, C.; Rubattu, S. Natriuretic peptides and cardiovascular damage in the metabolic syndrome: Molecular mechanisms and clinical implications. Clin. Sci. 2009, 118, 231–240. [Google Scholar] [CrossRef]
- Sakuma, M.; Nakamura, M.; Tanaka, F.; Onoda, T.; Itai, K.; Tanno, K.; Ohsawa, M.; Sakata, K.; Yoshida, Y.; Kawamura, K.; et al. Plasma B-type natriuretic peptide level and cardiovascular events in chronic kidney disease in a community-based population. Circ. J. 2010, 74, 792–797. [Google Scholar] [CrossRef]
- Takahama, H.; Nishikimi, T.; Takashio, S.; Hayashi, T.; Nagai-Okatani, C.; Asada, T.; Fujiwara, A.; Nakagawa, Y.; Amano, M.; Hamatani, Y.; et al. Change in the NT-proBNP/Mature BNP molar ratio precedes worsening renal function in patients with acute heart failure: A novel predictor candidate for cardiorenal syndrome. J. Am. Heart Assoc. 2019, 8, e011468. [Google Scholar] [CrossRef]
- Spanaus, K.S.; Kronenberg, F.; Ritz, E.; Schlapbach, R.; Fliser, D.; Hersberger, M.; Kollerits, B.; König, P.; von Eckardstein, A.; Mild-to-Moderate Kidney Disease Study Group. B-type natriuretic peptide concentrations predict the progression of nondiabetic chronic kidney disease: The Mild-to-Moderate Kidney Disease Study. Clin. Chem. 2007, 53, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- McCallum, W.; Tighiouart, H.; Kiernan, M.S.; Huggins, G.S.; Sarnak, M.J. Relation of kidney function decline and NT-proBNP with risk of mortality and readmission in acute decompensated heart failure. Am. J. Med. 2020, 133, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Palazzuoli, A.; Masson, S.; Ronco, C.; Maisel, A. Clinical relevance of biomarkers in heart failure and cardiorenal syndrome: The role of natriuretic peptides and troponin. Heart Fail. Rev. 2014, 19, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Van Kimmenade, R.R.; Januzzi, J.L.; Ellinor, P.T., Jr.; Sharma, U.C.; Bakker, J.A.; Low, A.F.; Martinez, A.; Crijns, H.J.; MacRae, C.A.; Menheere, P.P.; et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J. Am. Coll. Cardiol. 2006, 48, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, A.R.; Gullestad, L.; Ueland, T.; Aukrust, P.; Guo, Y.; Adourian, A.; Muntendam, P.; van Veldhuisen, D.J.; de Boer, R.A. Prognostic value of changes in galectin-3 levels over time in patients with heart failure: Data from CORONA and COACH. Circ. Heart Fail. 2013, 6, 219–226. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; Ruparel, K.; Sweitzer, N.K.; Fang, J.C.; Levy, W.C.; Sawyer, D.B.; Cappola, T.P. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J. Am. Coll. Cardiol. 2011, 58, 386–394. [Google Scholar] [CrossRef]
- Vorovich, E.; French, B.; Ky, B.; Goldberg, L.; Fang, J.C.; Sweitzer, N.K.; Cappola, T.P. Biomarker predictors of cardiac hospitalization in chronic heart failure: A recurrent event analysis. J. Card. Fail. 2014, 20, 569–576. [Google Scholar] [CrossRef][Green Version]
- Schmitz, J.; Owyang, A.; Oldhametal, E. IL-33,aninterleukin- 1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Daniels, L.B.; Bayes-Genis, A. Using ST2 in cardiovascular patients: A review. Future Cardiol. 2014, 10, 525–539. [Google Scholar] [CrossRef]
- Manzano-Fernndez, S.; Mueller, T.; Pascual-Figal, D.; Truong, Q.A.; Januzzi, J.L. Usefulness of soluble concentrations of interleukin family member ST2 as predictor of mortality in patients with acutely decompensated heart failure relative to left ventricular ejection fraction. Am. J. Cardiol. 2011, 107, 259–267. [Google Scholar] [CrossRef]
- Ky, B.; French, B.; McCloskey, K.; Rame, J.E.; McIntosh, E.; Shahi, P.; Dries, D.L.; Tang, W.W.; Wu, A.H.; Fang, J.C.; et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ. Heart Fail. 2011, 4, 180–187. [Google Scholar] [CrossRef]
- Shah, R.V.; Chen-Tournoux, A.A.; Picard, M.H.; van Kimmenade, R.R.; Januzzi, J.L. Serum levels of the interleukin-1 receptor family member ST2, cardiac structure and function, and long-term mortality in patients with acute dyspnea. Circ. Heart Fail. 2009, 2, 311–319. [Google Scholar] [CrossRef]
- Song, P.; Xu, J.; Song, Y.; Jiang, S.; Yuan, H.; Zhang, X. Association of plasma myeloperoxidase level with risk of coronary artery disease in patients with type 2 diabetes. Dis. Markers 2015, 2015, 761939. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gedikli, O.; Kiris, A.; Hosoglu, Y.; Karahan, C.; Kaplan, S. Serum myeloperoxidase level is associated with heart-type fatty acid-binding protein but not Troponin T in patients with chronic heart failure. Med. Princ. Pract. 2015, 24, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Afshinnia, F.; Zeng, L.; Byun, J.; Gadegbeku, C.A.; Magnone, M.C.; Whatling, C.; Valastro, B.; Kretzler, M.; Pennathur, S.; Michigan Kidney Translational Core CPROBE Investigator Group. Myeloperoxidase Levels and Its Product 3-Chlorotyrosine Predict Chronic Kidney Dis- ease Severity and Associated Coronary Artery Disease. Am. J. Nephrol. 2017, 46, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Grace, E.; Turner, R.M. Use of procalcitonin in patients with various degrees of chronic kidney disease including renal replacement therapy. Clin. Infect. Dis. 2014, 59, 1761–1767. [Google Scholar] [CrossRef]
- Martin, M.; Julia, S.; Alan, M. The role of procalcitonin in acute heart failure patients. SC Heart Fail. 2017, 4, 203–208. [Google Scholar]
- Fan, P.C.; Chang, C.H.; Chen, Y.C. Biomarkers for acute cardiorenal syndrome. Nephrology 2018, 23, 68–71. [Google Scholar] [CrossRef]
- Allen, L.A.; Felker, G.M. Multi-marker strategies in heart failure: Clinical and statistical approaches. Heart Fail. Rev. 2010, 15, 343–349. [Google Scholar] [CrossRef]
- Lena, A.; Anker, M.S.; Springer, J. Muscle wasting and sarcopenia in heart failure-the current state of science. Int. J. Mol. Sci. 2020, 21, 6549. [Google Scholar] [CrossRef]
- Lassus, J.P.; Nieminen, M.S.; Peuhkurinen, K.; Pulkki, K.; Siirilä-Waris, K.; Sund, R.; Harjola, V.P.; FINN-AKVA study group. Markers of renal function and acute kidney injury in acute heart failure: Definitions and impact on outcomes of the cardiorenal syndrome. Eur. Heart J. 2010, 31, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Jotoku, M.; Irita, J.; Enomoto, D.; Nagao, T.; Desilva, V.R.; Yamane, S.; Pei, Z.; Kojima, S.; Hamano, Y.; et al. Association between cystatin C and inflammation in patients with essential hypertension. Clin. Exp. Nephrol. 2010, 14, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Shlipak, M.G.; Mattes, M.D.; Peralta, C.A. Update on cystatin C: Incorporation into clinical practice. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 62, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.G.; Bruns, D.E.; Hortin, G.L.; Sandberg, S.; Aakre, K.M.; McQueen, M.J.; Itoh, Y.; Lieske, J.C.; Seccombe, D.W.; Jones, G.; et al. National Kidney Disease Education Program-IFCC Working Group on Standardization of Albumin in Urine Current issues in measurement and reporting of urinary albumin excretion. Clin. Chem. 2009, 55, 24–38. [Google Scholar] [CrossRef]
- Jackson, C.E.; Solomon, S.D.; Gerstein, H.C.; Zetterstrand, S.; Olofsson, B.; Michelson, E.L.; Granger, C.B.; Swedberg, K.; Pfeffer, M.A.; Yusuf, S.; et al. Albuminuria in chronic heart failure: Prevalence and prognostic importance. Lancet 2009, 374, 543–550. [Google Scholar] [CrossRef]
- Brisco, M.A.; Testani, J.M. Novel Renal Biomarkers to Assess Cardiorenal Syndrome. Curr. Heart Fail. Rep. 2014, 11, 485–499. [Google Scholar] [CrossRef]
- Chen, C.; Yang, X.; Lei, Y.; Zha, Y.; Liu, H.; Ma, C.; Tian, J.; Chen, P.; Yang, T.; Hou, F.F. Urinary biomarkers at the time of AKI diagnosis as predictors of progression of AKI among patients with acute cardiorenal syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1536–1544. [Google Scholar] [CrossRef]
- Coca, S.G.; Garg, A.X.; Thiessen-Philbrook, H.; Koyner, J.L.; Patel, U.D.; Krumholz, H.M.; Shlipak, M.G.; Parikh, C.R. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J. Am. Soc. Nephrol. 2014, 25, 1063–1071. [Google Scholar] [CrossRef]
- Driver, T.H.; Katz, R.; Ix, J.H.; Magnani, J.W.; Peralta, C.A.; Parikh, C.R.; Fried, L.; Newman, A.B.; Kritchevsky, S.B.; Sarnak, M.J.; et al. Urinary kidney injury molecule 1 (KIM-1) and interleukin 18 (IL-18) as risk markers for heart failure in older adults: The health, aging, and body composition (Health ABC) study. Am. J. Kidney Dis. 2014, 64, 49–56. [Google Scholar] [CrossRef]
- Medic, B.; Rovcanin, B.; Savic Vujovic, K.; Obradovic, D.; Duric, D.; Prostran, M. Evaluation of Novel Biomarkers of Acute Kidney Injury: The Possibilities and Limitations. Curr. Med. Chem. 2016, 23, 1981–1997. [Google Scholar]
- Damman, K.; Van Veldhuisen, D.J.; Navis, G.; Vaidya, V.S.; Smilde, T.D.; Westenbrink, B.D.; Bonventre, J.V.; Voors, A.A.; Hillege, H.L. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart 2010, 96, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Robles, N.R.; Lopez Gomez, J.; Garcia Pino, G.; Valladares, J.; Gallego, R.H.; Cerezo, I. Alpha-1-microglobulin: Prognostic value in chronic kidney disease. Med. Clin. 2020, S0025-7753, 30639–30640. [Google Scholar] [CrossRef]
- Mishra, J.; Dent, C.; Tarabishi, R.; Mitsnefes, M.M.; Ma, Q.; Kelly, C.; Ruff, S.M.; Zahedi, K.; Shao, M.; Bean, J.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005, 365, 1231–1238. [Google Scholar] [CrossRef]
- Van Deursen, V.M.; Damman, K.; Voorsetal, A.A. Prognostic value of plasma neutrophil gelatinase-associated lipocalin for mortality in patients with heart failure. Circ. Heart Fail. 2014, 7, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; van Veldhuisen, D.J.; Navis, G.; Voors, A.A.; Hillege, H.L. Urinary neutrophil gelatinase associated lipocalin (NGAL), a marker of tubular damage, is increased in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 997–1000. [Google Scholar] [CrossRef]
- Testani, M.; Tang, W.H.W. Biomarkers of acute kidney injury in chronic heart failure: What do the signals mean? JACC Heart Fail. 2013, 1, 425–426. [Google Scholar] [CrossRef]
- Jungbauer, C.G.; Birner, C.; Jung, B.; Buchner, S.; Lubnow, M.; von Bary, C.; Endemann, D.; Banas, B.; Mack, M.; Böger, C.A.; et al. Kidney injury molecule-1 and N-acetyl-ß-d-glucosaminidase in chronic heart failure: Possible biomarkers of cardiorenal syndrome. Eur. J. Heart Fail. 2011, 13, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Maatman, R.G.; Van Kuppevelt, T.H.; Veerkamp, J.H. Two types of fatty acid-binding protein in human kidney. Isolation, characterization and localization. Biochem. J. 1991, 273, 759–766. [Google Scholar] [CrossRef]
- Li, J.; Sheng, X.; Cheng, D.; Wang, F.; Jian, G.; Li, Y.; Xu, T.; Wang, X.; Fan, Y.; Wang, N. Is the mean platelet volume a predictive marker of a high in-hospital mortality of acute cardiorenal syndrome patients receiving continuous renal replacement therapy? Medicina 2018, 97, e11180. [Google Scholar] [CrossRef]
- Van den Brink, O.W.; Delbridge, L.M.; Rosenfeldt, F.L.; Penny, D.; Esmore, D.S.; Quick, D.; Kaye, D.M.; Pepe, S. Endogenous cardiac opioids: Enkephalins in adaptation and protection of the heart. Heart Lung Circ. 2003, 12, 178–187. [Google Scholar] [CrossRef]
- Ng, L.L.; Squire, I.B.; Jones, D.J.L.; Cao, T.H.; Chan, D.C.S.; Sandhu, J.K.; Quinn, P.A.; Davies, J.E.; Struck, J.; Hartmann, O.; et al. Proenkephalin, Renal Dysfunction, and Prognosis in Patients With Acute Heart Failure: A GREAT Network Study. J. Am. Coll. Cardiol. 2018, 69, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.L.; Hu, H.J.; Zhao, X.J.; Chi, W.W.; Liu, D.M.; Wang, Q.; Cui, W. Urine N-terminal pro-B-type natriuretic peptide and plasma proenkephalin are promising biomarkers for early diagnosis of cardiorenal syndrome type 1 in acute decompensated heart failure: A prospective, double-center, observational study in real-world. Ren. Fail. 2022, 44, 1486–1497. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.L.; Sandhu, J.K.; Narayan, H.; Quinn, P.A.; Squire, I.B.; Davies, J.E.; Bergmann, A.; Maisel, A.; Jones, D.J. Proenkephalin and prognosis after acute myocardial infarction. J. Am. Coll. Cardiol. 2014, 63, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, C.; Tian, J.; Zha, Y.; Xiong, Y.; Sun, Z.; Chen, P.; Li, J.; Yang, T.; Ma, C.; et al. Urinary Angiotensinogen Level Predicts AKI in Acute Decompensated Heart Failure: A Prospective, Two-Stage Study. J. Am. Soc. Nephrol. JASN 2015, 6, 2032–2041. [Google Scholar] [CrossRef]
- Wysocki, J.; Batlle, D. Urinary Angiotensinogen: A Promising Biomarker of AKI Progression in Acute Decompensated Heart Failure: What Does It Mean? Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 1515–1517. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M.; Ellison, D.H.; Mullens, W.; Cox, Z.L.; Testani, J.M. Diuretic Therapy for Patients With Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1178–1195. [Google Scholar] [CrossRef]
- Mullens, W.; Dauw, J.; Martens, P.; Verbrugge, F.H.; Nijst, P.; Meekers, E.; Tartaglia, K.; Chenot, F.; Moubayed, S.; Dierckx, R.; et al. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N. Engl. J. Med. 2022, 387, 1185–1195. [Google Scholar] [CrossRef]
- Brisco, M.A.; Zile, M.R.; Hanberg, J.S.; Wilson, F.P.; Parikh, C.R.; Coca, S.G.; Tang, W.H.; Testani, J.M. Relevance of Changes in Serum Creatinine During a Heart Failure Trial of Decongestive Strategies: Insights From the DOSE Trial. J. Card. Fail. 2016, 22, 753–760. [Google Scholar] [CrossRef]
- Grodin, J.L.; Carter, S.; Bart, B.A.; Goldsmith, S.R.; Drazner, M.H.; Tang, W.H.W. Direct comparison of ultrafiltration to pharmacological decongestion in heart failure: A per-protocol analysis of CARRESS-HF. Eur. J. Heart Fail. 2018, 20, 1148–1156. [Google Scholar] [CrossRef]
- Valente, M.A.; Voors, A.A.; Damman, K.; Van Veldhuisen, D.J.; Massie, B.M.; O’Connor, C.M.; Metra, M.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; et al. Diuretic response in acute heart failure: Clinical characteristics and prognostic significance. Eur. Heart J. 2014, 35, 1284–1293. [Google Scholar] [CrossRef] [PubMed]
- Testani, J.M.; Brisco, M.A.; Turner, J.M.; Spatz, E.S.; Bellumkonda, L.; Parikh, C.R.; Tang, W.H. Loop diuretic efficiency: A metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ. Heart Fail. 2014, 7, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Felker, G.M. Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2017, 377, 1964–1975. [Google Scholar] [CrossRef] [PubMed]
- Hanberg, J.S.; Rao, V.; Ter Maaten, J.M.; Laur, O.; Brisco, M.A.; Perry Wilson, F.; Grodin, J.L.; Assefa, M.; Samuel Broughton, J.; Planavsky, N.J.; et al. Hypochloremia and Diuretic Resistance in Heart Failure: Mechanistic Insights. Circ. Heart Fail. 2016, 9, e003180. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Steels, P.; Grieten, L.; Nijst, P.; Tang, W.H.; Mullens, W. Hyponatremia in acute decompensated heart failure: Depletion versus dilution. J. Am. Coll. Cardiol. 2015, 65, 480–492. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Testani, J.M.; Pitt, B. Pathophysiology of Diuretic Resistance and Its Implications for the Management of Chronic Heart Failure. Hypertension 2020, 76, 1045–1054. [Google Scholar] [CrossRef]
- Strobeck, J.E.; Feldschuh, J.; Miller, W.L. Heart Failure Outcomes With Volume-Guided Management. JACC Heart Fail. 2018, 6, 940–948. [Google Scholar] [CrossRef]
- Brisco-Bacik, M.A.; Ter Maaten, J.M.; Houser, S.R.; Vedage, N.A.; Rao, V.; Ahmad, T.; Wilson, F.P.; Testani, J.M. Outcomes Associated With a Strategy of Adjuvant Metolazone or High-Dose Loop Diuretics in Acute Decompensated Heart Failure: A Propensity Analysis. J. Am. Heart Assoc. 2018, 7, e009149. [Google Scholar] [CrossRef]
- Jaski, B.E.; Miller, D. Ultrafiltration in decompensated heart failure. Curr. Heart Fail. Rep. 2005, 2, 148–154. [Google Scholar] [CrossRef]
- Marenzi, G.; Lauri, G.; Grazi, M.; Assanelli, E.; Campodonico, J.; Agostoni, P. Circulatory response to fluid overload removal by extracorporeal ultrafiltration in refractory congestive heart failure. J. Am. Coll. Cardiol. 2001, 38, 963–968. [Google Scholar] [CrossRef]
- Costanzo, M.R.; Guglin, M.E.; Saltzberg, M.T.; Jessup, M.L.; Bart, B.A.; Teerlink, J.R.; Jaski, B.E.; Fang, J.C.; Feller, E.D.; Haas, G.J.; et al. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J. Am. Coll. Cardiol. 2007, 49, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Elkayam, U.; Hatamizadeh, P.; Janmohamed, M. The challenge of correcting volume overload in hospitalized patients with decompensated heart failure. J. Am. Coll. Cardiol. 2007, 49, 684–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fudim, M.; Brooksbank, J.; Giczewska, A.; Greene, S.J.; Grodin, J.L.; Martens, P.; Ter Maaten, J.M.; Sharma, A.; Verbrugge, F.H.; Chakraborty, H.; et al. Ultrafiltration in Acute Heart Failure: Implications of Ejection Fraction and Early Response to Treatment From CARRESS-HF. J. Am. Heart Assoc. 2020, 9, e015752. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Lin, M.H.; Chen, C.L.; Lai, Y.C.; Chen, C.Y.; Lin, Y.C.; Hung, C.C. Comprehensive Comparison of the Effect of Inotropes on Cardiorenal Syndrome in Patients with Advanced Heart Failure: A Network Meta-Analysis of Randomized Controlled Trials. J. Clin. Med. 2021, 10, 4120. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, L.I. Pharmacological bases for the use of dopamine and related drugs in the treatment of congestive heart failure. J. Cardiovasc. Pharmacol. 1989, 14 (Suppl. 8), S21–S28. [Google Scholar] [CrossRef]
- Friedrich, J.O.; Adhikari, N.; Herridge, M.S.; Beyene, J. Meta-analysis: Low-dose dopamine increases urine output but does not prevent renal dysfunction or death. Ann. Intern. Med. 2005, 142, 510–524. [Google Scholar] [CrossRef]
- Wan, S.H.; Stevens, S.R.; Borlaug, B.A.; Anstrom, K.J.; Deswal, A.; Felker, G.M.; Givertz, M.M.; Bart, B.A.; Tang, W.H.; Redfield, M.M.; et al. Differential Response to Low-Dose Dopamine or Low-Dose Nesiritide in Acute Heart Failure With Reduced or Preserved Ejection Fraction: Results From the ROSE AHF Trial (Renal Optimization Strategies Evaluation in Acute Heart Failure). Circ. Heart Fail. 2016, 9, e002593. [Google Scholar] [CrossRef]
- Badve, S.V.; Roberts, M.A.; Hawley, C.M.; Cass, A.; Garg, A.X.; Krum, H.; Tonkin, A.; Perkovic, V. Effects of beta-adrenergic antagonists in patients with chronic kidney disease: A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011, 58, 1152–1161. [Google Scholar] [CrossRef]
- Von Lueder, T.G.; Kotecha, D.; Atar, D.; Hopper, I. Neurohormonal Blockade in Heart Failure. Card. Fail. Rev. 2017, 3, 19–24. [Google Scholar] [CrossRef]
- Savoia, C.; Battistoni, A.; Calvez, V.; Cesario, V.; Montefusco, G.; Filippini, A. Microvascular Alterations in Hypertension and Vascular Aging. Curr. Hypertens Rev. 2017, 13, 16–23. [Google Scholar] [CrossRef]
- Savoia, C.; D’Agostino, M.; Lauri, F.; Volpe, M. Angiotensin type 2 receptor in hypertensive cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 125–132. [Google Scholar] [CrossRef]
- Bhandari, S.; Mehta, S.; Khwaja, A.; Cleland, J.G.F.; Ives, N.; Brettell, E.; Chadburn, M.; Cockwell, P.; STOP ACEi Trial Investigators. Renin-Angiotensin System Inhibition in Advanced Chronic Kidney Disease. N. Engl. J. Med. 2022, 387, 2021–2032. [Google Scholar] [CrossRef]
- Weir, M.R.; Lakkis, J.I.; Jaar, B.; Rocco, M.V.; Choi, M.J.; Kramer, H.J.; Ku, E. Use of Renin-Angiotensin System Blockade in Advanced CKD: An NKF-KDOQI Controversies Report. Am. J. Kidney Dis. 2018, 72, 873–884. [Google Scholar] [CrossRef]
- McCallum, W.; Tighiouart, H.; Ku, E.; Salem, D.; Sarnak, M.J. Trends in Kidney Function Outcomes Following RAAS Inhibition in Patients With Heart Failure With Reduced Ejection Fraction. Am. J. Kidney Dis. 2020, 75, 21–29. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Investigators P-H, Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Velazquez, E.J.; Morrow, D.A.; DeVore, A.D.; Duffy, C.I.; Ambrosy, A.P.; McCague, K.; Rocha, R.; Braunwald, E.; PIONEER-HF Investigators. Angiotensin-Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019, 380, 539–548. [Google Scholar] [CrossRef]
- Damman, K.; Gori, M.; Claggett, B.; Jhund, P.S.; Senni, M.; Lefkowitz, M.P.; Prescott, M.F.; Shi, V.C.; Rouleau, J.L.; Swedberg, K.; et al. Renal Effects and Associated Outcomes During Angiotensin-Neprilysin Inhibition in Heart Failure. JACC Heart Fail. 2018, 6, 489–498. [Google Scholar] [CrossRef]
- Damman, K.; Valente, M.A.; Voors, A.A.; O’Connor, C.M.; van Veldhuisen, D.J.; Hillege, H.L. Renal impairment, worsening renal function, and outcome in patients with heart failure: An updated meta-analysis. Eur. Heart J. 2014, 35, 455–469. [Google Scholar] [CrossRef]
- Haynes, R.; Judge, P.K.; Staplin, N.; Herrington, W.G.; Storey, B.C.; Bethel, A.; Bowman, L.; Brunskill, N.; Cockwell, P.; Hill, M.; et al. Effects of Sacubitril/Valsartan Versus Irbesartan in Patients with Chronic Kidney Disease. Circulation 2018, 138, 1505–1514. [Google Scholar] [CrossRef]
- Spannella, F.; Giulietti, F.; Filipponi, A.; Sarzani, R. Effect of sacubitril/valsartan on renal function: A systematic review and meta-analysis of randomized controlled trials. ESC Heart Fail. 2020, 7, 3487–3496. [Google Scholar] [CrossRef]
- Epstein, M.; Reaven, N.L.; Funk, S.E.; McGaughey, K.J.; Oestreicher, N.; Knispel, J. Evaluation of the treatment gap be- tween clinical guidelines and the utilization of renin-angiotensin-aldosterone system inhibitors. Am. J. Manag. Care 2015, 21 (Suppl. S11), S212–S220. [Google Scholar]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Kosiborod, M.; Cavender, M.A.; Fu, A.Z.; Wilding, J.P.; Khunti, K.; Holl, R.W.; Norhammar, A.; Birkeland, K.I.; Jørgensen, M.E.; Thuresson, M.; et al. Lower Risk of Heart Failure and Death in Patients Initiated on Sodium-Glucose Cotransporter-2 Inhibitors Versus Other Glucose-Lowering Drugs: The CVD-REAL Study (Comparative Effectiveness of Cardiovascular Outcomes in New Users of Sodium-Glucose Cotransporter-2 Inhibitors). Circulation 2017, 136, 249–259. [Google Scholar]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Docherty, K.F.; Claggett, B.L.; Jhund, P.S.; de Boer, R.A.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. SGLT-2 inhibitors in patients with heart failure: A comprehensive meta-analysis of five randomised controlled trials. Lancet 2022, 400, 757–767. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Stefánsson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; Toto, R.D.; et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: A prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021, 9, 22–31. [Google Scholar] [CrossRef]
- The EMPA-KIDNEY Collaborative Group; Herrington, W.G.; Staplin, N.; Wanner, C.; Green, J.B.; Hauske, S.J.; Emberson, J.R.; Preiss, D.; Judge, P.; Mayne, K.J.; et al. Empagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2023, 388, 117–127. [Google Scholar]
- Neuen, B.L.; Young, T.; Heerspink, H.J.L.; Neal, B.; Perkovic, V.; Billot, L.; Mahaffey, K.W.; Charytan, D.M.; Wheeler, D.C.; Arnott, C.; et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2019, 7, 845–854. [Google Scholar] [CrossRef]
- Wang, A.; Li, Z.; Zhuo, S.; Gao, F.; Zhang, H.; Zhang, Z.; Ren, G.; Ma, X. Mechanisms of Cardiorenal Protection With SGLT2 Inhibitors in Patients With T2DM Based on Network Pharmacology. Front. Cardiovasc. Med. 2022, 9, 857952. [Google Scholar] [CrossRef]
- Monzo, L.; Ferrari, I.; Cicogna, F.; Tota, C.; Calò, L. What proportion of patients with heart failure and preserved ejection fraction are eligible for empagliflozin? J. Cardiovasc. Med. 2022, 23, 567–569. [Google Scholar] [CrossRef]
- Monzo, L.; Ferrari, I.; Cicogna, F.; Tota, C.; Calò, L. Sodium-glucose co-transporter-2 inhibitors eligibility in patients with heart failure with reduced ejection fraction. Int. J. Cardiol. 2021, 341, 56–59. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. [Google Scholar] [CrossRef]
- Li, L.; Konishi, Y.; Morikawa, T.; Zhang, Y.; Kitabayashi, C.; Kobara, H.; Masaki, T.; Nakano, D.; Hitomi, H.; Kobori, H.; et al. Effect of a SGLT2 inhibitor on the systemic and intrarenal renin–angiotensin system in subtotally nephrectomized rats. J. Pharmacol. Sci. 2018, 137, 220–223. [Google Scholar] [CrossRef]
- van Raalte, D.H.; Bjornstad, P. Role of sodium-glucose cotransporter 2 inhibition to mitigate diabetic kidney disease risk in type 1 diabetes. Nephrol. Dial. Transplant. 2020, 35 (Suppl. 1), i24–i32. [Google Scholar] [CrossRef]
- Zhang, Y.; Nakano, D.; Guan, Y.; Hitomi, H.; Uemura, A.; Masaki, T.; Kobara, H.; Sugaya, T.; Nishiyama, A. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor–dependent pathway after renal injury in mice. Kidney Int. 2018, 94, 524–535. [Google Scholar] [CrossRef]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef]
- Kalra, S.; Aydin, H.; Sahay, M.; Ghosh, S.; Ruder, S.; Tiwaskar, M.; Kilov, G.; Kishor, K.; Nair, T.; Makkar, V.; et al. Cardiorenal Syndrome in Type 2 Diabetes Mellitus—Rational Use of Sodium-glucose Cotransporter-2 Inhibitors. Eur. Endocrinol. 2020, 16, 113–121. [Google Scholar] [CrossRef]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef]
- Wan, N.; Rahman, A.; Hitomi, H.; Nishiyama, A. The Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Sympathetic Nervous Activity. Front. Endocrinol. 2018, 9, 421. [Google Scholar] [CrossRef]
- Gheorghiade, M.; Konstam, M.A.; Burnett, J.C.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: The EVEREST Clinical Status Trials. JAMA 2007, 297, 1332–1343. [Google Scholar] [CrossRef]
- Felker, G.M.; Mentz, R.J.; Cole, R.T.; Adams, K.F.; Egnaczyk, G.F.; Fiuzat, M.; Patel, C.B.; Echols, M.; Khouri, M.G.; Tauras, J.M.; et al. Efficacy and Safety of Tolvaptan in Patients Hospitalized With Acute Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 1399–1406. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.; Chandler, A.; Dhingra, R.; Mody, F.V.; Eisen, H.; Haught, W.H.; Wagoner, L.; Gupta, D.; Patten, R.; et al. Short-Term Effects of Tolvaptan in Patients With Acute Heart Failure and Volume Overload. J. Am. Coll. Cardiol. 2017, 69, 1409–1419. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Silverberg, D.S.; Iovine, F.; Calabrò, A.; Campagna, M.S.; Gallotta, M.; Nuti, R. Effects of β-erythropoietin treatment on left ventricular remodeling, systolic function, and B-type natriuretic peptide levels in patients with the cardiorenal anemia syndrome. Am. Hear. J. 2007, 154, 645.e9–645.e15. [Google Scholar] [CrossRef]
- Edwards, N.C.; Price, A.M.; Steeds, R.P.; Ferro, C.J.; Townend, J.N.; Birmingham Cardio-Renal Group. Management of heart failure in patients with kidney disease—Updates from the 2021 ESC guidelines. Nephrol. Dial. Transplant. 2023, gfad011. [Google Scholar] [CrossRef]
- Kiage, J.N.; Latif, Z.; Craig, M.A.; Mansour, N.; Khouzam, R.N. Implantable Cardioverter Defibrillators and Chronic Kidney Disease. Curr. Probl. Cardiol. 2021, 46, 100639. [Google Scholar] [CrossRef]
- Jukema, J.W.; Timal, R.J.; Rotmans, J.I.; Hensen, L.C.R.; Buiten, M.S.; de Bie, M.K.; Putter, H.; Zwinderman, A.H.; van Erven, L.; Krol-van Straaten, M.J.; et al. Prophylactic Use of Implantable Cardioverter-Defibrillators in the Prevention of Sudden Cardiac Death in Dialysis Patients. Circulation 2019, 139, 2628–2638. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Definition | Description | Clinical Conditions | Main Pathophysiological Mechanisms of Heart and Kidney Damage |
|---|---|---|---|
| Type 1 CRS (Acute CRS) | Acute HF resulting in AKI | AKI in the setting of acute HF or cardiogenic shock | Venous congestion, Renal hypoperfusion, SNS/RAAS activation, Oxidative stress, Inflammation. |
| Type 2 CRS (Chronic CRS) | Chronic HF resulting in CKD | CKD in the setting of chronic HF The diagnosis of CKD is based on the Improving Global Outcomes Kidney Disease Outcomes (KDIGO) Quality Initiative (KDOQI) criteria: - albuminuria and/or glomerular filtration rate (GFR) < 60 mL/in/1.73 m2; - sustained decrease in GFR > 5 mL/min/1.73 m2/year; - decline of GFR > 10 mL/min/1.73 m in 2/5 years; - sustained increase in albuminuria along with suspected diagnosis of congestive HF before the onset or progression of CKD. | SNS/RAAS activation, Fibrosis, Oxidative stress, Inflammation. |
| Type 3 CRS (Acute renocardiac syndrome) | AKI resulting in acute HF | Acute HF in the setting of AKI. Acute HF is linked to acute worsening of renal function with consequent electrolyte imbalance, metabolic acidosis, and volume overload. | Volume overload, SNS/RAAS activation, Oxidative stress/ mitochondrial dysfunction, Inflammation, Electrolyte disorders and metabolic disorders (due to uremic condition). |
| Type 4 CRS (Chronic renocardiac syndrome) | CKD resulting in chronic HF | LVH/dysfunction and chronic HF in the setting of CKD (Uremic Cardiomyopathy) CKD is related to accelerated atherosclerosis, insulin resistance, lipid dysmetabolism, neurohormonal imbalance and consequently to the development of CVD. | SNS/RAAS activation, Inflammation/fibrosis Hyperphosphatemia, Secondary hyperparathyroidism, Increased levels of circulating erythropoiesis inhibitors, furans, phenols, beta-2-microglobulin, leptin and polyols. |
| Type 5 CRS (Secondary CRS) | Systemic processes resulting in both HF and kidney damage | Amyloidosis, Autoimmune Diseases (SLE), Sepsis, COVID-19, Advanced liver diseases, Hepatorenal syndrome, Cirrhosis. | Inflammatory and prothrombotic states, Secretion of proinflammatory Cytokines, Endothelial dysfunction, Impaired coronary and glomerular autoregulation. |
| Biomarkers | Characteristics | Clinical Utility |
|---|---|---|
| Cardiac Biomarkers | CRS Type | |
| cTn | Marker of myocardial injury that correlates with ventricular remodeling in HF. | 1, 2 |
| Natriuretic peptides | Markers of myocardial increased wall stretch. They are the most used and recognized biomarkers in chronic and acute HF and are also raised in patients with CKD. | 1, 2, 3, 4, 5 |
| sST2 | sST2 is a member of the IL-1 receptor family that affects the activation of Th2 cells and the production of Th2-related cytokines. sST2 correlates with cardiovascular events and mortality in patients with AHF and CHF. Moreover, sST2 correlates with the development of CKD, as well as to the risk of CV events and HF development in patients with renal dysfunction. | 2, 5 |
| Galectin-3 | It is a component of the beta-galactosidase-binding lectin family, it is released by activated macrophages and induces the activation and deposition of collagen in the extracellular matrix promoting fibrosis at renal and cardiac level. Patients with elevated Galectin-3 levels showed an accelerated decline of GFR. | 2, 4, 5 |
| VEGF | Involved in the regulation of endothelial function and angiopoiesis; it may affect myocardial afterload. It is elevated in patients with HF. | 2, 4, 5 |
| PDGF | Involved in regulation of myocardial and kidney fibrosis. It is elevated in patients with HF. | 2, 4, 5 |
| sFlt-1 | Soluble VEGF receptor associated with microvascular disease and impaired angiopoiesis. It is elevated in patients with HF. | 2, 4, 5 |
| Copeptin | It is considered a marker of activated hypothalamus pituitary-adrenals axis. There is evidence that copeptin could be associated with CVD in patients with CKD, as well as it could be considered as a marker of AHF and AKI. | 1, 2, 3 4, 5 |
| MR-proadrenomedullin | Involved in regulation of vascular leakage; it predicts the decline of renal function and morbidity in patients with HF. | 2, 4, 5 |
| Kidney biomarkers | ||
| Serum creatinine | Produced by skeletal muscle, its clearance its representative of renal function. | 1, 2, 3, 4, 5 |
| CysC | Cysteine proteinase inhibitor filtered through the glomerulus and then reabsorbed by tubular cells. It is an accurate surrogate marker of GFR. | 1, 2, 3, 4, 5 |
| Albuminuria | Marker of glomerular integrity/PCT function. | 2, 4 |
| TIMP/IGFBP7 | Involved in G1 cell cycle arrest; it increases in tubular cell injury as an early marker. | 1, 3, 5 |
| NGAL | Small protein freely filtered through the glomerulus and reabsorbed in the proximal tubule. It increases in case of tubular damage 24 h before the rise of creatinine. | 1, 3, 5 |
| NAG | NAG is a lysosomal protein excreted into urine in case of tubular damage. NAG is increased in patients with AKI, CKD, or HF and may predict prognosis in these patients. | 1, 3, 5 |
| KIM1 | KIM1 is expressed in proximal tubule cells after hypoxic injury and may identify the development of AKI or CKD in patients with HF. KIM1 is associated with HF, cardiovascular events, and deaths in patients with AKI and CKD. | 1, 3, 5 |
| IL-18 | It is a component of NLEP3 inflammasome, and it is elevated in AKI. IL-18 levels are also increased in HF. | 1, 3, 5 |
| L-FABP | L-FABP is expressed in tubular epithelial cells and is excreted into urine with cytotoxic lipids. Urinary L-FABP has been associated with ischemic tubular injury and risk for acute kidney failure in type 1 CRS. | 1, 3 |
| α-1 Microglobulin | It is filtered by glomerulus and is completely reabsorbed by the renal tubule. It can be found in urine in case of tubule damage. | 2, 4, 5 |
| Drugs | Mechanism of Action | Side Effects/Contraindications | Clinical Use |
|---|---|---|---|
| Beta Blockers | Beta adrenergic Receptor Antagonism | Bradycardia, AVB; asthma/bronchospasm, hypotension, unstable HF. | Predominantly in chronic HF with reduced and mid-range ejection fraction without cardiogenic shock to improve morbidity and mortality. (CRS 2-4) |
| ACEi/ARB | Inhibition of ACE or AT1 receptor antagonism | Hyperkalemia, Renal failure, Hypotension, Idiopathic angio-oedema; Pregnancy, breastfeeding | Predominantly in chronic HF with reduced and mid-range ejection fraction without cardiogenic Shock to improve morbidity and mortality. (CRS 2-4) |
| ARNI | AT1/Neprilysin Inhibitor | Hyperkalemia, Renal Failure, Hypotension | Predominantly in chronic heart failure with reduced and mid-range ejection fraction without cardiogenic Shock to improve morbidity and mortality. (CRS 2-4) |
| MRA | Antagonism of mineralocorticoid receptor | Hyperkalemia, Renal Failure, Hypotension | Predominantly in chronic heart failure with reduced and mid-range ejection fraction without cardiogenic Shock to improve morbidity and mortality. (CRS 2-4) |
| SGLT2i | Antagonism of the cotransporter SLC5A2 in the PT1 | Type 1 diabetes mellitus, Acute Metabolic Acidosis | Predominantly in chronic heart failure with reduced, ejection fraction without cardiogenic Shock to improve morbidity and mortality. (CRS 2-4) |
| Diuretics | NKCC, NCC, CA Antagonism | Hypotension, Hypokalemia, Hypo-/Hypercalcemia, Hyponatremia, Hypochloremia, Hypovolemia, Metabolic Alkalosis, Diuretic Resistance. | In Acute and Chronic heart failure with reduced, mid-range and preserved ejection fraction to improve symptoms and volume overload. (CRS 1-2-4) In acute and chronic kidney disease to maintain an effective diuresis (CRS 1-3-4) |
| Vaptans | Selective Antagonism of V2 Receptor | Pollakiuria, Nycturia, polydipsia, Hypernatremia, Signs of Liver Injury. | Advanced Heart Failure with hyponatremia (CRS 1-3) |
| Inotropic Drugs | Beta receptor agonism, Calcium sensitizers | Supraventricular and ventricular arrhythmias, increased myocardial oxygen consumption due to increased myocardial work, hypotension. | Acute heart failure and cardiogenic Shock (CRS 1) |
| UF/CRRT | Convection/Diffusion fluid and solute removal for the improvement of volume, osmolite and water balance | Hypotension, hypovolemia, reduced pre-load, thrombus formation, bleedings, vascular complications of the access site | Acute heart failure and cardiogenic shock in patients with volume overload in the absence of proper diuretic response (CRS 1-3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, G.; Lanza, O.; Savoia, C. New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. Int. J. Mol. Sci. 2023, 24, 5089. https://doi.org/10.3390/ijms24065089
Gallo G, Lanza O, Savoia C. New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. International Journal of Molecular Sciences. 2023; 24(6):5089. https://doi.org/10.3390/ijms24065089
Chicago/Turabian StyleGallo, Giovanna, Oreste Lanza, and Carmine Savoia. 2023. "New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy" International Journal of Molecular Sciences 24, no. 6: 5089. https://doi.org/10.3390/ijms24065089
APA StyleGallo, G., Lanza, O., & Savoia, C. (2023). New Insight in Cardiorenal Syndrome: From Biomarkers to Therapy. International Journal of Molecular Sciences, 24(6), 5089. https://doi.org/10.3390/ijms24065089