
An 8q24 Gain in Pancreatic Juice Is a Candidate Biomarker for the Detection of Pancreatic Cancer
 , , and
, , and 
Abstract
:1. Introduction
2. Results
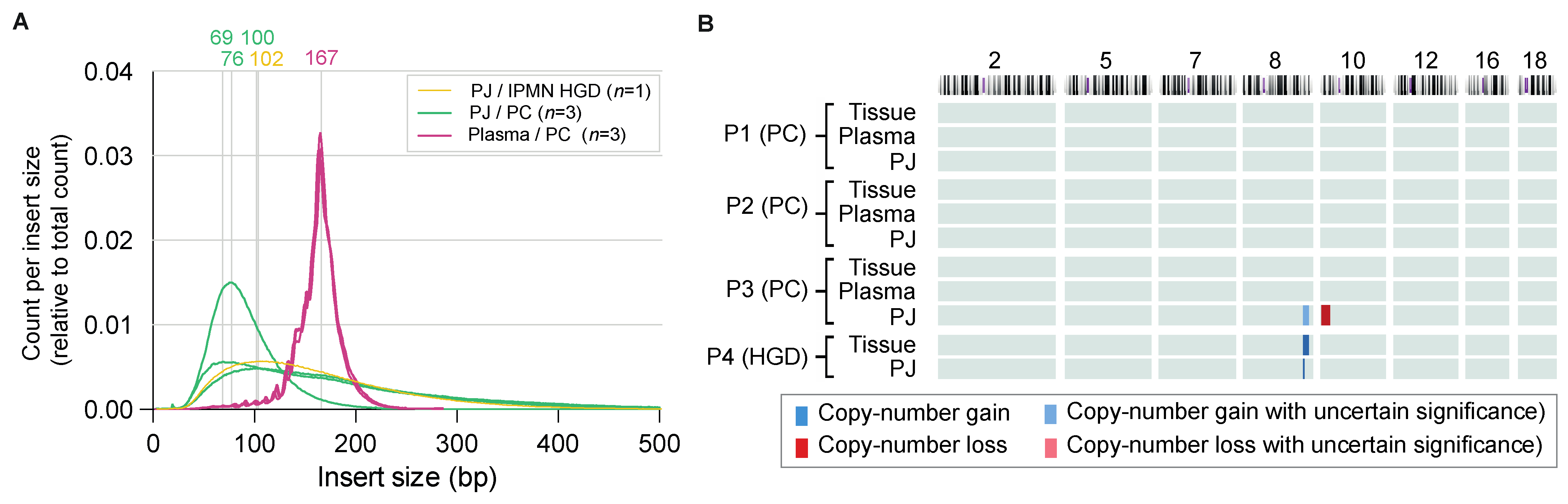
2.1. Feasibility Phase
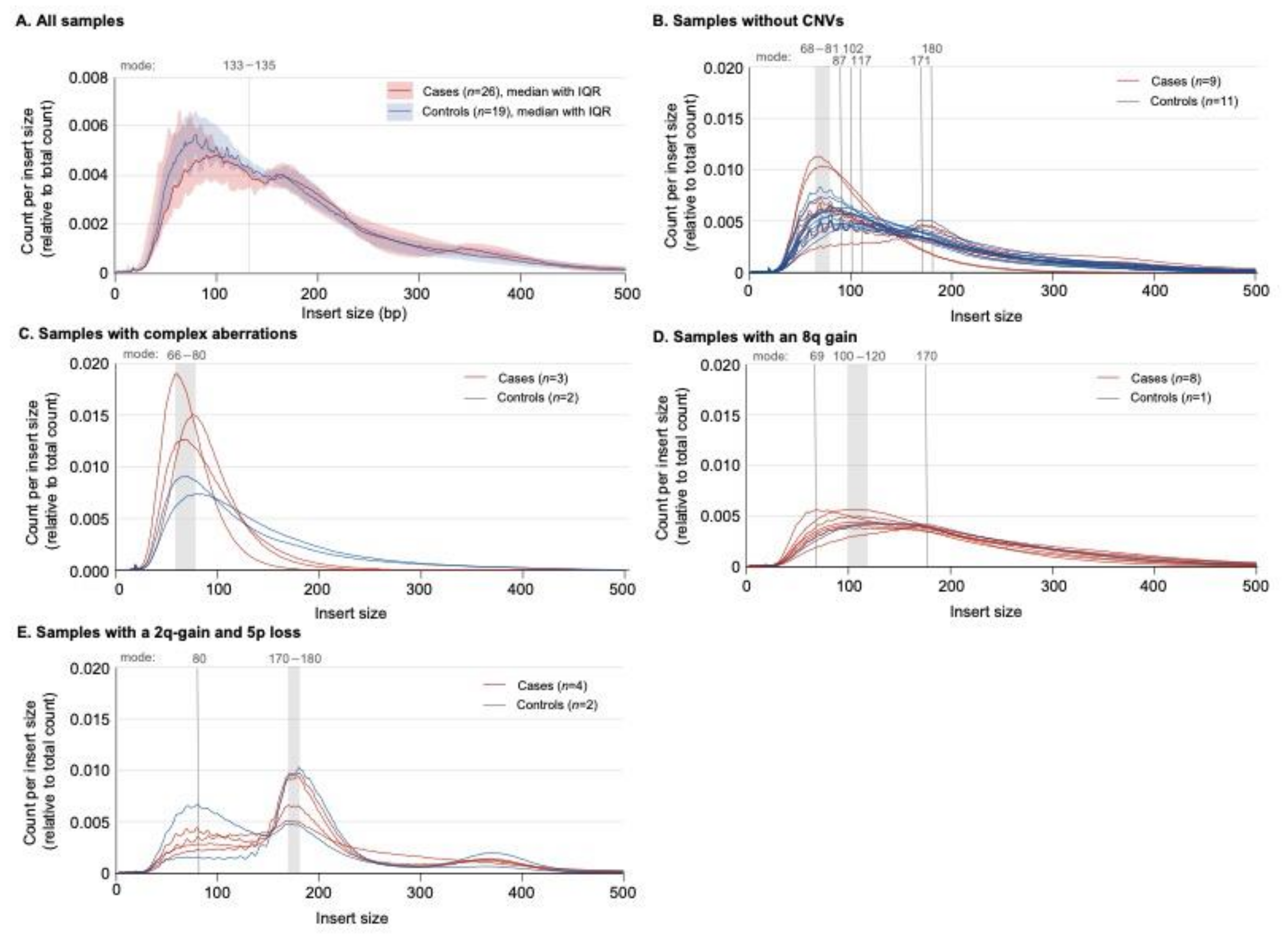
2.2. Experimental Phase
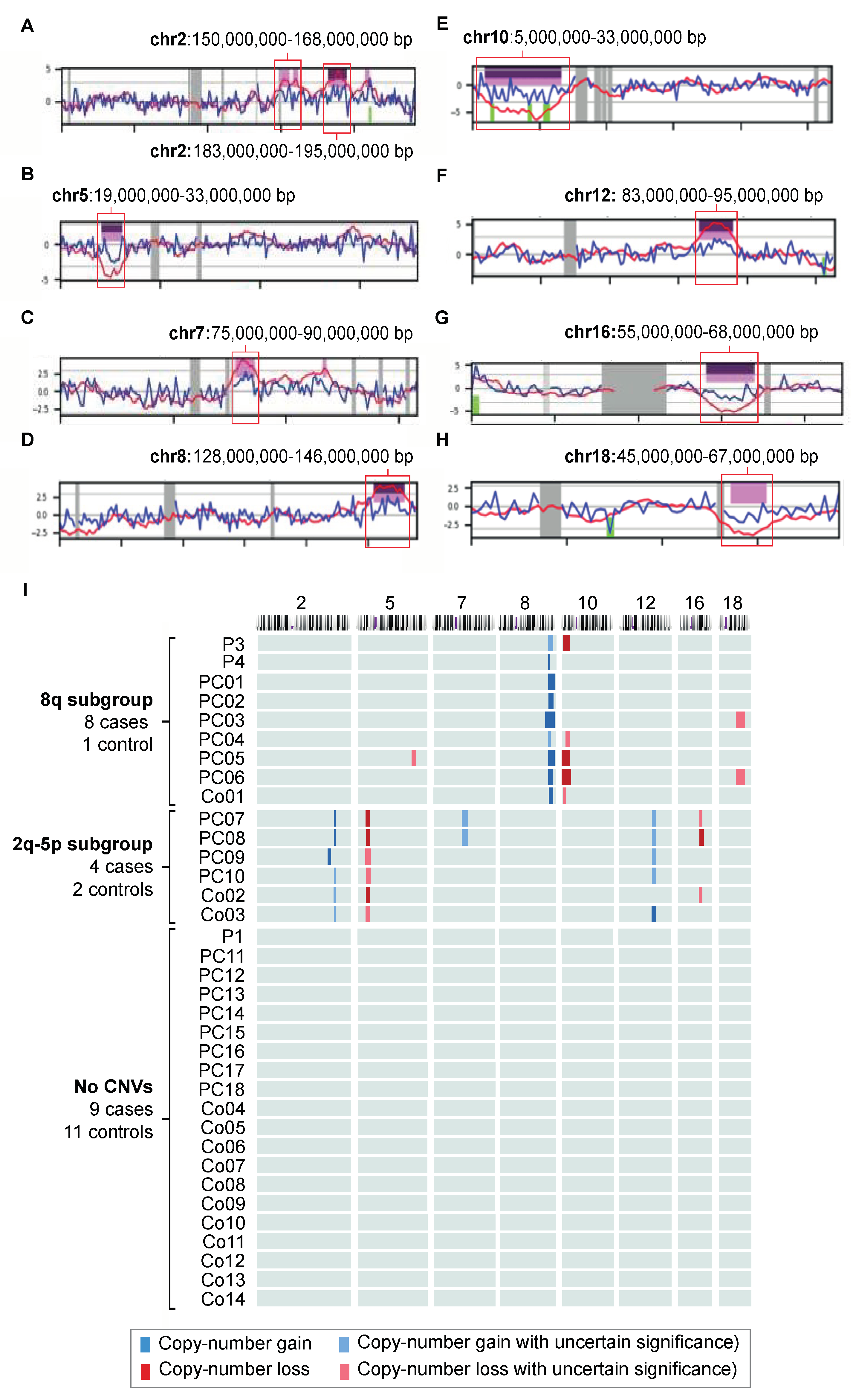
2.3. Copy Number Variants in Pancreatic Juice
2.3.1. The 8q24 Subgroup
2.3.2. The 2q-5p Subgroup
3. Discussion
4. Materials and Methods
4.1. Study Design and Patient Inclusion
4.2. Biomaterial Collection
4.3. cfDNA Analysis
4.4. Microarray Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | area under the curve |
| Bp | base pairs |
| CA19-9 | carbohydrate antigen 19.9 |
| cfDNA | cell-free DNA |
| ctDNA | circulating tumor DNA |
| CI | confidence interval |
| CNV | copy number variant |
| DNA | deoxyribonucleic acid |
| EUS | endoscopic ultrasound |
| ERP | endoscopic retrograde pancreatography |
| FNB | fine-needle biopsy |
| HGD | high-grade dysplasia |
| IPMN | intraductal papillary mucinous neoplasm |
| IQR | interquartile range |
| LGD | low-grade dysplasia |
| MCN | mucinous cystic neoplasm |
| MPD | main pancreatic duct |
| mtDNA | mitochondrial DNA |
| NIPT | noninvasive prenatal testing |
| NA | not applicable |
| PC | pancreatic cancer |
| PJ | pancreatic juice |
| ROC | receiver operating curve |
| SeqFF | fetal DNA fraction |
References
- Overbeek, K.A.; Levink, I.J.M.; Koopmann, B.D.M.; Harinck, F.; Konings, I.; Ausems, M.; Wagner, A.; Fockens, P.; van Eijck, C.H.; Groot Koerkamp, B.; et al. Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 2022, 71, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Wen, C.; Zhao, Y.; Fang, L.; Jin, Y.; Zhang, Z.; Zou, S.; Li, F.; Yang, Y.; Wu, L.; et al. Identification of copy number variation-driven molecular subtypes informative for prognosis and treatment in pancreatic adenocarcinoma of a Chinese cohort. eBioMedicine 2021, 74, 103716. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2021, 124, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Bonner, E.R.; Harrington, R.; Eze, A.; Bornhorst, M.; Kline, C.N.; Gordish-Dressman, H.; Dawood, A.; Das, B.; Chen, L.; Pauly, R.; et al. Circulating tumor DNA sequencing provides comprehensive mutation profiling for pediatric central nervous system tumors. npj Precis. Oncol. 2022, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Amant, F.; Verheecke, M.; Wlodarska, I.; Dehaspe, L.; Brady, P.; Brison, N.; Van Den Bogaert, K.; Dierickx, D.; Vandecaveye, V.; Tousseyn, T.; et al. Presymptomatic Identification of Cancers in Pregnant Women During Noninvasive Prenatal Testing. JAMA Oncol. 2015, 1, 814–819. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, D.W.; Chudova, D.; Sehnert, A.J.; Bhatt, S.; Murray, K.; Prosen, T.L.; Garber, J.E.; Wilkins-Haug, L.; Vora, N.L.; Warsof, S.; et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA 2015, 314, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Moellgaard, M.H.; Lund, I.C.B.; Becher, N.; Skytte, A.B.; Andreasen, L.; Srebniak, M.I.; Vogel, I. Incidental finding of maternal malignancy in an unusual non-invasive prenatal test and a review of similar cases. Clin. Case Rep. 2022, 10, e6280. [Google Scholar] [CrossRef]
- Heesterbeek, C.J.; Aukema, S.M.; Galjaard, R.H.; Boon, E.M.J.; Srebniak, M.I.; Bouman, K.; Faas, B.H.W.; Govaerts, L.C.P.; Hoffer, M.J.V.; den Hollander, N.S.; et al. Noninvasive Prenatal Test Results Indicative of Maternal Malignancies: A Nationwide Genetic and Clinical Follow-Up Study. J. Clin. Oncol. 2022, 40, 2426–2435. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. S5), v56–v68. [Google Scholar] [CrossRef]
- Levink, I.J.M.; Nesteruk, K.; Visser, D.I.; Sieuwerts, A.M.; Fernandes, C.J.C.; Jansen, M.; van Driel, L.; Poley, J.W.; Peppelenbosch, M.P.; Cahen, D.L.; et al. Optimization of Pancreatic Juice Collection: A First Step Toward Biomarker Discovery and Early Detection of Pancreatic Cancer. Am. J. Gastroenterol. 2020, 115, 2103–2108. [Google Scholar] [CrossRef]
- Klug, A.; Lutter, L.C. The helical periodicity of DNA on the nucleosome. Nucleic Acids Res. 1981, 9, 4267–4283. [Google Scholar] [CrossRef] [Green Version]
- Han, D.S.C.; Ni, M.; Chan, R.W.Y.; Chan, V.W.H.; Lui, K.O.; Chiu, R.W.K.; Lo, Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020, 106, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Schleger, C.; Verbeke, C.; Hildenbrand, R.; Zentgraf, H.; Bleyl, U. c-MYC Activation in Primary and Metastatic Ductal Adenocarcinoma of the Pancreas: Incidence, Mechanisms, and Clinical Significance. Mod. Pathol. 2002, 15, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Raimondo, M.; Guha, S.; Chen, J.; Diao, L.; Dong, X.; Wallace, M.B.; Killary, A.M.; Frazier, M.L.; Woodward, T.A.; et al. Circulating microRNAs in Pancreatic Juice as Candidate Biomarkers of Pancreatic Cancer. J. Cancer 2014, 5, 696–705. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, E.M.; Galvan, A.M.; Imamura-Kawasawa, Y.; Moldovan, G.L.; Nicolae, C.M. PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 2018, 46, 8908–8916. [Google Scholar] [CrossRef] [Green Version]
- Teng, K.-Y.; Mansour, A.G.; Zhu, Z.; Li, Z.; Tian, L.; Ma, S.; Xu, B.; Lu, T.; Chen, H.; Hou, D.; et al. Off-the-Shelf Prostate Stem Cell Antigen–Directed Chimeric Antigen Receptor Natural Killer Cell Therapy to Treat Pancreatic Cancer. Gastroenterology 2022, 162, 1319–1333. [Google Scholar] [CrossRef]
- Bausch, D.; Thomas, S.; Mino-Kenudson, M.; Fernández-del, C.C.; Bauer, T.W.; Williams, M.; Warshaw, A.L.; Thayer, S.P.; Kelly, K.A. Plectin-1 as a novel biomarker for pancreatic cancer. Clin. Cancer Res. 2011, 17, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Wernitznig, D.; Meier-Menches, S.M.; Cseh, K.; Theiner, S.; Wenisch, D.; Schweikert, A.; Jakupec, M.A.; Koellensperger, G.; Wernitznig, A.; Sommergruber, W.; et al. Plecstatin-1 induces an immunogenic cell death signature in colorectal tumour spheroids. Metallomics 2020, 12, 2121–2133. [Google Scholar] [CrossRef]
- Liang, W.; Liao, Y.; Zhang, J.; Huang, Q.; Luo, W.; Yu, J.; Gong, J.; Zhou, Y.; Li, X.; Tang, B.; et al. Heat shock factor 1 inhibits the mitochondrial apoptosis pathway by regulating second mitochondria-derived activator of caspase to promote pancreatic tumorigenesis. J. Exp. Clin. Cancer Res. 2017, 36, 64. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Gökmen-Polar, Y. HSF1 as a Cancer Biomarker and Therapeutic Target. Curr. Cancer Drug Targets 2019, 19, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Jakubison, B.L.; Schweickert, P.G.; Moser, S.E.; Yang, Y.; Gao, H.; Scully, K.; Itkin-Ansari, P.; Liu, Y.; Konieczny, S.F. Induced PTF1a expression in pancreatic ductal adenocarcinoma cells activates acinar gene networks, reduces tumorigenic properties, and sensitizes cells to gemcitabine treatment. Mol. Oncol. 2018, 12, 1104–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratyeva, L.G.; Chernov, I.P.; Zinovyeva, M.V.; Kopantzev, E.P.; Sverdlov, E.D. Expression of master regulatory genes of embryonic development in pancreatic tumors. Dokl. Biochem. Biophys. 2017, 475, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Krah, N.M.; De La, O.J.; Swift, G.H.; Hoang, C.Q.; Willet, S.G.; Chen Pan, F.; Cash, G.M.; Bronner, M.P.; Wright, C.V.; MacDonald, R.J.; et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. eLife 2015, 4, e07125. [Google Scholar] [CrossRef]
- Campos, M.L.; Sánchez-Arévalo Lobo, V.J.; Rodolosse, A.; Gottardi, C.J.; Mafficini, A.; Beghelli, S.; Scardoni, M.; Bassi, C.; Scarpa, A.; Real, F.X. ICAT is a novel Ptf1a interactor that regulates pancreatic acinar differentiation and displays altered expression in tumours. Biochem. J. 2013, 451, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 7101–7106. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, A.; Morris, J.P.t.; Hebrok, M. Bmi1 is required for regeneration of the exocrine pancreas in mice. Gastroenterology 2012, 143, 821–831.e822. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Tschen, S.I.; Bhushan, A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009, 23, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Bednar, F.; Schofield, H.K.; Collins, M.A.; Yan, W.; Zhang, Y.; Shyam, N.; Eberle, J.A.; Almada, L.L.; Olive, K.P.; Bardeesy, N.; et al. Bmi1 is required for the initiation of pancreatic cancer through an Ink4a-independent mechanism. Carcinogenesis 2015, 36, 730–738. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Barberá, V.M.; Martín, M.; Mariñoso, L.; Munné, A.; Carrato, A.; Real, F.X.; Fabre, M. The 18q21 region in colorectal and pancreatic cancer: Independent loss of DCC and DPC4 expression. Biochim. Biophys. Acta 2000, 1502, 283–296. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Wu, E.Y.; Kim, W.G.; Dillon, D.A.; Hirsch, M.S.; Sholl, L.M.; Agoston, A.T.; Setia, N.; Lauwers, G.Y.; Park, D.Y.; et al. Loss of SMAD4 protein expression in gastrointestinal and extra-gastrointestinal carcinomas. Histopathology 2019, 75, 546–551. [Google Scholar] [CrossRef]
- Wang, J.D.; Jin, K.; Chen, X.Y.; Lv, J.Q.; Ji, K.W. Clinicopathological significance of SMAD4 loss in pancreatic ductal adenocarcinomas: A systematic review and meta-analysis. Oncotarget 2017, 8, 16704–16711. [Google Scholar] [CrossRef] [Green Version]
- Hemann, M.T.; Lowe, S.W. The p53-Bcl-2 connection. Cell Death Differ. 2006, 13, 1256–1259. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Wang, B.; Gu, S.; Li, X.; Sun, S. Expression of Beclin 1 and Bcl-2 in pancreatic neoplasms and its effect on pancreatic ductal adenocarcinoma prognosis. Oncol. Lett. 2017, 14, 7849–7861. [Google Scholar] [CrossRef]
- Evans, J.D.; Cornford, P.A.; Dodson, A.; Greenhalf, W.; Foster, C.S.; Neoptolemos, J.P. Detailed tissue expression of bcl-2, bax, bak and bcl-x in the normal human pancreas and in chronic pancreatitis, ampullary and pancreatic ductal adenocarcinomas. Pancreatology 2001, 1, 254–262. [Google Scholar] [CrossRef]
- An, X.Z.; Zhao, Z.G.; Luo, Y.X.; Zhang, R.; Tang, X.Q.; Hao, D.; Zhao, X.; Lv, X.; Liu, D. Netrin-1 suppresses the MEK/ERK pathway and ITGB4 in pancreatic cancer. Oncotarget 2016, 7, 24719–24733. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Yu, D.H.; Chen, Y.; Zhao, C.Y.; Zhang, J.; Liu, Q.H.; Ni, C.R.; Zhu, M.H. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J. Gastroenterol. 2012, 18, 840–846. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2, e92232. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, F.; Liu, F.; Xu, G. Predicting STAT1 as a prognostic marker in patients with solid cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920917558. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, S.; Sun, N.; Chen, J. Differential expression of STAT1 and p21 proteins predicts pancreatic cancer progression and prognosis. Pancreas 2014, 43, 619–623. [Google Scholar] [CrossRef]
- Jinawath, N.; Shiao, M.S.; Norris, A.; Murphy, K.; Klein, A.P.; Yonescu, R.; Iacobuzio-Donahue, C.; Meeker, A.; Jinawath, A.; Yeo, C.J.; et al. Alterations of type II classical cadherin, cadherin-10 (CDH10), is associated with pancreatic ductal adenocarcinomas. Genes Chromosomes Cancer 2017, 56, 427–435. [Google Scholar] [CrossRef]
- Xu, Z.; Pang, T.C.Y.; Liu, A.C.; Pothula, S.P.; Mekapogu, A.R.; Perera, C.J.; Murakami, T.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: A key element of treatment that limits primary tumour growth and eliminates metastasis. Br. J. Cancer 2020, 122, 1486–1495. [Google Scholar] [CrossRef]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Biankin, A.V.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Hepatocyte growth factor inhibition: A novel therapeutic approach in pancreatic cancer. Br. J. Cancer 2016, 114, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Kidger, A.M.; Saville, M.K.; Rushworth, L.K.; Davidson, J.; Stellzig, J.; Ono, M.; Kuebelsbeck, L.A.; Janssen, K.-P.; Holzmann, B.; Morton, J.P.; et al. Suppression of mutant Kirsten-RAS (KRASG12D)-driven pancreatic carcinogenesis by dual-specificity MAP kinase phosphatases 5 and 6. Oncogene 2022, 41, 2811–2823. [Google Scholar] [CrossRef]
- Guo, F.; Cheng, X.; Jing, B.; Wu, H.; Jin, X. FGD3 binds with HSF4 to suppress p65 expression and inhibit pancreatic cancer progression. Oncogene 2022, 41, 838–851. [Google Scholar] [CrossRef]
- Franko, J.; Krasinskas, A.M.; Nikiforova, M.N.; Zarnescu, N.O.; Lee, K.K.; Hughes, S.J.; Bartlett, D.L.; Zeh, H.J., 3rd; Moser, A.J. Loss of heterozygosity predicts poor survival after resection of pancreatic adenocarcinoma. J. Gastrointest. Surg. 2008, 12, 1664–1672, discussion 1672–1663. [Google Scholar] [CrossRef]
- Kluzek, K.; Srebniak, M.I.; Majer, W.; Ida, A.; Milecki, T.; Huminska, K.; van der Helm, R.M.; Silesian, A.; Wrzesinski, T.M.; Wojciechowicz, J.; et al. Genetic characterization of Polish ccRCC patients: Somatic mutation analysis of PBRM1, BAP1 and KDMC5, genomic SNP array analysis in tumor biopsy and preliminary results of chromosome aberrations analysis in plasma cell free DNA. Oncotarget 2017, 8, 28558–28574. [Google Scholar] [CrossRef]
- Chi, J.; Chung, S.Y.; Parakrama, R.; Fayyaz, F.; Jose, J.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Therap. Adv. Gastroenterol. 2021, 14, 17562848211014818. [Google Scholar] [CrossRef]
- Emelyanova, M.; Pudova, E.; Khomich, D.; Krasnov, G.; Popova, A.; Abramov, I.; Mikhailovich, V.; Filipenko, M.; Menshikova, S.; Tjulandin, S.; et al. Platinum-based chemotherapy for pancreatic cancer: Impact of mutations in the homologous recombination repair and Fanconi anemia genes. Ther. Adv. Med. Oncol. 2022, 14, 17588359221083050. [Google Scholar] [CrossRef]
- Wang, M.; Lu, X.; Dong, X.; Hao, F.; Liu, Z.; Ni, G.; Chen, D. pERK1/2 silencing sensitizes pancreatic cancer BXPC-3 cell to gemcitabine-induced apoptosis via regulating Bax and Bcl-2 expression. World J. Surg. Oncol. 2015, 13, 66. [Google Scholar] [CrossRef] [Green Version]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [Green Version]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Nakahira, K.; Guo, X.; Choi, A.M.K.; Gu, Z. Very Short Mitochondrial DNA Fragments and Heteroplasmy in Human Plasma. Sci. Rep. 2016, 6, 36097. [Google Scholar] [CrossRef] [Green Version]
- Mair, R.; Mouliere, F.; Smith, C.G.; Chandrananda, D.; Gale, D.; Marass, F.; Tsui, D.W.Y.; Massie, C.E.; Wright, A.J.; Watts, C.; et al. Measurement of Plasma Cell-Free Mitochondrial Tumor DNA Improves Detection of Glioblastoma in Patient-Derived Orthotopic Xenograft Models. Cancer Res. 2019, 79, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Hata, T.; Suenaga, M.; Marchionni, L.; Macgregor-Das, A.; Yu, J.; Shindo, K.; Tamura, K.; Hruban, R.H.; Goggins, M. Genome-Wide Somatic Copy Number Alterations and Mutations in High-Grade Pancreatic Intraepithelial Neoplasia. Am. J. Pathol. 2018, 188, 1723–1733. [Google Scholar] [CrossRef] [Green Version]
- Schleger, C.; Arens, N.; Zentgraf, H.; Bleyl, U.; Verbeke, C. Identification of frequent chromosomal aberrations in ductal adenocarcinoma of the pancreas by comparative genomic hybridization (CGH). J. Pathol. 2000, 191, 27–32. [Google Scholar] [CrossRef]
- Mateos, R.N.; Nakagawa, H.; Hirono, S.; Takano, S.; Fukasawa, M.; Yanagisawa, A.; Yasukawa, S.; Maejima, K.; Oku-Sasaki, A.; Nakano, K.; et al. Genomic analysis of pancreatic juice DNA assesses malignant risk of intraductal papillary mucinous neoplasm of pancreas. Cancer Med. 2019, 8, 4565–4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Hannum, G.; Geis, J.; Tynan, J.; Hogg, G.; Zhao, C.; Jensen, T.J.; Mazloom, A.R.; Oeth, P.; Ehrich, M.; et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat. Diagn. 2015, 35, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Straver, R.; Sistermans, E.A.; Holstege, H.; Visser, A.; Oudejans, C.B.; Reinders, M.J. WISECONDOR: Detection of fetal aberrations from shallow sequencing maternal plasma based on a within-sample comparison scheme. Nucleic Acids Res. 2014, 42, e31. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Cases (n = 26) | Controls (n = 19) | p-Value | |
|---|---|---|---|
| Age, median (IQR) | 69 (9) | 60 (9) | <0.001 |
| Sex, n male (%) | 18 (69) | 7 (37) | 0.04 |
| BMI, median in kg/m2 (IQR) | 23 (4) | 24 (5) | 0.23 |
| Diabetes mellitus, n present (%) | 11 (42) | 0 (0) | <0.001 |
| Hereditary predisposition, n (%) | 0 (0) | 19 (100) | <0.001 |
| Member of FPC family | 7 (16) | ||
| CDKN2A germline mutation | 8 (18) | ||
| BRCA1/2 germline mutation | 3 (7) | ||
| PALB2 germline mutation | 1 (2) | ||
| History of malignancy, n (%) | 3 (12) | 8 (42) | 0.02 |
| Breast cancer | 0 (0) | 1 (5) | |
| Melanoma | 0 (0) | 7 (37) | |
| Other | 3 (12) | 0 (0) | |
| Any symptom, n (%) | 21 (81) | 0 (0) | <0.001 |
| Jaundice | 8 (31) | ||
| Epigastric pain | 15 (58) | ||
| Weight loss | 11 (42) | ||
| CA19.9 >37 kU/L, n (%) | 19 (73) | NA | NA |
| Treatment-naive, n (%) | 23 (88) | NA | NA |
| Resectability of PC, n (%) | NA | NA | |
| Resectable | 8 (31) | ||
| Borderline resectable | 2 (8) | ||
| Locally advanced PC | 16 (62) | ||
| Location mass, n (%) | NA | NA | |
| Uncinate/head | 15 (58) | ||
| Neck/corpus | 8 (31) | ||
| Tail | 3 (12) | ||
| CBD stent in situ, n (%) | 7 (27) | NA | NA |
| Surgery, n (%) | 11 (42) | NA | NA |
| Pancreaticoduodenectomy | 8 (31) | ||
| Distal pancreatectomy | 3 (12) |
| Present/Total | AUC (95% CI) | Sensitivity in % | Specificity in % | Accuracy in % | |
|---|---|---|---|---|---|
| 8q-10p profile | 9/40 | 0.64 (0.46–0.81) | 33 (16–55) | 94 (70–100) | 57 (41–73) |
| 2q-5p profile | 6/40 | 0.52 (0.34–0.70) | 17 (5–38) | 87 (62–98) | 45 (29–62) |
| 8q-10p or 2q-5p | 15/40 | 0.66 (0.48–-0.83) | 50 (29–71) | 81 (54–96) | 63 (46–77) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levink, I.J.M.; Srebniak, M.I.; De Valk, W.G.; van Veghel-Plandsoen, M.M.; Wagner, A.; Cahen, D.L.; Fuhler, G.M.; Bruno, M.J. An 8q24 Gain in Pancreatic Juice Is a Candidate Biomarker for the Detection of Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 5097. https://doi.org/10.3390/ijms24065097
Levink IJM, Srebniak MI, De Valk WG, van Veghel-Plandsoen MM, Wagner A, Cahen DL, Fuhler GM, Bruno MJ. An 8q24 Gain in Pancreatic Juice Is a Candidate Biomarker for the Detection of Pancreatic Cancer. International Journal of Molecular Sciences. 2023; 24(6):5097. https://doi.org/10.3390/ijms24065097
Chicago/Turabian StyleLevink, Iris J. M., Malgorzata I. Srebniak, Walter G. De Valk, Monique M. van Veghel-Plandsoen, Anja Wagner, Djuna L. Cahen, Gwenny M. Fuhler, and Marco J. Bruno. 2023. "An 8q24 Gain in Pancreatic Juice Is a Candidate Biomarker for the Detection of Pancreatic Cancer" International Journal of Molecular Sciences 24, no. 6: 5097. https://doi.org/10.3390/ijms24065097
APA StyleLevink, I. J. M., Srebniak, M. I., De Valk, W. G., van Veghel-Plandsoen, M. M., Wagner, A., Cahen, D. L., Fuhler, G. M., & Bruno, M. J. (2023). An 8q24 Gain in Pancreatic Juice Is a Candidate Biomarker for the Detection of Pancreatic Cancer. International Journal of Molecular Sciences, 24(6), 5097. https://doi.org/10.3390/ijms24065097